Abstract
Ca2+ is an integral mediator of intracellular signaling, impacting almost every aspect of cellular life. The Ca2+-conducting transporters located on the endoplasmic reticulum (ER) membrane shoulder the responsibility of constructing the global Ca2+ signaling landscape. These transporters gate the ER Ca2+ release and uptake, sculpt signaling duration and intensity, and compose the Ca2+ signaling rhythm to accommodate a plethora of biological activities. In this review, we explore the mechanisms of activation and functional regulation of ER Ca2+ transporters in the establishment of Ca2+ homeostasis. We also contextualize the aberrant alterations of these transporters in carcinogenesis, presenting Ca2+-based therapeutic interventions as a means to tackle malignancies.
1. Introduction
First revealed via light microscopy by French cytologist Garnier in 1897, the endoplasmic reticulum (ER) quickly became the topic of interest for many research endeavors [1]. Sharing a partial intersection with the nucleus and stretching to the cellular periphery, the ER consists of convoluted networks of cisternae that serve as specialized sites to facilitate the translation, modification, folding, sorting, and trafficking of proteins [2]. Perhaps the most important function of the ER, however, is its capacity to operate as the major Ca2+ storage organelle responsible for the maintenance of global Ca2+ homeostasis [3]. It has been long known that in order for eukaryotic cells to coordinate complex cellular events, they must employ specialized signaling molecules that warrant the transmission of extracellular signals into intracellular responses. Among these signaling molecules, Ca2+ represents the evolutionary choice of living cells [4]. Indeed, Ca2+ has been a conserved second messenger from the early days of prokaryotic existence and has evolved to virtually cover all essential functions in a cell [5]. This strategic positioning of Ca2+ within the eukaryotic signal transduction network is mainly due to its unique polarizability and coordination chemistry that accommodate reversible binding with Ca2+-sensing proteins [6]. The degree of subtlety encoded in the intensity, duration, amplitude, and downstream effector landscape of Ca2+ signals makes it one of the most versatile molecules supporting eukaryotic life [7]. Given the pivotal role of Ca2+ in maintaining cellular physiology, it is unsurprising that the systematic regulation of intracellular Ca2+ has become a hotspot for manipulation by various human pathologies, including cancer, a multifactorial disease seamlessly exemplifying such paradigm [8]. In their seminal review, Hanahan and Weinberg proposed the “hallmarks of cancer” in an effort to illustrate the defining features and mechanisms associated with oncogenic states [9]. Importantly, most of these hallmarks stem from alterations in the ER Ca2+ signaling milieu as they have been characterized in a broad range of clinical malignancies [10]. Here, we will examine the mechanisms and critical roles of homeostatic handling of ER Ca2+ by ER Ca2+ transporters and their dysregulations in cancer pathogenesis.
2. ER Topography and Ca2+ Handling
Morphological features distinguish the endoplasmic reticulum (ER) into the rough endoplasmic reticulum (RER) and the smooth endoplasmic reticulum (SER). Despite exhibiting morphological plasticity, RER presents itself in the form of flattened sheets whereas SER is mostly composed of tentacular tubules [11]. At the base of these morphological discrepancies are curvature-maintaining proteins. For instance, ER sheet formation involves the cytoskeleton-linking membrane protein 63 (CLIMP-63) and the transmembrane protein 170A (TMEM170A) while ER tubules are formed by reticulons (RTN1–4) and DP1/Yop1p family members [12]. Besides these morphological disparities, RER and SER execute distinct biological functions. The RER shares a common lumen with the nuclear membrane, which allows for the dynamic exchange of RNAs and proteins through nuclear pores [13]. Its cytoplasmic surface is “studded” with ribosomes where protein synthesis and modifications such as glycosylation occur [14]. Following entry into the RER through protein-conducting channels known as translocons, newly synthesized cytosolic proteins undergo extensive modifications such as folding, sorting, and even degradation in the case of protein misfolding [15]. Further away from the nucleus, the SER specializes in many metabolic processes, including lipid and steroid hormone synthesis, and cellular detoxification [2]. In addition, the SER operates as a major Ca2+ reservoir and is responsible for regulating ER Ca2+ dynamics which, in turn, ensure the optimal activity of both ER compartments. For instance, many chaperones exhibit high Ca2+ binding affinity and any perturbation in the concentration of ER Ca2+ impairs ER protein folding, leading to ER stress [16]. The SER Ca2+ dynamic affects the activation of both Ca2+ release (from the ER) and Ca2+ influx (from the extracellular space), generating cytosolic Ca2+ levels conducive to the activation of key Ca2+-dependent enzymes such as calcineurin, calmodulin-dependent kinases and/or binding proteins, all of which play important roles in proliferation, apoptosis, and motility [17]. Two defining features of the ER Ca2+ signaling network involve its ability to transmit localized, oscillatory Ca2+ signals throughout specific micro-domains within a cell and establish a compartmentalized Ca2+ gradient. Indeed, under resting conditions, the cytosolic Ca2+ concentration (≈100 nM) is 10,000 times lower than that of the extracellular space (≈1.5–2.5 mM) and 1000–5000-folds lower than that of the luminal ER (≈500 µM) (Figure 1) [18]. However, upon physiological stimulations, the cytosolic Ca2+ level rapidly increases from 100 nM to 1 mM either via Ca2+ release from its intracellular stores or via influx from the extracellular space [19]. Importantly, this rapid increase of cytosolic Ca2+ is followed by a timely Ca2+ removal system, re-setting the basis for Ca2+ homeostasis. The establishment of this Ca2+ gradient is vital in the functioning of all organisms and is coordinated by the activity of various ER Ca2+-releasing and-refiling transporters.
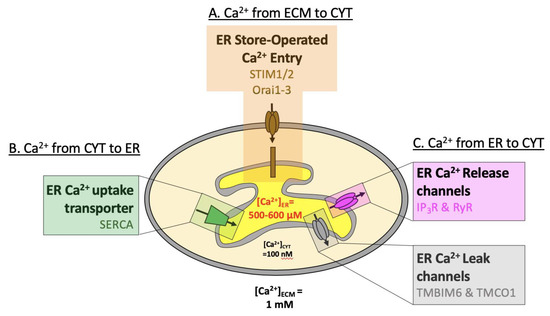
Figure 1.
Overview of ER Ca2+ handling. Schematic presentation of Ca2+ concentrations in the endoplasmic reticulum (ER, 0.6 mM) compared to the cytosol (CYT, 100 nM) and extracellular milieu (ECM, 1 mM). (A) Ca2+ flows down its electrochemical gradient from the ECM to CYT (through Orai) or (B) against its electrochemical gradient from the CYT to the ER (through SERCA). (C) Ca2+ flows from the ER to the CYT down its electrochemical gradient, either following the activation of IP3R & RyR, or through ER Ca2+-leak channels TMBIM6 & TMCO1.
3. ER Ca2+-Releasing Channels
ER Ca2+ release into the cytosol begins with signals generated at the plasma membrane. Stimulation of G-protein coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs) activates phospholipase C (PLC) beta and gamma, respectively. PLC then hydrolyzes the plasma membrane-enriched phosphatidylinositol 4,5-bisphosphate (PIP2) into 1,2-diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3) (Figure 2A). The newly generated IP3 diffuses into the cytoplasm and binds to its receptors (IP3 receptors, IP3Rs) on the ER membrane, causing them to open, ultimately leading to the release of ER Ca2+ into the cytosol (Figure 2B) [20]. The following sections will describe the functions and mechanisms of major ER Ca2+ transporters.
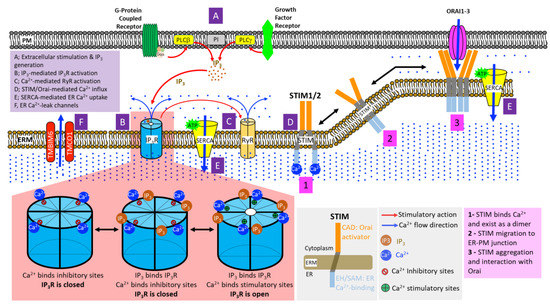
Figure 2.
Mechanisms of ER Ca2+ handling. (A) Stimulations of G-protein Coupled Receptors (GPCRs) and Receptor Tyrosine Kinases (RTKs) signal to phospholipase C-beta (PLC-beta) and PLC-gamma at the plasma membrane, respectively. This leads to PLC-mediated hydrolytic cleavage of phosphatidylinositol 4,5-bisphosphate, producing the Ca2+-mobilizing inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) (not shown in the figure) at the cell membrane. (B) Four molecules of IP3 bind to the tetrameric IP3 receptors (IP3Rs) on the ER membrane, exposing their stimulatory Ca2+ binding sites while simultaneously obstructing inhibitory Ca2+ binding sites. Upon the co-binding of Ca2+ and IP3, IP3R channel pore opens, initiating ER Ca2+ release. (C) Elevated cytosolic Ca2+ further induces the opening of ryanodine receptors (RyRs) on the ER membrane, causing rapid and massive influx of ER Ca2+ into the cytosol. (D) Dwindling luminal ER Ca2+ results in the oligomerization of EF-SAM domain of stromal interaction molecules (STIMs), which, in turn, induces the multimerization of cytoplasmic STIM domains followed by translocation and assembly of STIM clusters at the ER-plasma membrane (ER-PM) junctions. In direct physical association with Orai channels on the plasma membrane, STIM clusters induce the opening of Orai channel pore, allowing extracellular Ca2+ entry into the cytosol. (E) Powered by ATP hydrolysis, the Sarco/Endoplasmic Reticulum Ca2+-ATPases (SERCAs) shuttle the influx of extracellular Ca2+ into the ER or SR, restoring cellular Ca2+ homeostasis. (F) ER Ca2+-leak channels, such as TMBIM6 and TMCO1, prevent ER Ca2+ over-filling.
