RELMα Is Induced in Airway Epithelial Cells by Oncostatin M without Requirement of STAT6 or IL-6 in Mouse Lungs In Vivo



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Cell Culture
2.2. Airway Epithelial Cells
2.3. Sample Collection and Tissue Processing
2.4. Histology, Immunohistochemistry and Chromogenic In Situ Hybridization (CISH)
2.5. Flow Cytometry
2.6. Western Blot
2.7. Reverse Transcription Polymerase Chain Reaction (RT-PCR) / NanoString
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Statistical Analysis
3. Results
3.1. RELMα is Induced Upon Overexpression of OSM in Lungs of C57Bl/6 Mice and is Highly Expressed in Airway Epithelial Cells
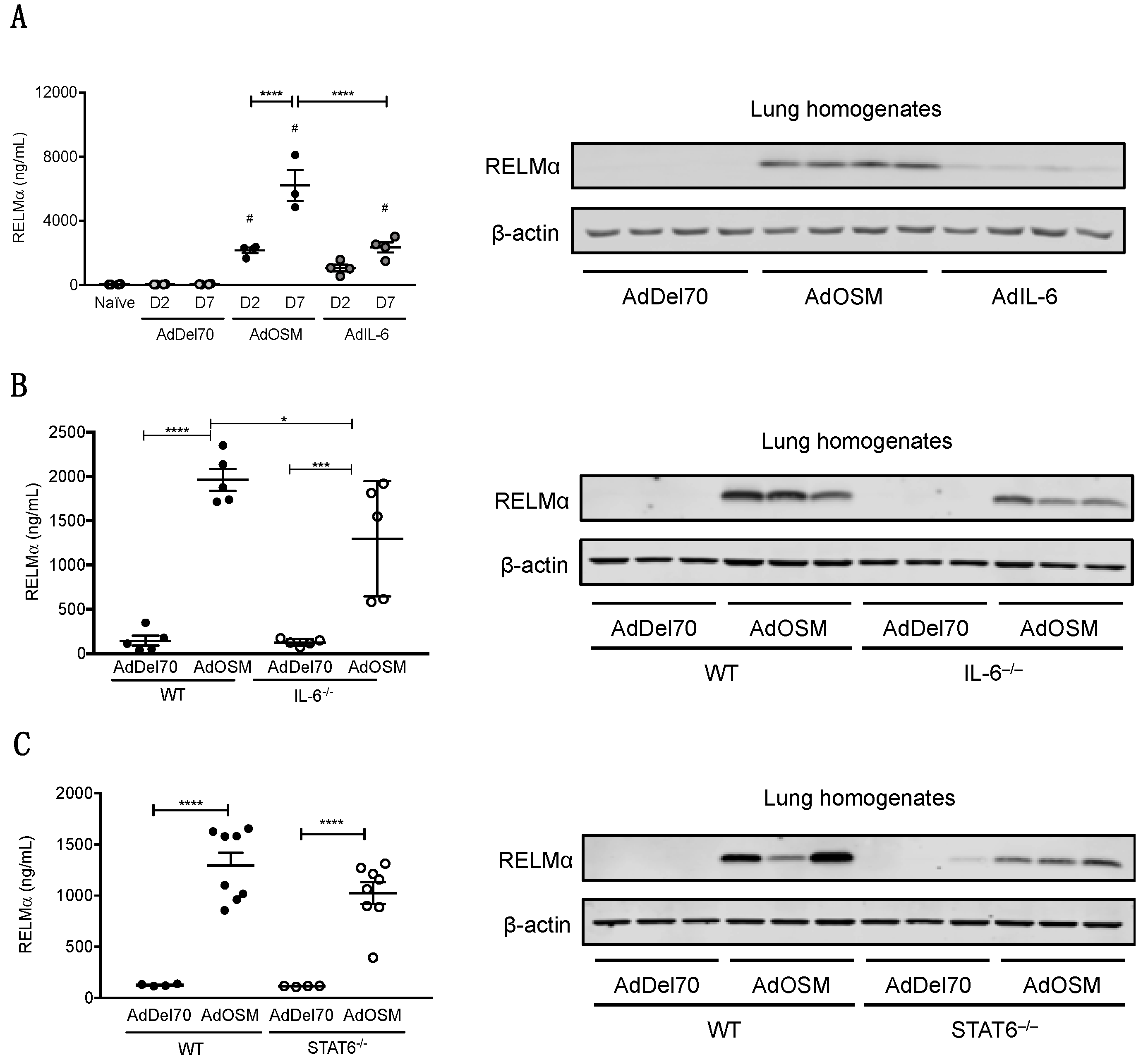
3.2. IL-6 and STAT3 are not Required for Airway Epithelial Cell Responses to OSM
3.3. YM-1 Is Induced by AdOSM
3.4. RELMα Supports Maximal CD206+ M2 Macrophage Numbers Induced by AdOSM
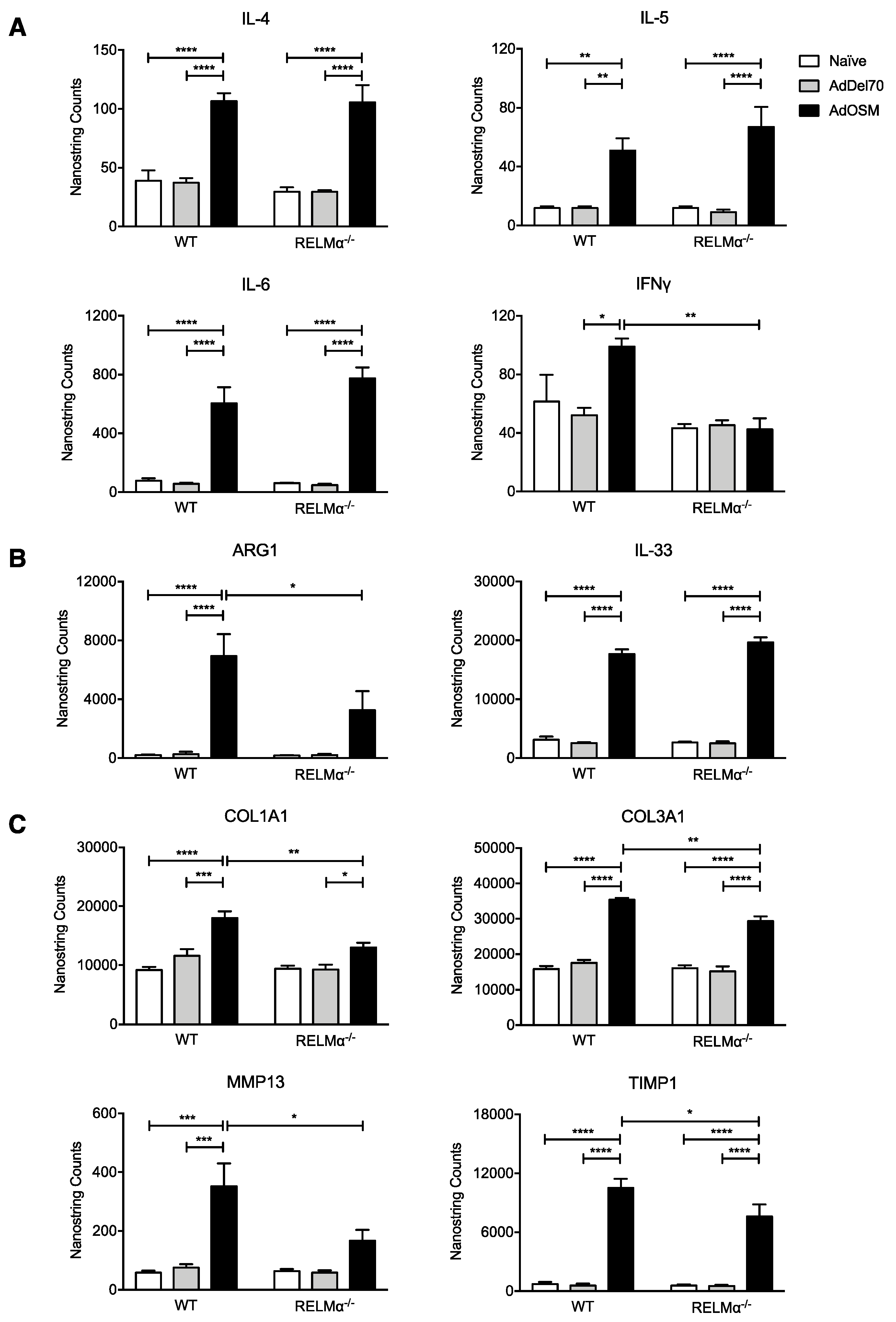
3.5. RELMα-Deficiency Does Not Alter Th2-Associated Cytokine Elevation but Reduces Arginase1 and Matrix Remodelling Gene Induction
3.6. RELMα-Deficiency Reduces Cell Proliferation and Parenchymal αSMA Accumulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burgess, J.K.; Mauad, T.; Tjin, G.; Karlsson, J.C.; Westergren-Thorsson, G. The extracellular matrix—the under-recognized element in lung disease? J. Pathol. 2016, 240, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Hogg, P.; Timens, W. The Pathology of Chronic Obstructive Pulmonary Disease. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Mauad, T.; Bel, E.H.; Sterk, P.J. Asthma therapy and airway remodeling. J. Allergy Clin. Immunol. 2007, 120, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- King, T.E. Idiopathic Pulmonary Fibrosis: Diagnosis and treatment. International consensus statement. Am J. Respir Crit Care Med. 2000, 161, 646–664. [Google Scholar]
- Wollin, L.; Bonella, F.; Stowasser, S. Idiopathic pulmonary fibrosis: Current treatment options and critical appraisal of nintedanib. Drug Des. Dev. Ther. 2015, 9, 6407–6419. [Google Scholar] [CrossRef] [PubMed]
- Re, S.L.; Lison, D.; Huaux, F. CD4+ T lymphocytes in lung fibrosis: Diverse subsets, diverse functions. J. Leukoc. Boil. 2013, 93, 499–510. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immun. 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Bonniaud, P.; Kolb, M.; Galt, T.; Robertson, J.; Robbins, C.; Stampfli, M.; Lavery, C.; Margetts, P.J.; Roberts, A.B.; Gauldie, J. Smad3 Null Mice Develop Airspace Enlargement and Are Resistant to TGF-β-Mediated Pulmonary Fibrosis. J. Immunol. 2004, 173, 2099–2108. [Google Scholar] [CrossRef]
