Parkin Overexpression Attenuates Sepsis-Induced Muscle Wasting

 ,
, 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Procedures
2.2. AAV Injections in Skeletal Muscle
2.3. Cecal Ligation and Perforation
2.4. Tissue Collection
2.5. Fiber Size Determination
2.6. Transmission Electron Microscopy (TEM)
2.7. Immunoblotting
2.8. Quantitative Real-Time PCR
2.9. Data Analysis and Statistics
3. Results
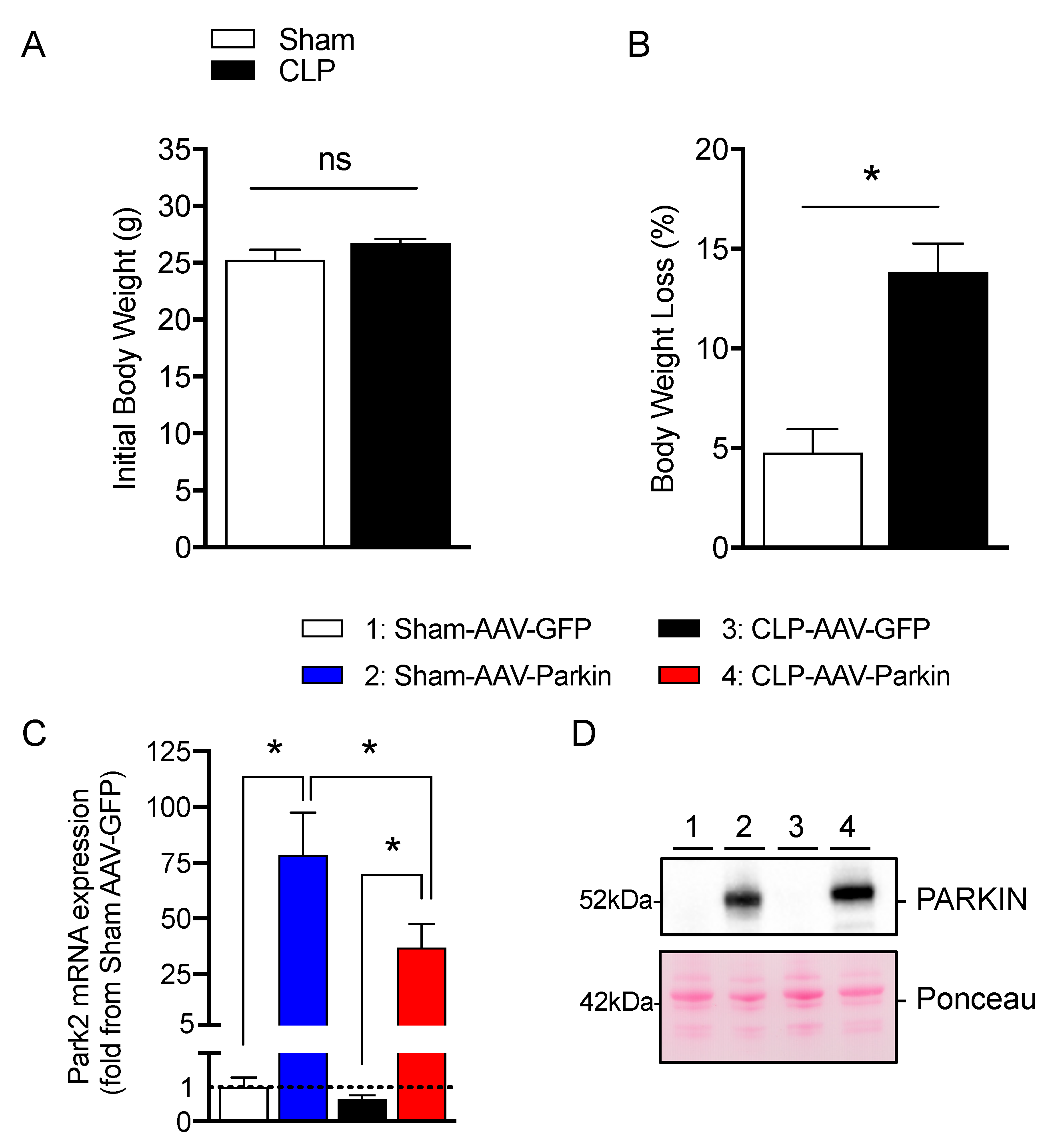
3.1. Successful Overexpression of Parkin in Skeletal Muscles of Sham and CLP Operated Mice
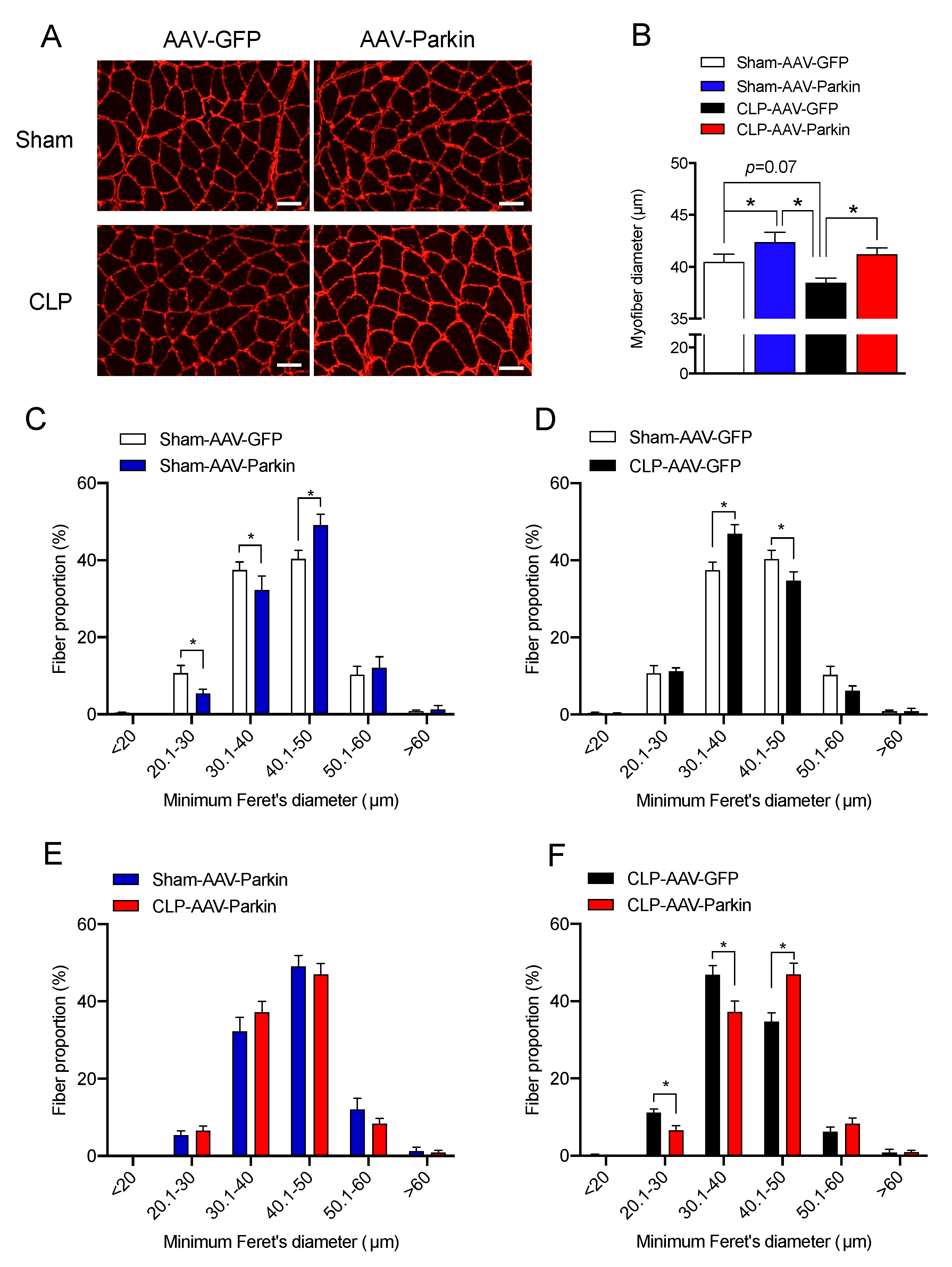
3.2. Parkin Overexpression Attenuates Sepsis-Induced Skeletal Muscle Atrophy
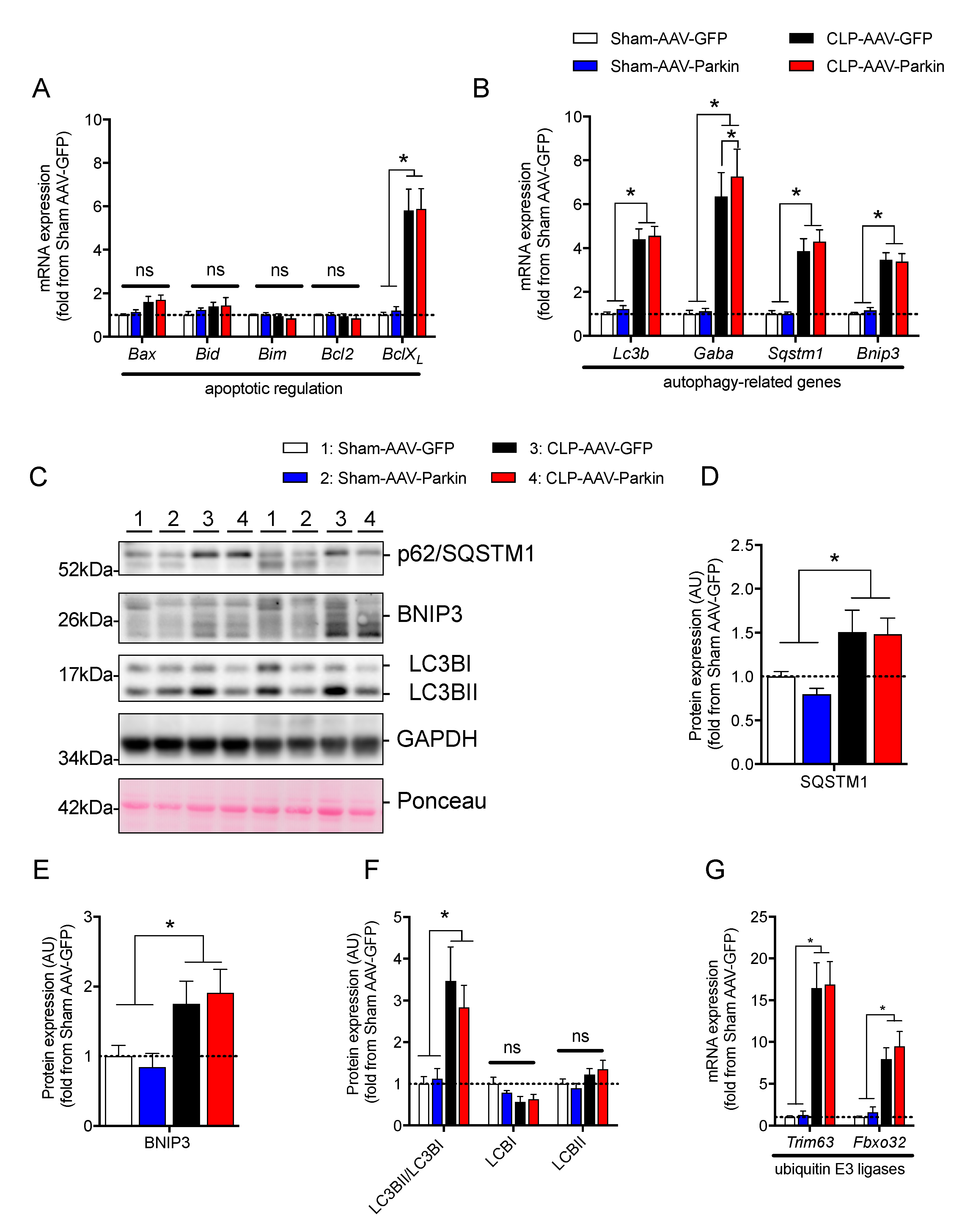
3.3. The Impact of Parkin Overexpression and Sepsis on Skeletal Muscle Catabolic Signaling
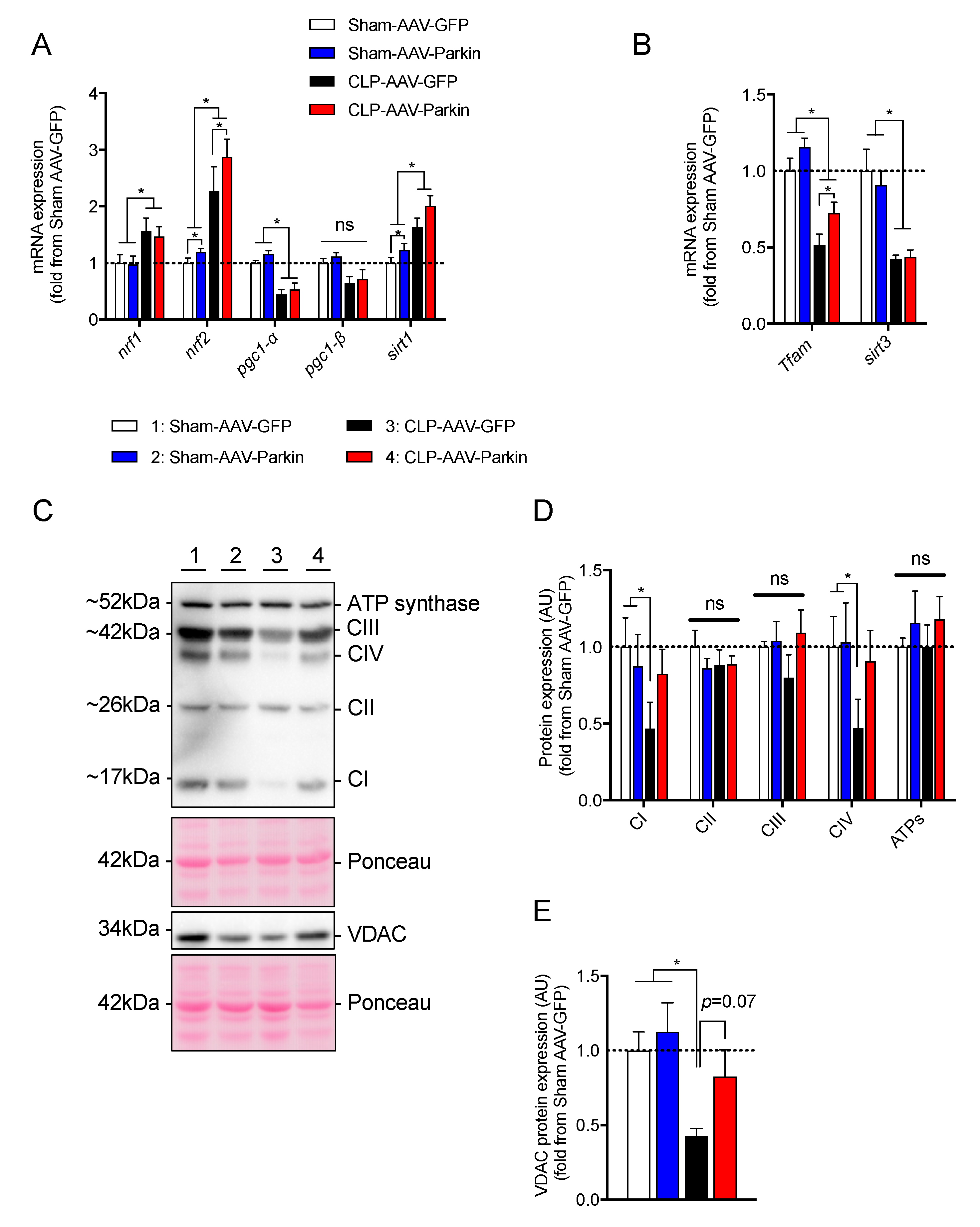
3.4. The Impact of Parkin Overexpression and Sepsis on the Expression of Genes and Proteins Regulating Mitochondrial Biology
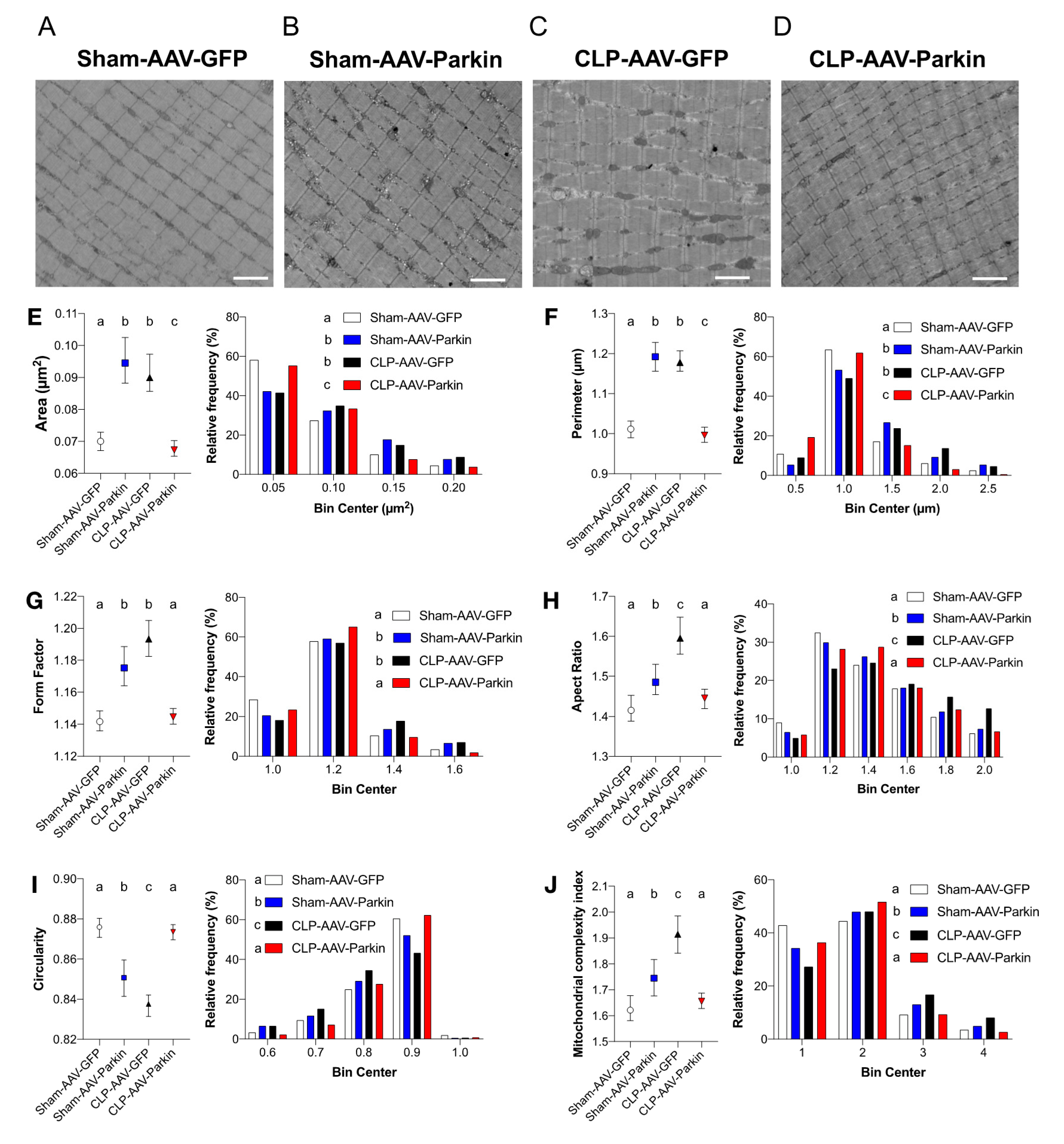