3.1. Inositol 1,4,5-Trisphosphate Receptors (IP3Rs)
IP3R is a macroscopic (~1.3 MDa), six-pass transmembrane ER Ca2+-release transporter, collectively functioning as a homo- or hetero-tetrameric assembly [21]. In humans, three genes sharing 70% sequence identity encode the three IP3R homologous isoforms, IP3R1s, IP3R2s, and IP3R3s [22]. IP3R1 is the most well-studied subtype and is ubiquitously expressed with the highest level detected in Purkinje neurons [23]. IP3R2 has the highest affinity for IP3 (IP3R2 > IP3R1 > IP3R3) and is predominantly expressed in hepatocytes [24,25,26]. Also broadly expressed, IP3R3 exhibits the highest expression level in gastric, salivary and pancreatic acinar cells [27]. Various mathematical and computational models have shed light on the gating kinetics of IP3Rs [28,29]. Despite those models being grounded on different assumptions, the prevailing view suggests that the tetrameric IP3R channels allow the binding of four IP3 molecules at the N-terminus and multiple Ca2+ ions at the C-terminus, differentially controlling the opening and closing of IP3Rs in a Ca2+ and IP3 concentration-dependent fashion [24,30,31]. Long-range allosteric regulation also exists to couple IP3-dependent conformational change at the N-terminus to the opening of Ca2+-conducting pore at the C-terminus [32]. Under resting conditions in the absence of bound IP3, IP3Rs direct the binding of Ca2+ to the inhibitory sites and remain closed. Upon stimulation, IP3 binds to IP3Rs and initiates channel opening by simultaneously exposing stimulatory Ca2+ binding sites and occluding the inhibitory Ca2+ binding sites, priming cytosolic Ca2+ to bind to the activating sites and facilitating ER Ca2+ release in a process known as Ca2+-induced Ca2+ release (CICR) (Figure 2B) [33]. Intriguingly, Yang and colleagues identified an IP3-independent caldendrin activation of all three IP3R channel isoforms [34]. Using gene editing to label endogenous IP3R1s and super-resolution microscopy, Thillaiappan et al. discovered that only a small population of immobilized IP3R clusters near the plasma membrane are licensed to respond to IP3 stimulation. These IP3R clusters initiate Ca2+ puffs that may serve as the origin and subsequent basis of both localized and distant propagation of CICR among the remainder dynamically-motile IP3Rs dispersed within the ER membrane [35,36]. Upon being expelled from the ER, IP3R-mediated Ca2+ transients are then selectively transported to various subcellular compartments including the cytosolic environment, the mitochondria through ER-mitochondria contact sites (ERMCS) (to which IP3R2 contributes the most), and the lysosomes via ER-lysosome contact sites [37,38,39]. Particularly, as cytosolic Ca2+ begins to rise, Ca2+ activates various downstream partners such as calmodulin, calcineurin and protein kinase C (PKC) which, in turn, modulate important cellular processes and functions, including transcriptional regulation, intracellular protein trafficking, differentiation, proliferation, adhesion and invasion. Ca2+ released to the vicinity of the plasma membrane can activate Ca2+-activated Cl- channels, such as anoctamin 1 for heat sensing [40,41]. Furthermore, IP3R-mediated Ca2+ delivery to the mitochondria at the ERMCS serves as a pivotal signal for apoptotic induction and facilitates metabolic reprogramming [42,43]. Moreover, in addition to fine-tuning cytosolic Ca2+ oscillations, lysosomal sequestration of Ca2+ released by IP3Rs may affect the biological behavior of lysosomes, such as endo-lysosomal membrane trafficking [44,45]. In spite of the vastly heterogeneous IP3R isoform expression in most animal cells, all IP3R isoforms seem to generate, at least at the most rudimentary level among all Ca2+ signals, localized Ca2+ puffs with almost unifying puff amplitudes and spatial-temporal puff kinetics [46]. However, during prolonged stimulation and despite serving as the receptor’s co-agonist along with IP3, Ca2+ can trigger IP3R ubiquitination and subsequent degradation as a preventative measure against toxic buildup of cytosolic Ca2+ [47,48]. Introducing further complexity to the IP3R-mediated Ca2+ signaling landscape are the exquisite sensitivities to co-agonist Ca2+ across IP3R isoforms. For instance, IP3R1 and IP3R2 are under biphasic regulation by Ca2+ where a moderate increase in cytosolic Ca2+ enhances the response to IP3 stimulation while high cytosolic Ca2+ inhibits such response [49,50]. On the other hand, IP3R3 produces monophasic Ca2+ transients [51,52]. This distinct susceptibility of each IP3R isoform to modulation by varying levels of cytosolic Ca2+ may serve as an underpinning molecular mechanism for the regenerative nature of spatiotemporal Ca2+ signals that display diverse intensity, amplitude, and duration in normal physiology and disease states. Yet, the complexity of factors involved and the clinical significance of the channel crosstalk, cell-type isoform expression equilibrium, cellular distribution and receptor conformation of IP3Rs are still not fully understood.
3.2. Ryanodine Receptors (RyRs)
Located on the ER membrane, RyRs mediate massive and rapid Ca2+ release via CICR [53,54]. RyRs are normally closed at low cytosolic [Ca2+] ranging from 100–200 nM. Once the rising cytosolic [Ca2+] reaches a certain threshold, it begins to act on the RyRs, triggering the opening of the homo-tetrameric channels. Extensively characterized in excitable tissues, RyRs exhibit optimal opening probability at sub-micromolar cytosolic Ca2+ concentration [55]. As a result, the IP3Rs-mediated increase in the resting cytosolic Ca2+ concentration paves the way for RyRs to reach their maximum functional capacity. Due to the critical nature of high-conductance RyRs in maintaining cellular electrophysiology, many proteins and molecules, such as calmodulin, calmodulin-dependent protein kinase II (CaMKII), Protein Kinase A (PKA), nicotinamide adenine dinucleotide hydrogen (NADH) and Mg2+, contribute to the precise functional modulation of this supramolecular assembly [56,57,58,59].
The roles of IP3Rs and RyRs are not confined to merely facilitating Ca2+ efflux. Seemingly trivial, the diverse forms of Ca2+ signaling are encoded in spikes, sparks, blips, puffs, and waves, often having profound biological implications. For instance, Ca2+ spikes at the micro-molar range are commonly observed in the apical pole of pancreatic acinar cells to assist in limited exocytosis and secretion of zymogen granules in response to low IP3 stimulation [60,61]. In cardiac myocytes, the opening of voltage-gated L-type Ca2+ channels caused by membrane depolarization increases intracellular Ca2+ level, triggering the opening of RyR2 and subsequent Ca2+ sparks, an essential element in maintaining excitation-contraction coupling in healthy cardiac functions [62]. Moreover, Ca2+ blips, also known as “triggering events” for Ca2+ puffs, are formed by small, transient Ca2+ elevations associated with the opening of a single IP3R channel. A multitude of single IP3R channels in an IP3R cluster evokes localized elevation of Ca2+ resulting in puffs which can affect nuclear Ca2+ signaling through fine-tuning Ca2+ delivery into nucleoplasm and potentially transcription [63]. With higher stimulation from IP3, Ca2+-induced Ca2+ release becomes an activating ligand for one cluster site to drive Ca2+ release at adjacent sites, leading to the generation of Ca2+ waves that propagate in a saltatory manner [64]. The generation of spatially confined Ca2+ waves has been linked to the modulation of the disassembly and turnover of focal adhesion sites, a process highly exploited during cancer metastasis [65]. A linear correlation between IP3R cluster size and Ca2+ puffs has also been established, suggesting that large clusters are potentially responsible for carrying out pacemaker activities [66]. Adding complexity to the already intricate network of Ca2+ signaling is the incorporation of yet another positive feedback mechanism that couples oscillations of Ca2+ to oscillations of IP3, all mediated by phospholipase C [28]. Together, IP3Rs fine-tune ER Ca2+ release whereas RyRs amplify such a response, effectively elevating cytosolic Ca2+ at a global scale.
4. ER Ca2+ Replenishment
As IP3Rs and RyRs act synergistically to increase the cytosolic Ca2+ concentration in order to mediate downstream signal transduction, the ER responds to its dwindling Ca2+ repository by activating store-operated Ca2+ entry (SOCE). The following paragraphs will detail the major players responsible for the ER cytoplasmic Ca2+ refill.
4.1. STIM-Orai
To mediate store-operated Ca2+ entry, the stromal interaction molecules (STIMs) located on the ER membrane physically interact with and activate Ca2+-selective Orai channels at the plasma membrane to mediate Ca2+ influx from the extracellular space. This phenomenon is known as Ca2+ release-activated channel (CRAC) [67,68]. STIM, a single-pass transmembrane protein, senses ER Ca2+ level using its luminal EF-hand and sterile alpha-motif (EF-SAM) domain and functions as the primary initiator of SOCE [69]. In the presence of ample ER Ca2+, the luminal EF-SAM of STIM1 is loaded with Ca2+ and exists in a monomeric state. Upon ER Ca2+ depletion, however, the luminal EF-SAM domain undergoes a conformational change that allows it to become aggregated and capable of directing the cytoplasmic portion of STIM to oligomerize and assemble at the ER-plasma membrane (ER-PM) junctions to be in close physical proximity with Orai channels [69]. Signaling through its STIM-Orai activating region (SOAR), STIM1 induces Orai channel opening and triggers Ca2+ influx from the extracellular space [70]. Considering the critical role of CRAC channel activity in maintaining the healthy dynamics of ER Ca2+ signaling, multiple safety mechanisms are established to ensure the functional regulation of CRACs (Figure 2E). For instance, STIM-induced ER-PM junctional domains contain regulatory proteins, such as CRAC regulatory protein 2A (CRACR2A), junctate, and partner of STIM1 (POST), to fine-tune Ca2+ mobilization into the cytosol [71]. To prevent excessive Ca2+ entry, STIM2.1, a naturally occurring STIM2 variant, hinders STIM-Orai cross-linking and decreases clustering of CRAC channels at the plasma membrane [72]. Furthermore, shifting away from over-reliance on the STIM-Orai mediated Ca2+ entry, transient receptor potential vanilloid 6 (TRPV6) has been reported to translocate from the ER to the plasma membrane to supply Orai-mediated Ca2+ influx [73]. Together, the existence of intricate regulatory networks for CRACs and the functional multiplicity underlying Ca2+ entry following ER Ca2+ depletion equip cells to battle Ca2+ fluctuations in times of stress.
4.2. SERCAs and ER Ca2+-Refilling
As STIM-Orai initiates Ca2+ influx from the extracellular space, the Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA) provides a means for excess cytoplasmic Ca2+ to be shuttled and stored into the ER, establishing a 1000-fold [Ca2+] gradient between the ER and cytosolic compartments [74]. A member of the P-type ATPase superfamily, SERCA utilizes the energy from ATP hydrolysis to alternate between two conformations, E1 and E2 each binding two Ca2+ with high specificity from the cytoplasmic side and releasing them into the luminal ER and SR, respectively [75]. In humans, the SERCA pump is encoded by three genes, ATP2A1, 2 and 3. Post-transcriptional modifications, mainly alternative splicing, generate at least 14 SERCA variants with diverse species-dependent cellular and tissue distributions throughout various stages of development [76,77,78,79]. The level of SERCA1a is the highest in adult slow-twitch skeletal muscles whereas SERCA1b is found predominantly in fetal fast-twitch muscles. In contrast, SERCA2a is expressed in cardiac tissues while SERCA2b is ubiquitously expressed. SERCA3 variants are often found co-expressed with the SERCA2b variant in a wide variety of tissues and cells, such as the salivary glands, lymphoid tissues, pancreatic cells and cerebellar Purkinje neurons [80,81]. The housekeeping SERCA2b protein, for instance, is composed of 3 cytosolic (A, N, and P) domains responsible for mediating ATP binding and hydrolysis, and one 11-helix transmembrane region involved in the regulation of Ca2+ transport [82,83]. As there seems to be notable differences in Ca2+-binding affinities across SERCA isoforms and amongst variants within the same SERCA isoform, the tissue-specific expression equilibrium of SERCA variants transmits differential Ca2+ rhythms required for the survival and function of that specific tissue [84,85]. Considering the crucial role of SERCA pumps in maintaining ER Ca2+ homeostasis, the intricate modulatory mechanisms and existence of various SERCA variants allow for a tight control of the molecular dynamics and kinetic behavior of this pump [86]. Furthermore, SERCA activity is modulated by various factors. For instance, curcumin presumably inhibits SERCA by preventing ATP binding leading to the inhibition of ATP-dependent ER Ca2+ uptake, whereas phospholamban (PLB) and its homolog sarcolipin act by reducing SERCA’s affinity for Ca2+ through direct interaction with SERCA at several ER transmembrane sites [87,88,89,90].
To prevent ER Ca2+ over-filling and ER stress, the transmembrane BAX inhibitor motif containing protein (TMBIM) and the transmembrane and coiled-coil domain 1 (TMCO1) act as Ca2+-leak channels. Among the six TMBIM protein family members, strong evidence points to TMBIM6 (or BI-1) being a seven-pass, pH-sensitive Ca2+-leak channel strictly localized to the ER in skeletal muscle, liver, kidney and spleen [91,92]. Structural insights on the bacterial homolog BsYetJ revealed that the distinct protonation states of Asp171 under acidic and alkaline pH environments affecting hydrogen bonding dynamics among Arg60 on the transmembrane 2 (TM2) region and the C-terminal di-aspartyl pH sensor Asp171 and Asp195 are responsible for altering the positioning of TM2 to mediate Ca2+ fluxes across membranes [93,94,95]. This is in agreement with previous finding stating the indispensable role of Asp213 (human equivalent of Asp195 in BsYetJ) in authorizing Ca2+ fluxes of synthetic human C-terminal peptide of BI-1 [96]. In addition to the passive Ca2+-leak channel TMBIM6, TMCO1 is a Ca2+ load-activated Ca2+ (CLAC) channel embedded across the ER membrane [97]. Mechanistically, TMCO1 undergoes reversible homo-tetramerization upon ethanol-induced elevation of ER Ca2+ content, forming a Ca2+-selective channel to allow the extrusion of excess Ca2+ before it rapidly dissembles upon restoration of the resting luminal ER [Ca2+]. The dynamic orchestration of Ca2+ uptake and release contributes to the maintenance of ER Ca2+ homeostasis, protecting the ionic integrity of the ER for cellular survival and physiological functions.