- Migliaccio, C.T.; Buford, M.C.; Jessop, F.; Holian, A. The IL-4Rα pathway in macrophages and its potential role in silica-induced pulmonary fibrosis. J. Leukoc. Boil. 2007, 83, 630–639. [Google Scholar] [CrossRef]
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair 2012, 5, 11. [Google Scholar] [CrossRef]
- Mozaffarian, A.; Brewer, A.W.; Trueblood, E.S.; Luzina, I.G.; Todd, N.W.; Atamas, S.P.; Arnett, H.A. Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis. J. Immunol. 2008, 181, 7243–7253. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Miyahima, A. Onconstatin M, a multifunctional cytokine. Rev. Physiol. Biochem. Pharmacol. 2003, 149, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Botelho, F.M.; Rodrigues, R.M.; Richards, C. Oncostatin M overexpression induces matrix deposition, STAT3 activation, and SMAD1 Dysregulation in lungs of fibrosis-resistant BALB/c mice. Lab. Investig. 2014, 94, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, R.J.J.; Knight, D.A.; Richards, C.; Prele, C.M.; Lau, H.L.; Jarnicki, A.G.; Jones, J.; Bozinovski, S.; Vlahos, R.; Thiem, S.; et al. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 2012, 4, 939–951. [Google Scholar] [CrossRef]
- Hermanns, H. Oncostatin M and interleukin-31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev. 2015, 26, 545–558. [Google Scholar] [CrossRef]
- Richards, C. The Enigmatic Cytokine Oncostatin M and Roles in Disease. ISRN Inflamm. 2013, 2013, 1–23. [Google Scholar] [CrossRef]
- West, N. Coordination of Immune-Stroma Crosstalk by IL-6 Family Cytokines. Front. Immunol. 2019, 10, 1093. [Google Scholar] [CrossRef]
- Richards, C.; Izakelian, L.; Dubey, A.; Zhang, G.; Wong, S.; Kwofie, K.; Qureshi, A.; Botelho, F. Regulation of IL-33 by Oncostatin M in Mouse Lung Epithelial Cells. Mediat. Inflamm. 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- Simpson, J.L.; Baines, K.J.; Boyle, M.J.; Scott, R.J.; Gibson, P.G. Oncostatin M (OSM) is increased in asthma with incompletely reversible airflow obstruction. Exp. Lung Res. 2009, 35, 781–794. [Google Scholar] [CrossRef]