5. ER Ca2+ Transporters and Cancer Pathophysiology
The pitfalls of abnormal levels of activity of the ER Ca2+ transporters are manifested clinically in a diverse array of human cancers. As tumor pathogenesis varies with each malignancy, it is important to be aware of the highly context-dependent nature of the methods through which cancer cells hijack the ER Ca2+ signaling. The rest of this review will cover some of the mechanisms employed by cancer cells to sabotage ER Ca2+ signaling and the current therapeutic strategies being investigated as potential treatments for cancer patients.
5.1. IP3Rs in Cancer
The strategic positioning and close proximity of the ER to key organelles (e.g., mitochondria, lysosomes and nucleus) have allowed IP3Rs to emerge as crucial determinants of cell fate [98,99]. As a result, IP3Rs must strike a meticulous balance among allocating and transferring appropriate levels of Ca2+ into the mitochondria to ramp up cellular bioenergetic supplies, into the lysosomes to modulate autophagy, and into the nucleus to regulate transcription. Complex regulations of IP3Rs have been documented, preponderant insights of which come from the pro-apoptotic and anti-apoptotic members of the B-cell lymphoma 2 (Bcl-2) family proteins that act primarily by affecting mitochondrial membrane permeability. Within the human Bcl-2 family, pro-apoptotic members (Bax, Bak, Bok, Bid, BAD, Bik, Bim, Noxa, PUMA) can be distinguished by the acquisition of the Bcl-2 homology 3 (BH3) domain, whereas the anti-apoptotic proteins (Bcl-2, Bcl-XL, Mcl-1, Bcl-W, BFL-1, Bcl-B) not only include this BH3 domain but also harbor the Bcl-2 homology 4 (BH4) domain at the N terminus to keep cellular apoptosis at bay [100,101]. Some of the prominent ways in which anti-apoptotic Bcl-2 proteins hijack ER Ca2+ signaling to minimize the production of apoptotic Ca2+ transients are based on protein-protein interactions. Using Fluorescence Resonance Energy Transfer (FRET) and GST-IP3R1 fragment pulldowns, Rong and colleagues precisely pinpointed that endogenous Bcl-2 binds to amino acid residues 1389–1408 in the regulatory and coupling domain of IP3R1 to inhibit its apoptosis-inducing Ca2+ release in Jurkat cells [102]. In a subsequent study, Rong et al. defined the BH4 domain of the Bcl-2 protein to be a functional unit that conferred anti-apoptotic protection against IP3R1 activity [103]. Furthermore, using bioinformatics and site-directed mutagenesis, Monaco and colleagues discovered that a single amino acid difference in the BH4 domain of Bcl-2 and Bcl-XL may account for the differential binding of the proteins to IP3R1 and the distinctive regulation of IP3-induced Ca2+ release (IICR) [104]. Recent evidence suggests that, in addition to binding to the modulatory region of the IP3R1 as Rong et al. proposed in 2008, purified BH4 domain of Bcl-2 is also capable of forming a physical complex with and participating in competitive binding to the ligand-binding domain (LBD) of IP3R1 with receptor agonist IP3 to either activate or inhibit the IP3R1 channel activity in concordance with the extent of IP3-evoked receptor stimulation [105]. As Bcl-2-IP3R interaction was established, scientists began to search for the potential involvements and mechanistic understandings of other anti-apoptotic Bcl-2 family members in the modulation of IP3R Ca2+ release. Interestingly, in stark contrast to Bcl-2 inhibition of IP3R channel activity, Bcl-XL sensitizes all three IP3R isoforms to IP3 stimulation while promoting ER- Ca2+-mediated mitochondrial bioenergetics and enhancing spontaneous cytosolic Ca2+ signaling in conferring apoptotic resistance [106,107]. However, new structural evidence uncovers that the biphasic regulation of IP3R channel gating kinetics in the maintenance of cell viability occurs through the binding of BH3-like domain on the carboxyl terminus of IP3R by the BH3 domain-binding pocket of Bcl-XL [106]. Indeed, structurally similar anti-apoptotic Bcl-2 proteins, such as Mcl-1, have been reported to bind to the carboxyl termini of all three mammalian IP3R isoforms with comparable affinity and increase spontaneous IP3R-dependent Ca2+ oscillations as necessary steps to maintaining cellular survival in response to cytotoxic agents [108]. Nonetheless, these complexes of anti-apoptotic Bcl-2 proteins and IP3R channels may open the door for innovative therapeutic interventions. As a matter of fact, recent years have witnessed the tremendous breakthrough in the use of synthetic peptides to disrupt the Bcl-2-IP3R complex in chronic lymphocytic leukemia, multiple myeloma, follicular lymphoma and small cell lung cancer either alone or with other mimetics to potentiate anti-neoplastic effects and / or tackle chemo-resistance [109,110,111,112,113]. On the other end of the spectrum, a growing body of evidence suggests that IP3R activity is subject to regulation by tumor suppressors. For instance, tumor suppressor proteins phosphatase and tensin homolog (PTEN) in human prostate cancer and BRCA1-associated protein-1 (BAP1) in asbestos-induced malignant transformation partially act through stabilizing IP3R3s against receptor ubiquitination; thus, potentiating Ca2+ transport into the mitochondria to drive apoptosis [114,115]. Furthermore, in colorectal cancer cell lines, abrogation of oncogenic K-Ras unleashed IP3R3 activity, enhancing IP3R3-mediated Ca2+ release and inducing cellular sensitization to apoptosis [116]. As a result, the systematic coordination of these effector regulators of IP3Rs carries profound impacts on cell fate.
Besides the functional regulation of IP3Rs by the oncoprotein-tumor suppressor crosstalk, the selective expression of individual IP3R isoform has also been tampered with in several clinical malignancies. For instance, IP3R3 is up-regulated in gastric cancer, glioblastoma and renal cell carcinoma [117,118,119]. Additionally, IP3R3 expression level has been found increased and positively correlated with the migratory and invasive capacities of breast cancer and glioblastoma cell lines and that caffeine-mediated IP3R3 inhibition abrogated proliferative and invasive phenotypes in glioblastoma and extended survival rate [120,121]. As migration is associated with cell shape, IP3R3 likely remodels cytoskeletal structure to support breast cancer cell migration and invasion [122]. Moreover, surgically resected colorectal carcinomas indicated elevated IP3R3 expression, in proportion to the depth of invasion, lymph node and liver metastases [123]. Collectively, these evidence makes elevated IP3R3 level a reliable diagnostic marker for various clinical malignancies. Unlike IP3R3, whose expression pattern has been well documented in human carcinomas across multiple tissues, the expression profiles of IP3R1 and IP3R2 remain elusive in pathophysiology as they seem to draw diverse implications on various aspects of tumorigenesis, such as tumor initiation, migration, survival, and even drug resistance. For instance, heightened IP3R1 activity promotes prostate cancer cell survival and resistance to hormonal deprivation therapy [123]. Conversely, increased IP3R1 level is shown to potentiate melatonin-induced apoptosis among ovarian cancer and colorectal cancer cell lines while simultaneously conferring attenuated antioxidant responses [124]. Similarly, this pro-apoptotic effect of IP3R1 has also been studied in vitro and in vivo after subjecting ovarian carcinoma cells to cytotoxic agent sulforaphane [125]. Moreover, IP3R1 expression is markedly reduced in cisplatin-resistant bladder cancer cell lines and that transient induced over-expression of IP3R1 in resistant cells restored chemo-sensitivity to cisplatin [126]. Exerting similarly broad impacts as IP3R1s, IP3R2-mediated Ca2+ oscillation plays extensive roles ranging from lung cancer cell migration, maintenance of the regenerative capacity of liver cancer stem cells and to the induction of senescence [127,128,129]. Moreover, recent study shows that, diffuse large B-cell lymphoma (DLBCL) cells with constitutive IP3 signaling and addiction to Bcl-2-mediated attenuation of IP3R2 Ca2+ release are sensitive to apoptotic induction by Bcl-2/IP3R Disruptor-2 (BIRD2), which is compatible with the previous finding in DLBCL that increased IP3R2 protein level is associated with high sensitivity to apoptosis among SU-DHL-4 cells subsequent to treatment with BIRD2 [130,131]. Collectively, rampant manipulations of the IP3R expression profile throughout cancer development epitomize the notion that many malignancies have harbored the increasingly diversifying capacity to sabotage IP3R-mediated Ca2+ transients and therefore, global Ca2+ signaling to stimulate oncogenesis at the genetic level.
5.2. RyRs in Cancer
Encoded by three separate human genes and composed of homo-tetrameric supramolecular assemblies, RyRs (I, II, and III) are mostly expressed and studied in the context of excitable tissues, including skeletal muscle, cardiac tissues, and the brain, respectively [132,133,134]. However, emerging clinical and empirical evidence from oncological studies has described the functional expression of RyRs as highly diverse across a vast array of human malignancies. For instance, Abdul et al. examined the total RyR protein expression in patient-derived ductal breast cancer epithelium and found that the overall RyR expression is positively correlated with tumor grade, suggesting the involvement of RyRs in breast cancer survival. However, the addition of RyR agonist, 4-chloro-m-cresol, inhibited breast cancer cell proliferation [135]. Furthermore, in comparison with normal thyroid tissues, tissues derived from thyroid carcinoma exhibit decreased expression of RyR2, the down-regulation of which is tightly associated with decreased patient survival rate, lymphatic metastasis, extracapsular extension, and bleak clinical prognosis [136]. On the other hand, RyR2 is over-expressed in melanoma tissues as compared to melanocytes. However, the reported increase of RyR2 expression is not concomitant with an increase in RyR-mediated Ca2+-release [137]. Similar results were reported by Bennett and colleagues, who demonstrated that neither ryanodine nor caffeine (RyR agonist) elicited a measurable RyR2-mediated Ca2+ transient in cervical cancer epithelial cell line HeLa, suggesting aberrant functional properties of RyR2s in the survival of cancer cells [138]. Besides aberrant RyR2 expression levels in giving rise to malignancy, several mutations of RyR2s have been linked to lung cancer [139]. Furthermore, RyR3 over-expression is detected in breast cancer where RyR3s play an essential role in proliferation and migration [140]. Nevertheless, studying RyRs through the lens of cellular apoptosis, Mariot et al. demonstrated that the functional expression and activation of RyR1s and RyR2s by caffeine led to apoptosis of prostate cancer LNCaP cells, whereas inhibition of these receptors with ryanodine protected against apoptosis [140]. Furthermore, even with apoptosis-resistant cancer cells, RyR-mediated Ca2+ release has been shown to facilitate Neferine-induced autophagic cell death [141].
In addition to mediating cancer progression, RyRs have also been linked to chemo-resistance. For instance, RyR1s contribute to acquired chemo-resistance by executing non-enzymatic interactions with chemotherapy-induced GSTO1 (glutathione S-transferase omega 1) to fine-tune cytosolic Ca2+ levels needed for the enrichment of the tumor-initiating breast cancer stem cells (BCSCs) [142]. Though recognizing RyR1′s role in driving BCSCs seems promising in tackling chemo-resistance, the feasibility of achieving pharmacological inhibition of RyR1s remains low due to limitations imposed by drug delivery, resultant toxicity, and target specificity across RyR subtypes [143]. While current findings hold promise for the derivation of a future RyR-based anti-neoplastic therapy, more research is needed to understand the underlying mechanisms of the differential regulation of these receptors in physiology and pathophysiology.
5.3. STIM-Orai Channels in Cancer
Ca2+ signaling sets the fundamental basis for metastatic dissemination of tumors to distant tissues through activation of proliferative and invasive pathways, such as nuclear factor of activated T-cells (NFAT) and extracellular signal-regulated kinases (ERKs) [144,145]. The activity of these oncogenic pathways is often dictated by pathological modifications of Ca2+ release and influx channels, in particular, at the level of store operated Ca2+ entry (SOCE). Since STIM1-Orai1 signaling axis encompasses the predominant mechanism underlying SOCE, it is often the target of oncogenic manipulations at both genetic and functional levels. For example, STIM1 is over-expressed in colorectal cancer and its expression level is positively correlated with tumor size, depth of invasion, and lymph node metastasis [146]. In glioblastoma multiforme, STIM1 and Orai1 knockdown decreased cancer cell invasion and proliferation, respectively [147]. STIM1 is also a crucial mediator for cell proliferation, migration as well as angiogenesis in cervical cancer and invasion in melanoma [148,149]. Furthermore, in breast cancer cell line MDA-MB-231, STIM1 and Orai1 remodel focal adhesion turnovers and are required for tumor invasion and metastasis [147]. Similar observations were ascertained in pancreatic ductal adenocarcinoma where STIM1-mediated ER-PM junction formation was found to be re-distributed during epithelial-mesenchymal transition, underscoring the essential role of altered state of SOCE in cellular migration and malignant transformation [150]. Furthermore, revealed by time-lapse imaging, esophageal squamous cell carcinoma (ESCC) KYSE-150 cells showed hyperactive spontaneous intracellular Ca2+ oscillations, potentially due to the elevated expression of Orai1 [151]. To corroborate this, McAndrew further demonstrated that Orai1 siRNA knockdown not only attenuated cytosolic Ca2+ influx in breast cancer MDA-MB-231 and MCF-7 cell lines in the presence of invasive stimulus PAR-2, but also reduced their viability [152]. As STIM1 and Orai1 over-expression has been observed across a multitude of malignancies, they are inarguably among the most enticing drug targets in anti-cancer therapy.
In addition to adjusting the expression and activity of the canonical STIM1-Orai1 signaling axis, cancer cells also have developed the ability to switch to store-independent Ca2+ entry to escape a potentially “doomed fate.” It was not until the 1990s that an alternative “store-independent Ca2+ entry” model was proposed to provide a more accurate depiction of Ca2+ entry under a physiological level of agonists. The proposed mechanism suggested that, instead of sustained elevated intracellular Ca2+, subtle periodic oscillations of intracellular Ca2+ take over during SOCE [153,154]. Although Orai1 was the most well understood ion channel at the time, the possibility of alternative mechanisms responsible for such periodic oscillations of Ca2+ entry led scientists to examine the functions and roles of other members of the Orai family channels. Motiani et al. explored the selective requirement of many breast cancer cell lines for the use of Orai3 as opposed to the canonical Orai1-mediated SOCE based on the presence or absence of plasma membrane estrogen receptors [155]. Later, in 2013, Motiani et al. further demonstrated the selective use of Orai3 Ca2+ channels in mediating SOCE in estrogen receptor α-expressing (ERα+) breast cancer cells. Conversely, Orai3 knockdown led to decreased ERα+ MCF7 cell proliferation and invasion [156]. Another independent study led by Faouzi also demonstrated that Orai3 knockdown impaired breast cancer MCF-7 cell proliferation and arrested cell cycle progression at the G1 phase without affecting the proliferation and survival of wild-type mammary MCF-10A cells [157]. As the role of Orai3 in facilitating tumorigenicity became more prominent, Dubois et al. discovered increased endogenous expression of Orai3 protein and increased reliance on the use of Orai3-Orai1-jointly-mediated store-independent, arachidonic-acid-regulated channels among prostate cancer cells. This selective utilization of Orai3 by prostate cancer cells can partially be attributed to greater evasion of apoptotic signals closely associated with sole Orai1 functioning [158]. This change in the Orai3/ Orai1 expression dynamic created a shift from the use of the canonical Orai1-based SOCE and marks the oncogenic switch that facilitates prostate cancer tumor progression. The remarkable capacity of cancer cells to adjust their receptor expression equilibrium to enhance survival while achieving the same biological signaling outputs is a truly fascinating area for scientific investigation and a promising realm for drug discovery.
5.4. SERCAs in Cancer
SERCA activity represents a nodal point of cellular survival and has been extensively exploited in carcinogenesis [159]. The expression profile of SERCA I, II, and III isoforms is highly diverse across human malignancies. Mounting evidence indicates that many SERCAs are down-regulated in cancer. For instance, the SERCA1 isoform is decreased in cisplatin-resistant epithelial ovarian cancer cell line MDAH-2774 [160]. SERCA2b expression is significantly reduced in small cell lung cancer, thyroid cancer, oral squamous cell carcinoma and colon cancer [161,162,163,164]. Additionally, highlighting the interplay between SERCA2 deficiency to malignancy came the finding of Prasad et al. that haploinsufficiency of Atp2a2, which encodes the SERCA2 isoform, leads to increased likelihood of developing squamous cell papillomas [165]. Furthermore, the level of SERCA3 isoform plummets in breast carcinomas and colon adenocarcinomas [166,167]. An in-depth mechanistic explanation as to why SERCA down-regulation takes prevalence in these types of cancer is, nevertheless, still lacking. Considering SERCAs function by selectively replenishing the ER Ca2+ store, a pivotal biological implication connecting decreased luminal ER Ca2+ re-filling and cancer cell apoptotic resistance suggests that reduced ER Ca2+ store, despite exerting pleiotropic effects on intracellular Ca2+ handling, may translate into low cytosolic Ca2+ release, therefore, decreased activity of Ca2+-induced opening of the mitochondrial permeability transition pore (PTP), hence greater cell survival [168,169,170,171]. Supporting this notion, many research endeavors have found that the anti-apoptotic Bcl-2 protein upregulated in numerous malignancies inhibits the activity of various SERCA isoforms, leading to reduced ER Ca2+ uptake and attenuated pro-apoptotic mitochondrial Ca2+ influx [172,173]. Similarly, Scorrano et al. demonstrated that double knockout of pro-apoptotic Bcl-2 family members, Bax and Bak in mouse embryonic fibroblasts resulted in the inhibition of SERCA activity and decreased mitochondrial Ca2+ uptake, depicting a delicate balance between anti-apoptotic and pro-apoptotic Bcl-2 family members in fine tuning ER Ca2+ release [174]. Nonetheless, the complete and irreversible abolishment of SERCA activity by thapsigargin (TG) drives intrinsic apoptosis through the induction of prolonged ER stress [175,176]. Of note, many chemotherapeutics act through tumor suppressors to modulate Ca2+ signaling. A prominent example of this is that in response to Adriamycin challenge, the master tumor suppressor protein p53 localizes to the ER/mitochondria associated membranes and promotes SERCA activity by reducing its oxidation. This gives rise to ER Ca2+ overload and elicits ER Ca2+ release as a means of apoptotic induction [177]. Importantly, revealed by intravital fluorescent microscopy, this critical crosstalk between the SERCA pump and p53 in generating apoptotic signals is also substantiated in vivo in cancer photodynamic therapy using light-activated photosensitizer phthalocyanine, linking p53 sensitization of cellular apoptosis to ER Ca2+-overload and increased mitochondrial Ca2+ uptake [178]. Intriguingly, an alternative paradigm argues that SERCA over-expression has also become a hallmark in a variety of cancers. For instance, SERCA2b expression is positively correlated with colorectal malignancy as SERCA2b over-expression promotes pro-survival mitogen-activated protein kinase (MAPK) and protein kinase B (also known as AKT) signaling and drives proliferation and migration of human colorectal adenocarcinoma SW480 cells [179]. Moreover, SERCA2b is found over-expressed in epithelial prostate cancer cells and that knockdown of SERCA2b decreases prostate cancer proliferation [180]. In comparison with normal cells treated with curcumin, curcumin inhibition of SERCA2 activity selectively inhibits ovarian cancer cell viability [181]. Furthermore, upregulation of the SERCA3 isoform is detected in gastric carcinomas [182]. Mechanistically, by increasing luminal ER [Ca2+] via SERCA over-expression, rapidly proliferating cancer cells strategically endure and alleviate cytotoxic stress associated with their hyperactive protein synthesis and folding machineries [183]. Since cancer is a multifactorial disease, it is not surprising that even a combination of SERCA2(b) up-regulation and SERCA3 isoform down-regulation exists in the case of epidermal growth factor-induced epithelial mesenchymal transition in breast cancer MDA-MB-468 cells, further solidifying the link between aberrant SERCA activity and malignancy [184]. The purpose of reprogramming the expression pattern of SERCA isoforms in various malignant lesions is to confer cancer cells the ability to tailor the amplitude, duration and frequency of ER Ca2+ re-uptake to sustain their specific oncogenic needs. Hence, it is within reason that different SERCA isoforms demonstrate varying expression kinetics throughout distinct stages of tumorigenicity [185].
6. Targeting ER Ca2+ Signaling in Anti-Cancer Therapy
As ER Ca2+ signaling is indispensable for cell development, movement, metabolism, survival, and signal transduction, this, therefore, poses a challenge for a specific and efficacious Ca2+-based drug design. Similar to the “undruggable” Ras and MAPK, targeting ER Ca2+ alone seems impractical due to its ubiquitous presence and integral contribution to cellular physiology [186]. However, targeting proteins that interact with Ca2+ at the levels of channels/transporters/pumps and downstream effector molecules that decipher Ca2+-encoded messages seems to be more feasible [187]. Rather than targeting the IP3R channel activity alone as an isolated molecular entity, research is now directed towards gaining collective understandings of the fate-determining pathways following IP3R -mediated Ca2+ release, such as the IP3R-VDAC1-MCU-signaling axis bridging ER Ca2+ release and mitochondrial Ca2+ uptake [188]. Indeed, many chemotherapeutic agents, such as cisplatin and doxorubicin, fine tune ER-mitochondria crosstalk and alter oncogene-tumor suppressor function dynamics to elicit potent apoptotic Ca2+ signals, inhibiting tumor cell survival [189]. It is not the intention of this review to cover all channel inhibitors governing ER Ca2+ signaling, however, we will briefly describe the use of ER Ca2+ transporter-based chemical drug conjugate, computational pharmacology and extrapolate the immunotherapeutic potential of CRAC channels in the design of novel anti-neoplastic therapy.
Conjugating tumor-specific marker with seemingly unlikely drug target offers new hope in drug delivery. For instance, thapsigargin (TG) is widely used in research laboratories to deplete ER Ca2+ through prolonged inhibition of SERCA activity. This depletion of the ER Ca2+ store, in and of itself, induces ER stress, and causes elevated cytoplasmic Ca2+ that can activate intrinsic apoptotic pathways through calmodulin/calcineurin-mediated signal transduction [190]. Despite being shown to potentiate taxane-mediated tumor killing, TG has not been widely adopted in clinical settings due to its non-selective cytotoxicity [191]. Recognizing the heterogeneous molecular signatures of a tumor would vastly boost our chance of designing targeted therapies. An example of such attempt was documented by Denmeade and Isaacs: “Chemical modification and coupling of thapsigargin to a PSA-cleavable peptide sequence carrier seems to be a promising approach to target both normal and malignant prostate cancers” [192]. This pro-drug construct allows for the specific delivery of TG to prostate cancer cells, disrupting ER Ca2+ signaling and generating ER stress to induce apoptosis.
As an alternative investigative tool, computational pharmacology has been utilized to explore Ca2+ binding kinetics during SOCE. Found up-regulated in glioblastoma multiforme (GBM), STIM1 and Orai1 are positively associated with GBM invasiveness [193]. Through the use of in-silico models, such as molecular dynamic simulations and structure-based virtual screening, Sampath and Sankaranarayanan identified SB01990, SPB06836, and KM06293 as drug leads capable of disrupting Ca2+ binding to the active sites of Orai1, inhibiting ORAI-mediated Ca2+ influx with relatively ideal pharmacokinetics [194]. However, further in vivo testing is required to characterize the pharmacodynamic and pharmacokinetic properties of those inhibitors.