- Baines, K.J.; Simpson, J.L.; Gibson, P.G. Innate Immune Responses Are Increased in Chronic Obstructive Pulmonary Disease. PLoS ONE 2011, 6, e18426. [Google Scholar] [CrossRef]
- Pothoven, K.L.; Norton, J.E.; Hulse, K.; Suh, L.A.; Carter, R.G.; Rocci, E.; Harris, K.E.; Shintani-Smith, S.; Conley, D.B.; Chandra, R.K.; et al. Oncostatin M promotes mucosal epithelial barrier dysfunction, and its expression is increased in patients with eosinophilic mucosal disease. J. Allergy Clin. Immunol. 2015, 136, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.; Izakelian, L.; A Ayaub, E.; Ho, L.; Stephenson, K.; Wong, S.; Kwofie, K.; Austin, R.C.; Botelho, F.; Ask, K.; et al. Separate roles of IL-6 and oncostatin M in mouse macrophage polarizationin vitroandin vivo. Immunol. Cell Boil. 2017, 96, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, I.N.; Kabakoff, R.C.; Chan, B.; Baker, T.W.; Gurney, A.; Henzel, W.; Nelson, C.; Lowman, H.B.; Wright, B.D.; Skelton, N.J.; et al. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 2000, 19, 4046–4055. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Brown, E.J.; Wright, C.M.; Bhat, S.; Banerjee, R.R.; Dai, C.Y.; Enders, G.H.; Silberg, D.G.; Wen, X.; Wu, G.D.; et al. A family of tissue-specific resistin-like molecules. Proc. Natl. Acad. Sci. USA 2001, 98, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Munitz, A.; Waddell, A.; Seidu, L.; Cole, E.T.; Ahrens, R.; Hogan, S.P.; E Rothenberg, M. Resistin-like molecule α enhances myeloid cell activation and promotes colitis. J. Allergy Clin. Immunol. 2008, 122, 1200–1207. [Google Scholar] [CrossRef]
- Nair, M.G.; Du, Y.; Perrigoue, J.G.; Zaph, C.; Taylor, J.J.; Goldschmidt, M.; Swain, G.P.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; et al. Alternatively activated macrophage-derived RELM-α is a negative regulator of type 2 inflammation in the lung. J. Exp. Med. 2009, 206, 937–952. [Google Scholar] [CrossRef]
- Chen, F.; Wu, W.; Jin, L.; Millman, A.; Palma, M.; El-Naccache, D.; Lothstein, K.E.; Dong, C.; Edelblum, K.L.; Gause, W.C. B Cells Produce the Tissue-Protective Protein RELMα during Helminth Infection, which Inhibits IL-17 Expression and Limits Emphysema. Cell Rep. 2018, 25, 2775–2783. [Google Scholar] [CrossRef]
- Kumamoto, Y.; Camporez, J.P.G.; Jurczak, M.J.; Shanabrough, M.; Horvath, T.; Shulman, G.I.; Iwasaki, A. CD301b + Mononuclear Phagocytes Maintain Positive Energy Balance through Secretion of Resistin-like Molecule Alpha. Immun. 2016, 45, 583–596. [Google Scholar] [CrossRef]