Another interesting area for clinical implementation of CRAC-channel based drug design is immunotherapy. Within a tumor mass, the tumor microenvironment (TME) is produced by the functional crosstalk among miscellaneous cell types, such as the cytotoxic T-lymphocytes (CTLs), B-lymphocytes, Natural Killer (NK) cells, tumor-associated macrophages (TAMs), tumor-associated neutrophils, regulatory T cells, pericytes, vascular endothelial cells, and cancer-associated fibroblasts [195,196,197]. Among these cells, CTLs and NK cells primarily exert their anti-tumor effect by secreting granzymes and perforin directly into the tumor cells resulting in cell lysis. Considering the critical involvement of STIM-Orai channels in the production of Ca2+ transients required for the proliferation, migration, recruitment of T lymphocytes as well as the subsequent degranulation of lytic vesicles into the tumor cell, CRAC channel activity is key to the initiation and maintenance of a potent anti-tumor immune response [198,199,200]. Indeed, loss-of-function mutations in human ORAI1 or STIM1 lead to increased susceptibility of developing tumors [201]. However, considering that CRAC channel activity is essential for both anti-tumor immunity and oncogenesis (as discussed in Section 5.3), it is, therefore, essential to acknowledge the double-edged-sword effect of utilizing STIM and Orai proteins as a therapeutic axis and that a comprehensive understanding of tumor-specific channel regulators and downstream signaling pathways is needed before therapeutic design.
Adding to the complexity of targeting ER Ca2+ signaling for cancer therapy are the different facets of drug design. What are the precise pharmacophore and mechanisms of action? Are there any non-specific interactions with other drug molecules, targets, or enzymes? What are the pharmacokinetic properties, namely absorption, distribution, metabolism, and excretion associated with the drug? Are there adverse side effects? How to reconcile and fit thermodynamic stability, drug bioavailability, and solubility all into the diverse pharmacogenomics of the patients? Nonetheless, we are entering an exciting era of biomedical research where basic mechanistic understandings of ER Ca2+ and its homeostatic regulation are vigorously pursued for the development of new anti-cancer therapies.
7. Closing Remarks
In this review, we have summarized the major ER Ca2+ transporters and their aberrant functional alterations in cancer. We described the homeostatic regulation of ER Ca2+ store and its connection with the global Ca2+ signaling transduction network. We also appreciated the many ways ER Ca2+ signaling manifests itself through its receptor distribution, isoform expression, downstream effector landscape and how these processes could be hijacked in malignancies. We examined the dynamic modulation of these transporters in the context of organellar crosstalk as well as endogenous regulation by oncoproteins and tumor suppressors. We then culminated the review with pharmacological interventions of ER Ca2+ transporters. As future design of anti-cancer therapy continues, it is awe-inspiring to reflect on the width and depth of Ca2+ signaling as its regulatory networks have evolved since early prokaryotic life. Without a doubt, learning to comprehend and communicate in a beautiful yet universal language spoken by both prokaryotic and eukaryotic lives through the intricate flow of Ca2+ across cellular compartments is a useful and powerful way in combating cancer and more.
Author Contributions
X.Z., A.M.S., Y.E.H. conceived the review; X.Z. wrote the original draft of the review; A.M.S. edited and contributed to the writing of the original draft of the review. Y.E.H. edited and approved the final version of the review. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by ResearchNS Establishment grant number 2019–2174 and NSERC Discovery grant 2018–05528.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sjöstrand, F.S. The endoplasmic reticulum. In Cytology and Cell Physiology, 3rd ed.; Academic Press: London, UK, 1964; pp. 311–375. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The endoplasmic reticulum. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Filadi, R.; Zampese, E.; Pozzan, T.; Pizzo, P.; Fasolato, C. Endoplasmic reticulum-mitochondria connections, calcium cross-talk and cell fate: A closer inspection. In Endoplasmic Reticulum Stress in Health and Disease; Springer Netherlands: Dordrecht, The Netherlands, 2012; pp. 75–106. [Google Scholar]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Cai, X.; Wang, X.; Patel, S.; Clapham, D.E. Insights into the early evolution of animal calcium signaling machinery: A unicellular point of view. Cell Calcium 2015, 57, 166–173. [Google Scholar] [CrossRef]
- Carafoli, E.; Krebs, J. Why calcium? How calcium became the best communicator. J. Biol. Chem. 2016, 291, 20849–20857. [Google Scholar] [CrossRef]
- Li, L.; Stefan, M.I.; Le Novère, N. Calcium input frequency, duration and amplitude differentially modulate the relative activation of calcineurin and CaMKII. PLoS ONE 2012, 7, e43810. [Google Scholar] [CrossRef]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium–cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Bong, A.H.; Monteith, G.R. Calcium signaling and the therapeutic targeting of cancer cells. Biochim. Biophys. Acta 2018, 1865, 1786–1794. [Google Scholar] [CrossRef]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough sheets and smooth tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef]
- Lin, S.; Sun, S.; Hu, J. Molecular basis for sculpting the endoplasmic reticulum membrane. Int. J. Biochem. Cell Biol. 2012, 44, 1436–1443. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The transport of molecules between the nucleus and the cytosol. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Protein glycosylation in the ER and Golgi complex. In Molecular Cell Biology, 4th ed.; WH Freeman: New York, NY, USA, 2000. [Google Scholar]
- Fewell, S.W.; Brodsky, J.L. Entry into the endoplasmic reticulum: Protein translocation, folding and quality control. In Trafficking Inside Cells; Springer: New York, NY, USA, 2009; pp. 119–142. [Google Scholar]
- Coe, H.; Michalak, M. Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 2009, 28, F96–F103. [Google Scholar]
- Kang, M.; Othmer, H.G. The variety of cytosolic calcium responses and possible roles of PLC and PKC. Phys. Biol. 2007, 4, 325. [Google Scholar] [CrossRef] [PubMed]
- Laude, A.J.; Simpson, A.W. Compartmentalized signalling: Ca2+ compartments, microdomains and the many facets of Ca2+ signalling. FEBS J. 2009, 276, 1800–1816. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C.; Bootman, M.D.; Scott, J.D. Second messengers. Cold Spring Harb. Perspect. Biol. 2016, 8, a005926. [Google Scholar] [CrossRef] [PubMed]
- Serysheva, I.I. Toward a high-resolution structure of IP3R channel. Cell Calcium 2014, 56, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Serysheva, I.I.; Baker, M.R.; Fan, G. Structural insights into IP 3 R function. In Membrane Dynamics and Calcium Signaling; Springer International Publishing: Cham, Switzerland, 2017; pp. 121–147. [Google Scholar]
- Sharp, A.H.; Nucifora, F.C., Jr.; Blondel, O.; Sheppard, C.A.; Zhang, C.; Snyder, S.H.; Russell, J.T.; Ryugoand, D.K.; Ross, C.A. Differential cellular expression of isoforms of inositol 1, 4, 5-triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 1999, 406, 207–220. [Google Scholar] [CrossRef]
- Ivanova, H.; Vervliet, T.; Missiaen, L.; Parys, J.B.; De Smedt, H.; Bultynck, G. Inositol 1, 4, 5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta 2014, 1843, 2164–2183. [Google Scholar] [CrossRef]
- Hirata, K.; Pusl, T.; O’Neill, A.F.; Dranoff, J.A.; Nathanson, M.H. The type II inositol 1, 4, 5-trisphosphate receptor can trigger Ca2+ waves in rat hepatocytes. Gastroenterology 2002, 122, 1088–1100. [Google Scholar] [CrossRef]
- Iwai, M.; Michikawa, T.; Bosanac, I.; Ikura, M.; Mikoshiba, K. Molecular basis of the isoform-specific ligand-binding affinity of inositol 1, 4, 5-trisphosphate receptors. J. Biol. Chem. 2007, 282, 12755–12764. [Google Scholar] [CrossRef]
- Fujino, I.; Yamada, N.; Miyawaki, A.; Hasegawa, M.; Furuichi, T.; Mikoshiba, K. Differential expression of type 2 and type 3 inositol 1, 4, 5-trisphosphate receptor mRNAs in various mouse tissues: In situ hybridization study. Cell Tissue Res. 1995, 280, 201–210. [Google Scholar]
- De Young, G.W.; Keizer, J. A single-pool inositol 1, 4, 5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc. Natl. Acad. Sci. USA 1992, 89, 9895–9899. [Google Scholar] [CrossRef] [PubMed]
- Othmer, H.G.; Tang, Y. Oscillations and waves in a model of InsP 3-controlled calcium dynamics. In Experimental and Theoretical Advances in Biological Pattern Formation; Springer: Boston, MA, USA, 1993; pp. 277–300. [Google Scholar]
- Tang, Y.; Stephenson, J.L.; Othmer, H.G. Simplification and analysis of models of calcium dynamics based on IP3-sensitive calcium channel kinetics. Biophys. J. 1996, 70, 246–263. [Google Scholar] [CrossRef][Green Version]
- Alzayady, K.J.; Wang, L.; Chandrasekhar, R.; Wagner, L.E.; Van Petegem, F.; Yule, D.I. Defining the stoichiometry of inositol 1, 4, 5-trisphosphate binding required to initiate Ca2+ release. Sci. Signal. 2016, 9, ra35. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Miyatake, H.; Terauchi, A.; Mikoshiba, K. IP3-mediated gating mechanism of the IP3 receptor revealed by mutagenesis and X-ray crystallography. Proc. Natl. Acad. Sci. USA 2017, 114, 4661–4666. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.W.; Tovey, S.C. IP3 receptors: Toward understanding their activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a004010. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; McBride, S.; Mak, D.-O.D.; Vardi, N.; Palczewski, K.; Haeseleer, F.; Foskett, J.K. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc. Natl. Acad. Sci. USA 2002, 99, 7711–7716. [Google Scholar] [CrossRef] [PubMed]
- Thillaiappan, N.B.; Chavda, A.P.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Ca 2+ signals initiate at immobile IP 3 receptors adjacent to ER-plasma membrane junctions. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Prole, D.L.; Taylor, C.W. Structure and function of IP3 receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035063. [Google Scholar] [CrossRef]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic reticulum–mitochondrial contactology: Structure and signaling functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Bartok, A.; Weaver, D.; Golenár, T.; Nichtova, Z.; Katona, M.; Bánsághi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP 3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 receptors preferentially associate with ER-lysosome contact sites and selectively deliver Ca2+ to lysosomes. Cell Rep. 2018, 25, 3180–3193. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.Y.; Jha, A.; Ahuja, M.; Muallem, S. Ca2+ influx at the ER/PM junctions. Cell Calcium 2017, 63, 29–32. [Google Scholar] [CrossRef]
- Cho, H.; Yang, Y.D.; Lee, J.; Lee, B.; Kim, T.; Jang, Y.; Back, S.K.; Na, H.S.; Harfe, B.D.; Wang, F.; et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat. Neurosci. 2012, 15, 1015. [Google Scholar] [CrossRef] [PubMed]
- Hanson, C.J.; Bootman, M.D.; Roderick, H.L. Cell signalling: IP3 receptors channel calcium into cell death. Curr. Biol. 2004, 14, R933–R935. [Google Scholar] [CrossRef] [PubMed]
- Bustos, G.; Cruz, P.; Lovy, A.; Cárdenas, C. Endoplasmic reticulum–mitochondria calcium communication and the regulation of mitochondrial metabolism in cancer: A novel potential target. Front. Oncol. 2017, 7, 199. [Google Scholar] [CrossRef]
- López-Sanjurjo, C.I.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Lysosomes shape Ins (1, 4, 5) P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J. Cell Sci. 2013, 126, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Gray, S.R.; Bright, N.A. Endosome–lysosome fusion. In Proceedings of the Lysosomes in Health and Disease, Charles Darwin House, London, UK, 13–14 May 2010; pp. 1413–1416. [Google Scholar]
- Lock, J.T.; Alzayady, K.J.; Yule, D.I.; Parker, I. All three IP3 receptor isoforms generate Ca2+ puffs that display similar characteristics. Sci. Signal. 2018, 11, eaau0344. [Google Scholar]
- Alzayady, K.J.; Wojcikiewicz, R.J. The role of Ca2+ in triggering inositol 1, 4, 5-trisphosphate receptor ubiquitination. Biochem. J. 2005, 392, 601–606. [Google Scholar] [CrossRef]
- Wojcikiewicz, R.J. Regulated ubiquitination of proteins in GPCR-initiated signaling pathways. Trends Pharmacol. Sci. 2004, 25, 35–41. [Google Scholar] [CrossRef]
- Vervloessem, T.; Yule, D.I.; Bultynck, G.; Parys, J.B. The type 2 inositol 1, 4, 5-trisphosphate receptor, emerging functions for an intriguing Ca2+-release channel. Biochim. Biophys. Acta 2015, 1853, 1992–2005. [Google Scholar] [CrossRef]
- Mak, D.-O.D.; McBride, S.M.; Petrenko, N.B.; Foskett, J.K. Novel regulation of calcium inhibition of the inositol 1, 4, 5-trisphosphate receptor calcium-release channel. J. Gen. Physiol. 2003, 122, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, T.; Maeda, A.; Yamazawa, T.; Hirose, K.; Kurosaki, T.; Iino, M. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 1999, 18, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Hegg, C.C.; Jia, C.; Chick, W.S.; Restrepo, D.; Hansen, A. Microvillous cells expressing IP3 receptor type 3 in the olfactory epithelium of mice. Eur. J. Neurosci. 2010, 32, 1632–1645. [Google Scholar] [CrossRef] [PubMed]
- Fellner, S.K.; Arendshorst, W.J. Voltage-gated Ca2+ entry and ryanodine receptor Ca2+-induced Ca2+ release in preglomerular arterioles. Am. J. Physiol. Renal Physiol. 2007, 292, F1568–F1572. [Google Scholar] [CrossRef]
- Endo, M. Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 2009, 89, 1153–1176. [Google Scholar] [CrossRef]
- Meissner, G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium 2004, 35, 621–628. [Google Scholar] [CrossRef]
- Balshaw, D.M.; Xu, L.; Yamaguchi, N.; Pasek, D.A.; Meissner, G. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J. Biol. Chem. 2001, 276, 20144–20153. [Google Scholar] [CrossRef]
- Camors, E.; Valdivia, H.H. CaMKII regulation of cardiac ryanodine receptors and inositol triphosphate receptors. Front. Pharmacol. 2014, 5, 101. [Google Scholar] [CrossRef]
- Zima, A.V.; Copello, J.A.; Blatter, L.A. Differential modulation of cardiac and skeletal muscle ryanodine receptors by NADH. FEBS Lett. 2003, 547, 32–36. [Google Scholar] [CrossRef]
- Laver, D.; Baynes, T.; Dulhunty, A. Magnesium inhibition of ryanodine-receptor calcium channels: Evidence for two independent mechanisms. J. Membr. Biol. 1997, 156, 213–229. [Google Scholar] [CrossRef]
- Yao, Y.; Choi, J.; Parker, I. Quantal puffs of intracellular Ca2+ evoked by inositol trisphosphate in Xenopus oocytes. J. Physiol. 1995, 482, 533–553. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Miyashita, Y.; Kasai, H. Micromolar and submicromolar Ca2+ spikes regulating distinct cellular functions in pancreatic acinar cells. EMBO J. 1997, 16, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Hoang-Trong, T.M.; Ullah, A.; Jafri, M.S. Calcium sparks in the heart: Dynamics and regulation. Res. Rep. Biol. 2015, 6, 203. [Google Scholar]
- Lipp, P.; Thomas, D.; Berridge, M.J.; Bootman, M.D. Nuclear calcium signalling by individual cytoplasmic calcium puffs. EMBO J. 1997, 16, 7166–7173. [Google Scholar] [CrossRef] [PubMed]
- Foskett, J.K.; White, C.; Cheung, K.-H.; Mak, D.-O.D. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef]
- Giannone, G.; Rondé, P.; Gaire, M.; Beaudouin, J.; Haiech, J.; Ellenberg, J.; Takeda, K. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J. Biol. Chem. 2004, 279, 28715–28723. [Google Scholar] [CrossRef]
- Dickinson, G.D.; Swaminathan, D.; Parker, I. The probability of triggering calcium puffs is linearly related to the number of inositol trisphosphate receptors in a cluster. Biophys. J. 2012, 102, 1826–1836. [Google Scholar] [CrossRef][Green Version]
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef]
- Prakriya, M. The molecular physiology of CRAC channels. Immunol. Rev. 2009, 231, 88–98. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Do Heo, W.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Nwokonko, R.M.; Cai, X.; Loktionova, N.A.; Abdulqadir, R.; Xin, P.; Niemeyer, B.A.; Wang, Y.; Trebak, M.; Gill, D.L. Cross-linking of Orai1 channels by STIM proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E3398–E3407. [Google Scholar] [CrossRef] [PubMed]
- Raphaël, M.; Lehen’kyi, V.y.; Vandenberghe, M.; Beck, B.; Khalimonchyk, S.; Abeele, F.V.; Farsetti, L.; Germain, E.; Bokhobza, A.; Mihalache, A.; et al. TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc. Natl. Acad. Sci. USA 2014, 111, E3870–E3879. [Google Scholar] [CrossRef] [PubMed]
- Elaib, Z.; Saller, F.; Bobe, R. The calcium entry-calcium refilling coupling. In Calcium Entry Pathways in Non-Excitable Cells; Springer International Publishing: Cham, Switerland, 2016; pp. 333–352. [Google Scholar]
- Møller, J.V.; Olesen, C.; Winther, A.-M.L.; Nissen, P. The sarcoplasmic Ca 2+-ATPase: Design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 2010, 43, 501–566. [Google Scholar] [CrossRef]
- Gélébart, P.; Martin, V.; Enouf, J.; Papp, B. Identification of a new SERCA2 splice variant regulated during monocytic differentiation. Biochem. Biophys. Res. Commun. 2003, 303, 676–684. [Google Scholar] [CrossRef]
- Altshuler, I.; Vaillant, J.J.; Xu, S.; Cristescu, M.E. The evolutionary history of sarco (endo) plasmic calcium ATPase (SERCA). PLoS ONE 2012, 7, e52617. [Google Scholar] [CrossRef]
- Dally, S.; Corvazier, E.; Bredoux, R.; Bobe, R.; Enouf, J. Multiple and diverse coexpression, location, and regulation of additional SERCA2 and SERCA3 isoforms in nonfailing and failing human heart. J. Mol. Cell. Cardiol. 2010, 48, 633–644. [Google Scholar] [CrossRef]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Bobe, R.; Bredoux, R.; Corvazier, E.; Lacabaratz-Porret, C.; Martin, V.; Kovacs, T.; Enouf, J. How many Ca2+ ATPase isoforms are expressed in a cell type? A growing family of membrane proteins illustrated by studies in platelets. Platelets 2005, 16, 133–150. [Google Scholar] [CrossRef]
- Corvazier, E.; Bredoux, R.; Kovács, T.; Enouf, J. Expression of sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) 3 proteins in two major conformational states in native human cell membranes. Biochim. Et Biophys. Acta 2009, 1788, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Sakuta, N.; Watanabe, S.; Zhang, Y.; Yoshikaie, K.; Tanaka, Y.; Ushioda, R.; Kato, Y.; Takagi, J.; Tsukazaki, T.; et al. Structural Basis of Sarco/Endoplasmic Reticulum Ca2+-ATPase 2b Regulation via Transmembrane Helix Interplay. Cell Rep. 2019, 27, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Vandecaetsbeek, I.; Trekels, M.; De Maeyer, M.; Ceulemans, H.; Lescrinier, E.; Raeymaekers, L.; Wuytack, F.; Vangheluwe, P. Structural basis for the high Ca2+ affinity of the ubiquitous SERCA2b Ca2+ pump. Proc. Natl. Acad. Sci. USA 2009, 106, 18533–18538. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekera, P.C.; Kargacin, M.E.; Deans, J.P.; Lytton, J. Determination of apparent calcium affinity for endogenously expressed human sarco (endo) plasmic reticulum calcium-ATPase isoform SERCA3. Am. J. Physiol. Cell Physiol. 2009, 296, C1105–C1114. [Google Scholar] [CrossRef]
- Martin, V.; Bredoux, R.; Corvazier, E.; Van Gorp, R.; Kovàcs, T.; Gélébart, P.; Enouf, J. Three novel sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) 3 isoforms expression, regulation, and function of the members of the SERCA3 family. J. Biol. Chem. 2002, 277, 24442–24452. [Google Scholar] [CrossRef]
- Chen, J.; Sitsel, A.; Benoy, V.; Sepúlveda, M.R.; Vangheluwe, P. Primary active Ca2+ transport systems in health and disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035113. [Google Scholar] [CrossRef]
- Bilmen, J.G.; Khan, S.Z.; Javed, M.u.H.; Michelangeli, F. Inhibition of the SERCA Ca2+ pumps by curcumin: Curcumin putatively stabilizes the interaction between the nucleotide-binding and phosphorylation domains in the absence of ATP. Eur. J. Biochem. 2001, 268, 6318–6327. [Google Scholar] [CrossRef]
- Asahi, M.; Green, N.M.; Kurzydlowski, K.; Tada, M.; MacLennan, D.H. Phospholamban domain IB forms an interaction site with the loop between transmembrane helices M6 and M7 of sarco (endo) plasmic reticulum Ca2+ ATPases. Proc. Natl. Acad. Sci. USA 2001, 98, 10061–10066. [Google Scholar] [CrossRef]
- Dicke, A.A.; Gopinath, T.; Vostrikov, V.V.; Veglia, G. The Effects of Sarcolipin Phosphorylation on SERCA Regulation. Biophys. J. 2016, 110, 395a. [Google Scholar] [CrossRef]
- MacLENNAN, D.H.; Toyofuku, T.; Kimura, Y. Sites of regulatory interaction between calcium ATPases and phospholamban. In Alterations of Excitation-Contraction Coupling in the Failing Human Heart; Steinkopff-Verlag: Heidelberg, Germany, 1998; pp. 17–24. [Google Scholar]
- Lisak, D.A.; Schacht, T.; Enders, V.; Habicht, J.; Kiviluoto, S.; Schneider, J.; Henke, N.; Bultynck, G.; Methner, A.; Nickel, N. The transmembrane Bax inhibitor motif (TMBIM) containing protein family: Tissue expression, intracellular localization and effects on the ER CA2+-filling state. Biochim. Biophys. Acta 2015, 1853, 2104–2114. [Google Scholar] [CrossRef]
- Liu, Q. TMBIM-mediated Ca2+ homeostasis and cell death. Biochim. Biophys. Acta 2017, 1864, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Bultynck, G.; Kiviluoto, S.; Methner, A. Bax inhibitor-1 is likely a pH-sensitive calcium leak channel, not a H+/Ca2+ exchanger. Sci. Signal. 2014, 7, pe22. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Bruni, R.; Kloss, B.; Assur, Z.; Kloppmann, E.; Rost, B.; Hendrickson, W.A.; Liu, Q. Structural basis for a pH-sensitive calcium leak across membranes. Science 2014, 344, 1131–1135. [Google Scholar] [CrossRef]