- Liu, T.; Jin, H.; Ullenbruch, M.; Hu, B.; Hashimoto, N.; Moore, B.B.; McKenzie, A.; Lukacs, N.W.; Phan, S.H. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: Role of IL-4/IL-13 and mediation via STAT-6. J. Immunol. 2004, 173, 3425–3431. [Google Scholar] [CrossRef]
- Nair, M.G.; Cochrane, D.W.; Allen, J.E. Macrophages in chronic type 2 inflammation have a novel phenotype characterized by the abundant expression of Ym1 and Fizz1 that can be partly replicated in vitro. Immunol. Lett. 2003, 85, 173–180. [Google Scholar] [CrossRef]
- Khorram, N.; Sugimoto, K.; Sheppard, D.; Rosenthal, P.; Cho, J.; Pham, A.; Miller, M.; Zuraw, B.; Croft, M.; Broide, D.; et al. Alternaria Induces Stat-6 Dependent Acute Airway Eosinophilia And Epithelial Fizz1 Expression That Promotes Airway Fibrosis And Epithelial Thickness. J. Allergy Clin. Immunol. 2012, 129, AB54. [Google Scholar] [CrossRef]
- Lee, M.-R.; Shim, D.; Yoon, J.; Jang, H.S.; Oh, S.-W.; Suh, S.H.; Choi, J.-H.; Oh, G.T. Retnla Overexpression Attenuates Allergic Inflammation of the Airway. PLoS ONE 2014, 9, e112666. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yu, H.; Ullenbruch, M.; Jin, H.; Ito, T.; Wu, Z.; Liu, J.; Phan, S.H. The In Vivo Fibrotic Role of FIZZ1 in Pulmonary Fibrosis. PLoS ONE 2014, 9, e88362. [Google Scholar] [CrossRef] [PubMed]
- Knipper, J.A.; Willenborg, S.; Brinckmann, J.; Bloch, W.; Maaß, T.; Wagener, R.; Krieg, T.; Sutherland, T.E.; Munitz, A.; Rothenberg, M.E.; et al. Interleukin-4 Receptor α Signaling in Myeloid Cells Controls Collagen Fibril Assembly in Skin Repair. Immun. 2015, 43, 803–816. [Google Scholar] [CrossRef]
- Chen, G.; Wang, S.H.; Jang, J.; Odegaard, J.I.; Nair, M.G. Comparison of RELMα and RELMβ Single- and Double-Gene-Deficient Mice Reveals that RELMα Expression Dictates Inflammation and Worm Expulsion in Hookworm Infection. Infect. Immun. 2016, 84, 1100–1111. [Google Scholar] [CrossRef]
- Fritz, D.K.; Kerr, C.; Fattouh, R.; Llop-Guevara, A.; Khan, W.I.; Jordana, M.; Richards, C. A Mouse Model of Airway Disease: Oncostatin M-Induced Pulmonary Eosinophilia, Goblet Cell Hyperplasia, and Airway Hyperresponsiveness Are STAT6 Dependent, and Interstitial Pulmonary Fibrosis Is STAT6 Independent. J. Immunol. 2010, 186, 1107–1118. [Google Scholar] [CrossRef]