- Guo, G.; Xu, M.; Chang, Y.; Luyten, T.; Seitaj, B.; Liu, W.; Zhu, P.; Bultynck, G.; Shi, L.; Quick, M.; et al. Ion and pH Sensitivity of a TMBIM Ca2+ Channel. Structure 2019, 27, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Bultynck, G.; Kiviluoto, S.; Henke, N.; Ivanova, H.; Schneider, L.; Rybalchenko, V.; Luyten, T.; Nuyts, K.; De Borggraeve, W.; Bezprozvanny, I.; et al. The C terminus of Bax inhibitor-1 forms a Ca2+-permeable channel pore. J. Biol. Chem. 2012, 287, 2544–2557. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 is an ER Ca2+ load-activated Ca2+ channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef]
- Lam, D.; Kosta, A.; Luciani, M.-F.; Golstein, P. The inositol 1, 4, 5-trisphosphate receptor is required to signal autophagic cell death. Mol. Biol. Cell 2008, 19, 691–700. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Huang, J.; Liu, W.; Kou, X.; Tang, H.; Wang, H.; Yu, X.; Gao, S.; Ouyang, K.; Yang, H.-T. IP3R-mediated Ca2+ signals govern hematopoietic and cardiac divergence of Flk1+ cells via the calcineurin–NFATc3–Etv2 pathway. J. Mol. Cell Biol. 2017, 9, 274–288. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef]
- Shimizu, S.; Konishi, A.; Kodama, T.; Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 3100–3105. [Google Scholar] [CrossRef]
- Rong, Y.-P.; Aromolaran, A.S.; Bultynck, G.; Zhong, F.; Li, X.; McColl, K.; Matsuyama, S.; Herlitze, S.; Roderick, H.L.; Bootman, M.D.; et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol. Cell 2008, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.-P.; Bultynck, G.; Aromolaran, A.S.; Zhong, F.; Parys, J.B.; De Smedt, H.; Mignery, G.A.; Roderick, H.L.; Bootman, M.D.; Distelhorst, C.W. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 14397–14402. [Google Scholar] [CrossRef]
- Monaco, G.; Decrock, E.; Akl, H.; Ponsaerts, R.; Vervliet, T.; Luyten, T.; De Maeyer, M.; Missiaen, L.; Distelhorst, C.; De Smedt, H.; et al. Selective regulation of IP 3-receptor-mediated Ca 2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012, 19, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, H.; Wagner, L.E.; Tanimura, A.; Vandermarliere, E.; Luyten, T.; Welkenhuyzen, K.; Alzayady, K.J.; Wang, L.; Hamada, K.; Mikoshiba, K.; et al. Mikoshiba, K. Bcl-2 and IP 3 compete for the ligand-binding domain of IP 3 Rs modulating Ca 2+ signaling output. Cell. Mol. Life Sci. 2019, 76, 3843–3859. [Google Scholar] [CrossRef]
- White, C.; Li, C.; Yang, J.; Petrenko, N.B.; Madesh, M.; Thompson, C.B.; Foskett, J.K. The endoplasmic reticulum gateway to apoptosis by Bcl-X L modulation of the InsP 3 R. Nat. Cell Biol. 2005, 7, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, X.; Vais, H.; Thompson, C.B.; Foskett, J.K.; White, C. Apoptosis regulation by Bcl-xL modulation of mammalian inositol 1, 4, 5-trisphosphate receptor channel isoform gating. Proc. Natl. Acad. Sci. USA 2007, 104, 12565–12570. [Google Scholar] [CrossRef] [PubMed]
- Eckenrode, E.F.; Yang, J.; Velmurugan, G.V.; Foskett, J.K.; White, C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1, 4, 5-trisphosphate receptor-dependent Ca2+ signaling. J. Biol. Chem. 2010, 285, 13678–13684. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Harr, M.W.; Bultynck, G.; Monaco, G.; Parys, J.B.; De Smedt, H.; Rong, Y.-P.; Molitoris, J.K.; Lam, M.; Ryder, C.; et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2–IP3 receptor interaction. Blood J. Am. Soc. Hematol. 2011, 117, 2924–2934. [Google Scholar] [CrossRef]
- Lavik, A.R.; Zhong, F.; Chang, M.-J.; Greenberg, E.; Choudhary, Y.; Smith, M.R.; McColl, K.S.; Pink, J.; Reu, F.J.; Matsuyama, S.; et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget 2015, 6, 27388. [Google Scholar] [CrossRef]
- Greenberg, E.; McColl, K.; Zhong, F.; Wildey, G.; Dowlati, A.; Distelhorst, C. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1, 4, 5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis. 2015, 6, e2034. [Google Scholar] [CrossRef]
- Distelhorst, C.W. Targeting Bcl-2-IP3 receptor interaction to treat cancer: A novel approach inspired by nearly a century treating cancer with adrenal corticosteroid hormones. Biochim. Biophys. Acta 2018, 1865, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Distelhorst, C.W.; Bootman, M.D. Creating a new cancer therapeutic agent by targeting the interaction between Bcl-2 and IP3 receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035196. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3-and Ca 2+-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca 2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Pierro, C.; Cook, S.J.; Foets, T.C.; Bootman, M.D.; Roderick, H.L. Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodelling of the isoform composition of IP3Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J. Cell Sci. 2014, 127, 1607–1619. [Google Scholar] [CrossRef]
- Sakakura, C.; Miyagawa, K.; Fukuda, K.; Shimomura, K.; Takemura, M.; Takagi, T.; Kin, S.; Nakase, Y.; Fujiyama, J.; Mikoshiba, K.; et al. Possible involvement of inositol 1, 4, 5-trisphosphate receptor type 3 (IP3R3) in the peritoneal dissemination of gastric cancers. Gan Kagaku Ryoho. Cancer Chemother. 2003, 30, 1784–1787. [Google Scholar]
- Kang, S.S.; Han, K.-S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Inhibition of the Ca2+ release channel, IP3R subtype 3 by caffeine slows glioblastoma invasion and migration and extends survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef]
- Rezuchova, I.; Hudecova, S.; Soltysova, A.; Matuskova, M.; Durinikova, E.; Chovancova, B.; Zuzcak, M.; Cihova, M.; Burikova, M.; Penesova, A.; et al. Type 3 inositol 1, 4, 5-trisphosphate receptor has antiapoptotic and proliferative role in cancer cells. Cell Death Dis. 2019, 10, 186. [Google Scholar] [CrossRef]
- Mound, A.; Vautrin-Glabik, A.; Foulon, A.; Botia, B.; Hague, F.; Parys, J.B.; Ouadid-Ahidouch, H.; Rodat-Despoix, L. Downregulation of type 3 inositol (1, 4, 5)-trisphosphate receptor decreases breast cancer cell migration through an oscillatory Ca2+ signal. Oncotarget 2017, 8, 72324. [Google Scholar] [CrossRef]
- Kang, S.S.; Han, K.-S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-mediated inhibition of calcium release channel inositol 1, 4, 5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef]
- Vautrin-Glabik, A.; Botia, B.; Kischel, P.; Ouadid-Ahidouch, H.; Rodat-Despoix, L. IP3R3 silencing induced actin cytoskeletal reorganization through ARHGAP18/RhoA/mDia1/FAK pathway in breast cancer cell lines. Biochim. Biophys. Acta 2018, 1865, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Shibao, K.; Fiedler, M.J.; Nagata, J.; Minagawa, N.; Hirata, K.; Nakayama, Y.; Iwakiri, Y.; Nathanson, M.H.; Yamaguchi, K. The type III inositol 1, 4, 5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 2010, 48, 315–323. [Google Scholar] [CrossRef]
- Chovancova, B.; Hudecova, S.; Lencesova, L.; Babula, P.; Rezuchova, I.; Penesova, A.; Grman, M.; Moravcik, R.; Zeman, M.; Krizanova, O. Melatonin-induced changes in cytosolic calcium might be responsible for apoptosis induction in tumour cells. Cell. Physiol. Biochem. 2017, 44, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Hudecova, S.; Markova, J.; Simko, V.; Csaderova, L.; Stracina, T.; Sirova, M.; Fojtu, M.; Svastova, E.; Gronesova, P.; Pastorek, M.; et al. Sulforaphane-induced apoptosis involves the type 1 IP3 receptor. Oncotarget 2016, 7, 61403–61418. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsunoda, T.; Koga, H.; Yokomizo, A.; Tatsugami, K.; Eto, M.; Inokuchi, J.; Hirata, A.; Masuda, K.; Okumura, K.; Naito, S. Inositol 1, 4, 5-trisphosphate (IP 3) receptor type1 (IP 3 R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene 2005, 24, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Shui, B.; Zhao, W.; Liu, H.; Li, W.; Lee, J.C.; Doran, R.; Lee, F.K.; Sun, T.; Shen, Q.S.; et al. Central role of IP 3 R2-mediated Ca 2+ oscillation in self-renewal of liver cancer stem cells elucidated by high-signal ER sensor. Cell Death Dis. 2019, 10, 396. [Google Scholar] [CrossRef]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calvé, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 5, 3792. [Google Scholar] [CrossRef]
- Huang, X.; Jin, M.; Chen, Y.-X.; Wang, J.; Zhai, K.; Chang, Y.; Yuan, Q.; Yao, K.-T.; Ji, G. ERP44 inhibits human lung cancer cell migration mainly via IP3R2. Aging 2016, 8, 1276–1285. [Google Scholar] [CrossRef][Green Version]
- Bittremieux, M.; La Rovere, R.M.; Akl, H.; Martines, C.; Welkenhuyzen, K.; Dubron, K.; Baes, M.; Janssens, A.; Vandenberghe, P.; Laurenti, L.; et al. Constitutive IP 3 signaling underlies the sensitivity of B-cell cancers to the Bcl-2/IP 3 receptor disruptor BIRD-2. Cell Death Differ. 2019, 26, 531–547. [Google Scholar] [CrossRef]
- Akl, H.; Monaco, G.; La Rovere, R.; Welkenhuyzen, K.; Kiviluoto, S.; Vervliet, T.; Molgó, J.; Distelhorst, C.; Missiaen, L.; Mikoshiba, K.; et al. IP 3 R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP 3 R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013, 4, e632. [Google Scholar] [CrossRef]
- Witherspoon, J.W.; Meilleur, K.G. Review of RyR1 pathway and associated pathomechanisms. Acta Neuropathol. Commun. 2016, 4, 121. [Google Scholar] [CrossRef] [PubMed]
- Ather, S.; Respress, J.L.; Li, N.; Wehrens, X.H. Alterations in ryanodine receptors and related proteins in heart failure. Biochim. Biophys. Acta 2013, 1832, 2425–2431. [Google Scholar] [CrossRef]
- Murayama, T.; Ogawa, Y. Properties of Ryr3 ryanodine receptor isoform in mammalian brain. J. Biol. Chem. 1996, 271, 5079–5084. [Google Scholar] [PubMed]
- Abdul, M.; Ramlal, S.; Hoosein, N. Ryanodine receptor expression correlates with tumor grade in breast cancer. Pathol. Oncol. Res. 2008, 14, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Zhang, D.; Chen, J.; He, G.; Gao, L. Low expression of ryanodine receptor 2 is associated with poor prognosis in thyroid carcinoma. Oncol. Lett. 2019, 18, 3605–3612. [Google Scholar] [CrossRef]
- Deli, T.; Varga, N.; Ádám, A.; Kenessey, I.; Rásó, E.; Puskás, L.G.; Tóvári, J.; Fodor, J.; Fehér, M.; Szigeti, G.P.; et al. Functional genomics of calcium channels in human melanoma cells. Int. J. Cancer 2007, 121, 55–65. [Google Scholar] [CrossRef]
- Bennett, D.L.; Cheek, T.R.; Berridge, M.J.; De Smedt, H.; Parys, J.B.; Missiaen, L.; Bootman, M.D. Expression and function of ryanodine receptors in nonexcitable cells. J. Biol. Chem. 1996, 271, 6356–6362. [Google Scholar] [CrossRef]
- Wang, C.; Liang, H.; Lin, C.; Li, F.; Xie, G.; Qiao, S.; Shi, X.; Deng, J.; Zhao, X.; Wu, K.; et al. Molecular subtyping and prognostic assessment based on tumor mutation burden in patients with lung adenocarcinomas. Int. J. Mol. Sci. 2019, 20, 4251. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Y.; Song, F.; Zheng, H.; Hu, L.; Lu, H.; Liu, P.; Hao, X.; Zhang, W.; Chen, K. Functional SNP in the microRNA-367 binding site in the 3′ UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc. Natl. Acad. Sci. USA 2011, 108, 13653–13658. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Michelangeli, F.; Qu, Y.Q.; Xu, S.-W.; Han, Y.; Mok, S.W.F.; Dias, I.R.D.S.R.; Javed, M.-U.-H.; Chan, W.-K.; Xue, W.-W.; et al. Neferine induces autophagy-dependent cell death in apoptosis-resistant cancers via ryanodine receptor and Ca 2+-dependent mechanism. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef]
- Lu, H.; Chen, I.; Shimoda, L.A.; Park, Y.; Zhang, C.; Tran, L.; Zhang, H.; Semenza, G.L. Chemotherapy-Induced Ca2+ release stimulates breast cancer stem cell enrichment. Cell Rep. 2017, 18, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Mackrill, J.J. Ryanodine receptor calcium channels and their partners as drug targets. Biochem. Pharmacol. 2010, 79, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Toker, A. NFAT proteins: Emerging roles in cancer progression. Nat. Rev. Cancer 2009, 9, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Chuderland, D.; Seger, R. Calcium regulates ERK signaling by modulating its protein-protein interactions. Commun. Integr. Biol. 2008, 1, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Sun, J.; Huang, M.-Y.; Wang, Y.-S.; Hou, M.-F.; Sun, Y.; He, H.; Krishna, N.; Chiu, S.-J.; Lin, S.; et al. STIM1 overexpression promotes colorectal cancer progression, cell motility and COX-2 expression. Oncogene 2015, 34, 4358–4367. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.-Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Chiu, W.-T.; Chen, Y.-T.; Lin, P.-Y.; Huang, H.-J.; Chou, C.-Y.; Chang, H.-C.; Tang, M.-J.; Shen, M.-R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.-C.; Kim, M.; et al. STIM1-and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef]
- Okeke, E.; Parker, T.; Dingsdale, H.; Concannon, M.; Awais, M.; Voronina, S.; Molgó, J.; Begg, M.; Metcalf, D.; Knight, A.E.; et al. Epithelial–mesenchymal transition, IP3 receptors and ER–PM junctions: Translocation of Ca2+ signalling complexes and regulation of migration. Biochem. J. 2016, 473, 757–767. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Shuttleworth, T. What drives calcium entry during [Ca2+] ioscillations?–challenging the capacitative model. Cell Calcium 1999, 25, 237–246. [Google Scholar] [CrossRef]
- Shuttleworth, T.J. Arachidonic acid activates the noncapacitative entry of Ca2+ during [Ca2+] i oscillations. J. Biol. Chem. 1996, 271, 21720–21725. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A Novel Native Store-operated Calcium Channel Encoded by Orai3 selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor α-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef]
- Dubois, C.; Abeele, F.V.; Lehen’kyi, V.y.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Kucukkaya, B.; Basoglu, H.; Erdag, D.; Akbas, F.; Susgun, S.; Yalcintepe, L. Calcium homeostasis in cisplatin resistant epithelial ovarian cancer. Gen. Physiol. Biophys. 2019, 38, 353–363. [Google Scholar] [CrossRef]
- Bergner, A.; Kellner, J.; Tufman, A.; Huber, R.M. Endoplasmic reticulum Ca 2+-homeostasis is altered in small and non-small cell lung cancer cell lines. J. Exp. Clin. Cancer Res. 2009, 28, 25. [Google Scholar] [CrossRef] [PubMed]
- Korošec, B.; Glavač, D.; Rott, T.; Ravnik-Glavač, M. Alterations in the ATP2A2 gene in correlation with colon and lung cancer. Cancer Genet. Cytogenet. 2006, 171, 105–111. [Google Scholar] [CrossRef]
- Pacifico, F.; Ulianich, L.; De Micheli, S.; Treglia, S.; Leonardi, A.; Vito, P.; Formisano, S.; Consiglio, E.; Di Jeso, B. The expression of the sarco/endoplasmic reticulum Ca2+-ATPases in thyroid and its down-regulation following neoplastic transformation. J. Mol. Endocrinol. 2003, 30, 399–409. [Google Scholar] [CrossRef]
- Varga, K.; Hollósi, A.; Pászty, K.; Hegedűs, L.; Szakács, G.; Tímár, J.; Papp, B.; Enyedi, Á.; Padányi, R. Expression of calcium pumps is differentially regulated by histone deacetylase inhibitors and estrogen receptor alpha in breast cancer cells. BMC Cancer 2018, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Boivin, G.P.; Miller, M.L.; Liu, L.H.; Erwin, C.R.; Warner, B.W.; Shull, G.E. Haploinsufficiency of Atp2a2, encoding the sarco (endo) plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump, predisposes mice to squamous cell tumors via a novel mode of cancer susceptibility. Cancer Res. 2005, 65, 8655–8661. [Google Scholar] [CrossRef] [PubMed]
- Papp, B.; Brouland, J.-P. Altered endoplasmic reticulum calcium pump expression during breast tumorigenesis. Breast Cancer Basic Clin. Res. 2011, 5, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Brouland, J.-P.; Gélébart, P.; Kovacs, T.; Enouf, J.; Grossmann, J.; Papp, B. The loss of sarco/endoplasmic reticulum calcium transport ATPase 3 expression is an early event during the multistep process of colon carcinogenesis. Am. J. Pathol. 2005, 167, 233–242. [Google Scholar] [CrossRef]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca 2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed]
- Szalai, G.; Krishnamurthy, R.; Hajnóczky, G. Apoptosis driven by IP3-linked mitochondrial calcium signals. EMBO J. 1999, 18, 6349–6361. [Google Scholar] [CrossRef]
- Vanoverberghe, K.; Abeele, F.V.; Mariot, P.; Lepage, G.; Roudbaraki, M.; Bonnal, J.; Mauroy, B.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Ca 2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2004, 11, 321–330. [Google Scholar] [CrossRef]
- Schoneich, C.; Dremina, E.; Hewarathna, A. Bcl-2 modulates ER/SR calcium uptake by interaction with SERCA and heat shock proteins. Free Radic. Biol. Med. 2017, 108, S73. [Google Scholar] [CrossRef]
- Dremina, E.S.; Sharov, V.S.; Kumar, K.; Zaidi, A.; Michaelis, E.K.; Schöneich, C. Anti-apoptotic protein Bcl-2 interacts with and destabilizes the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). Biochem. J. 2004, 383, 361–370. [Google Scholar] [CrossRef]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Masud, A.; Mohapatra, A.; Lakhani, S.A.; Ferrandino, A.; Hakem, R.; Flavell, R.A. Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. J. Biol. Chem. 2007, 282, 14132–14139. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Ramirez, F.G.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Fan, L.; Li, A.; Li, W.; Cai, P.; Yang, B.; Zhang, M.; Gu, Y.; Shu, Y.; Sun, Y.; Shen, Y.; et al. Novel role of Sarco/endoplasmic reticulum calcium ATPase 2 in development of colorectal cancer and its regulation by F36, a curcumin analog. Biomed. Pharmacother. 2014, 68, 1141–1148. [Google Scholar] [CrossRef]
- Crépin, A.; Bidaux, G.; Vanden-Abeele, F.; Dewailly, E.; Goffin, V.; Prevarskaya, N.; Slomianny, C. Prolactin stimulates prostate cell proliferation by increasing endoplasmic reticulum content due to SERCA 2b over-expression. Biochem. J. 2007, 401, 49–55. [Google Scholar] [CrossRef]
- Seo, J.-A.; Kim, B.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Curcumin induces apoptosis by inhibiting sarco/endoplasmic reticulum Ca2+ ATPase activity in ovarian cancer cells. Cancer Lett. 2016, 371, 30–37. [Google Scholar] [CrossRef]
- Xu, X.-Y.; Gou, W.-F.; Yang, X.; Wang, G.-L.; Takahashi, H.; Yu, M.; Mao, X.-Y.; Takano, Y.; Zheng, H.-C. Aberrant SERCA3 expression is closely linked to pathogenesis, invasion, metastasis, and prognosis of gastric carcinomas. Tumor Biol. 2012, 33, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.; Rao, R. Calcium-ATPases: Gene disorders and dysregulation in cancer. Biochim. Biophys. Acta 2016, 1863, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Parsonage, M.T.; Cabot, P.J.; Parat, M.-O.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Assessment of gene expression of intracellular calcium channels, pumps and exchangers with epidermal growth factor-induced epithelial-mesenchymal transition in a breast cancer cell line. Cancer Cell Int. 2013, 13, 76. [Google Scholar] [CrossRef] [PubMed]
- Arbabian, A.; Brouland, J.P.; Gélébart, P.; Kovàcs, T.; Bobe, R.; Enouf, J.; Papp, B. Endoplasmic reticulum calcium pumps and cancer. Biofactors 2011, 37, 139–149. [Google Scholar] [CrossRef]
- Xu, M.; Seas, A.; Kiyani, M.; Ji, K.S.; Bell, H.N. A temporal examination of calcium signaling in cancer-from tumorigenesis, to immune evasion, and metastasis. Cell Biosci. 2018, 8, 25. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Carbone, M.; Amelio, I.; Affar, E.B.; Brugarolas, J.; Cannon-Albright, L.A.; Cantley, L.C.; Cavenee, W.K.; Chen, Z.; Croce, C.M.; D’Andrea, A.; et al. Consensus report of the 8 and 9th Weinman Symposia on Gene x Environment Interaction in carcinogenesis: Novel opportunities for precision medicine. Cell Death Differ. 2018, 25, 1885–1904. [Google Scholar] [CrossRef]
- Kerkhofs, M.; Bittremieux, M.; Morciano, G.; Giorgi, C.; Pinton, P.; Parys, J.B.; Bultynck, G. Emerging molecular mechanisms in chemotherapy: Ca 2+ signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Tombal, B.; Weeraratna, A.T.; Denmeade, S.R.; Isaacs, J.T. Thapsigargin induces a calmodulin/calcineurin-dependent apoptotic cascade responsible for the death of prostatic cancer cells. Prostate 2000, 43, 303–317. [Google Scholar] [CrossRef]
- Wu, Y.; Fabritius, M.; Ip, C. Chemotherapeutic sensitization by endoplasmic reticulum stress: Increasing the efficacy of taxane against prostate cancer. Cancer Biol. Ther. 2009, 8, 146–152. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. The SERCA pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol. Ther. 2005, 4, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.-H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflügers Arch. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Sampath, B.; Sankaranarayanan, K. Glu106 targeted inhibitors of ORAI1 as potential Ca2+ release-activated Ca2+ (CRAC) channel blockers–molecular modeling and docking studies. J. Recept. Signal Transduct. 2016, 36, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.C.; Qu, B.; Hoth, M. Calcium, cancer and killing: The role of calcium in killing cancer cells by cytotoxic T lymphocytes and natural killer cells. Biochim. Biophys. Acta 2013, 1833, 1603–1611. [Google Scholar] [CrossRef]
- Hoth, M. CRAC channels, calcium, and cancer in light of the driver and passenger concept. Biochim. Biophys. Acta 2016, 1863, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Maul-Pavicic, A.; Chiang, S.C.; Rensing-Ehl, A.; Jessen, B.; Fauriat, C.; Wood, S.M.; Sjöqvist, S.; Hufnagel, M.; Schulze, I.; Bass, T.; et al. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc. Natl. Acad. Sci. USA 2011, 108, 3324–3329. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Lin, P.-C.; Yeh, Y.-M.; Chen, L.-H.; Shen, M.-R. Store-operated Ca2+ entry in tumor progression: From molecular mechanisms to clinical implications. Cancers 2019, 11, 899. [Google Scholar] [CrossRef] [PubMed]
- Trebak, M.; Kinet, J.-P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019, 19, 154–169. [Google Scholar] [CrossRef]
- Weidinger, C.; Shaw, P.J.; Feske, S. STIM1 and STIM2-mediated Ca2+ influx regulates antitumour immunity by CD8+ T cells. EMBO Mol. Med. 2013, 5, 1311–1321. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).