- Botelho, F.M.; Rangel-Moreno, J.; Fritz, D.; Randall, T.D.; Xing, Z.; Richards, C. Pulmonary expression of oncostatin M (OSM) promotes inducible BALT formation independently of IL-6, despite a role for IL-6 in OSM-driven pulmonary inflammation. J. Immunol. 2013, 191, 1453–1464. [Google Scholar] [CrossRef]
- Munitz, A.; Cole, E.T.; Karo-Atar, D.; Finkelman, F.D.; E Rothenberg, M. Resistin-Like Molecule–α Regulates IL-13–Induced Chemokine Production but Not Allergen-Induced Airway Responses. Am. J. Respir. Cell Mol. Boil. 2012, 46, 703–713. [Google Scholar] [CrossRef]
- Fritz, D.K.; Kerr, C.; Botelho, F.; Stampfli, M.; Richards, C. Oncostatin M (OSM) primes IL-13- and IL-4-induced eotaxin responses in fibroblasts: Regulation of the type-II IL-4 receptor chains IL-4Rα and IL-13Rα1. Exp. Cell Res. 2009, 315, 3486–3499. [Google Scholar] [CrossRef]
- Krljanac, B.; Schubart, C.; Naumann, R.; Wirtz, S.; Culemann, S.; Krönke, G.; Voehringer, D. RELMa-expressing macrophages protect against fatal lung damage and reduce parasite burden during helminth infection. Sci. Immunol. 2019, 4, 1–11. [Google Scholar] [CrossRef]
- Stütz, A.M.; Pickart, L.A.; Trifilieff, A.; Baumruker, T.; Prieschl-Strassmayr, E.; Woisetschläger, M. The Th2 Cell Cytokines IL-4 and IL-13 Regulate Found in Inflammatory Zone 1/Resistin-Like Molecule Gene Expression by a STAT6 and CCAAT/Enhancer-Binding Protein-Dependent Mechanism. J. Immunol. 2003, 170, 1789–1796. [Google Scholar] [CrossRef]
- Sutherland, T.E.; Rückerl, D.; Logan, N.; Duncan, S.; Wynn, T.A.; Allen, J.E. Ym1 induces RELMα and rescues IL-4Rα deficiency in lung repair during nematode infection. PLoS Pathog. 2018, 14, e1007423. [Google Scholar] [CrossRef]
- Ayaub, E.A.; Dubey, A.; Imani, J.; Botelho, F.; Kolb, M.R.J.; Richards, C.; Ask, K. Overexpression of OSM and IL-6 impacts the polarization of pro-fibrotic macrophages and the development of bleomycin-induced lung fibrosis. Sci. Rep. 2017, 7, 13281. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, M.A.; MacKinnon, A.C.; Ramachandran, P.; Dhaliwal, K.; Duffin, R.; Phythian-Adams, A.; Van Rooijen, N.; Haslett, C.; Howie, S.E.M.; Simpson, A.J.; et al. Ly6C hi Monocytes Direct Alternatively Activated Profibrotic Macrophage Regulation of Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Zhou, Y.; Johns, R.A. Bruton’s tyrosine kinase (BTK) is a binding partner for hypoxia induced mitogenic factor (HIMF/FIZZ1) and mediates myeloid cell chemotaxis. FASEB J. 2007, 21, 1376–1382. [Google Scholar] [CrossRef]
- Liu, T.; Hu, B.; Choi, Y.Y.; Chung, M.; Ullenbruch, M.; Yu, H.; Lowe, J.B.; Phan, S.H. Notch1 Signaling in FIZZ1 Induction of Myofibroblast Differentiation. Am. J. Pathol. 2009, 174, 1745–1755. [Google Scholar] [CrossRef]
- Martins, V.; Santos, F.G.D.L.; Wu, Z.; Capelozzi, V.; Phan, S.H.; Liu, T. FIZZ1-induced myofibroblast transdifferentiation from adipocytes and its potential role in dermal fibrosis and lipoatrophy. Am. J. Pathol. 2015, 185, 2768–2776. [Google Scholar] [CrossRef]
- Pine, G.M.; Batugedara, H.; Nair, M. Here, there and everywhere: Resistin-like molecules in infection, inflammation, and metabolic disorders. Cytokine 2018, 110, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Baek, H.A.; Yu, H.; Lee, H.J.; Park, B.H.; Ullenbruch, M.; Liu, J.; Nakashima, T.; Choi, Y.Y.; Wu, G.D.; et al. FIZZ2/RELM-β Induction and Role in Pulmonary Fibrosis. J. Immunol. 2011, 187, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.L.; Yin, L.J.; Sharma, S.; Kierstein, S.; Wu, H.F.; Eid, G.; Haczku, A.; Corrigan, C.J.; Ying, S. Resistin-like molecule-β (RELM-β) targets airways fibroblasts to effect remodelling in asthma: From mouse to man. Clin. Exp. Allergy 2015, 45, 940–952. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, L.; Yip, A.; Lao, F.; Botelho, F.; Richards, C.D. RELMα Is Induced in Airway Epithelial Cells by Oncostatin M without Requirement of STAT6 or IL-6 in Mouse Lungs In Vivo. Cells 2020, 9, 1338. https://doi.org/10.3390/cells9061338
Ho L, Yip A, Lao F, Botelho F, Richards CD. RELMα Is Induced in Airway Epithelial Cells by Oncostatin M without Requirement of STAT6 or IL-6 in Mouse Lungs In Vivo. Cells. 2020; 9(6):1338. https://doi.org/10.3390/cells9061338
Chicago/Turabian StyleHo, Lilian, Ashley Yip, Francis Lao, Fernando Botelho, and Carl D. Richards. 2020. "RELMα Is Induced in Airway Epithelial Cells by Oncostatin M without Requirement of STAT6 or IL-6 in Mouse Lungs In Vivo" Cells 9, no. 6: 1338. https://doi.org/10.3390/cells9061338
APA StyleHo, L., Yip, A., Lao, F., Botelho, F., & Richards, C. D. (2020). RELMα Is Induced in Airway Epithelial Cells by Oncostatin M without Requirement of STAT6 or IL-6 in Mouse Lungs In Vivo. Cells, 9(6), 1338. https://doi.org/10.3390/cells9061338