3.5. Effects of Parkin Overexpression and Sepsis on Mitochondrial Morphology and Dynamics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Harrison, T.B.; Rich, M.M.; Moss, M. Early development of critical illness myopathy and neuropathy in patients with severe sepsis. Neurology 2006, 67, 1421–1425. [Google Scholar] [CrossRef] [PubMed]
- Tennila, A.; Salmi, T.; Pettila, V.; Roine, R.O.; Varpula, T.; Takkunen, O. Early signs of critical illness polyneuropathy in icu patients with systemic inflammatory response syndrome or sepsis. Intensive Care Med. 2000, 26, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Supinski, G.S.; Callahan, L.A. Diaphragm weakness in mechanically ventilated critically ill patients. Crit. Care 2013, 17, R120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonghe, B.; Bastuji-Garin, S.; Durand, M.C.; Malissin, I.; Rodrigues, P.; Cerf, C.; Outin, H.; Sharshar, T. Respiratory weakness is associated with limb weakness and delayed weaning in critical illness. Crit. Care Med. 2007, 35, 2007–2015. [Google Scholar] [CrossRef]
- Ali, N.A.; O’Brien, J.M., Jr.; Hoffmann, S.P.; Phillips, G.; Garland, A.; Finley, J.C.; Almoosa, K.; Hejal, R.; Wolf, K.M.; Lemeshow, S.; et al. Acquired weakness, handgrip strength, and mortality in critically ill patients. Am. J. Respir. Crit. Care Med. 2008, 178, 261–268. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 2010, 304, 1787–1794. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, O.; Reid, M.; Van den Berghe, G.; Vanhorebeek, I.; Hermans, G.; Rich, M.; Larsson, L. The sick and the weak: Neuropathies/myopathies in the critically ill. Physiol. Rev. 2015, 95, 1025–1109. [Google Scholar] [CrossRef] [Green Version]
- Callahan, L.A.; Supinski, G.S. Sepsis-induced myopathy. Crit. Care Med. 2009, 37, S354–S367. [Google Scholar] [CrossRef] [Green Version]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Brealey, D.; Karyampudi, S.; Jacques, T.S.; Novelli, M.; Stidwill, R.; Taylor, V.; Smolenski, R.T.; Singer, M. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R491–R497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredriksson, K.; Hammarqvist, F.; Strigard, K.; Hultenby, K.; Ljungqvist, O.; Wernerman, J.; Rooyackers, O. Derangements in mitochondrial metabolism in intercostal and leg muscle of critically ill patients with sepsis-induced multiple organ failure. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1044–E1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooyackers, O.E.; Kersten, A.H.; Wagenmakers, A.J. Mitochondrial protein content and in vivo synthesis rates in skeletal muscle from critically ill rats. Clin. Sci. (Lond.) 1996, 91, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, L.A.; Supinski, G.S. Sepsis induces diaphragm electron transport chain dysfunction and protein depletion. Am. J. Respir Crit. Care Med. 2005, 172, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouser, E.D.; Julian, M.W.; Blaho, D.V.; Pfeiffer, D.R. Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit. Care Med. 2002, 30, 276–284. [Google Scholar] [CrossRef]
- Protti, A.; Carre, J.; Frost, M.T.; Taylor, V.; Stidwill, R.; Rudiger, A.; Singer, M. Succinate recovers mitochondrial oxygen consumption in septic rat skeletal muscle. Crit. Care Med. 2007, 35, 2150–2155. [Google Scholar] [CrossRef]
- Alvarez, S.; Boveris, A. Mitochondrial nitric oxide metabolism in rat muscle during endotoxemia. Free Radic Biol. Med. 2004, 37, 1472–1478. [Google Scholar] [CrossRef]
- Vanasco, V.; Cimolai, M.C.; Evelson, P.; Alvarez, S. The oxidative stress and the mitochondrial dysfunction caused by endotoxemia are prevented by alpha-lipoic acid. Free Radic. Res. 2008, 42, 815–823. [Google Scholar] [CrossRef]
- Welty-Wolf, K.E.; Simonson, S.G.; Huang, Y.C.; Fracica, P.J.; Patterson, J.W.; Piantadosi, C.A. Ultrastructural changes in skeletal muscle mitochondria in gram-negative sepsis. Shock 1996, 5, 378–384. [Google Scholar] [CrossRef]
- Mofarrahi, M.; Sigala, I.; Guo, Y.; Godin, R.; Davis, E.C.; Petrof, B.; Sandri, M.; Burelle, Y.; Hussain, S.N. Autophagy and skeletal muscles in sepsis. PLoS ONE 2012, 7, e47265. [Google Scholar] [CrossRef]
- Owen, A.M.; Patel, S.P.; Smith, J.D.; Balasuriya, B.K.; Mori, S.F.; Hawk, G.S.; Stromberg, A.J.; Kuriyama, N.; Kaneki, M.; Rabchevsky, A.G.; et al. Chronic muscle weakness and mitochondrial dysfunction in the absence of sustained atrophy in a preclinical sepsis model. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates pink1 import and proteolytic destabilization by parl. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. Pink1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. Pink1-dependent recruitment of parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F. Pink1 stabilized by mitochondrial depolarization recruits parkin to damaged mitochondria and activates latent parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Piquereau, J.; Godin, R.; Deschenes, S.; Bessi, V.L.; Mofarrahi, M.; Hussain, S.N.; Burelle, Y. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy 2013, 9, 1837–1851. [Google Scholar] [CrossRef]
- Stana, F.; Vujovic, M.; Mayaki, D.; Leduc-Gaudet, J.P.; Leblanc, P.; Huck, L.; Hussain, S.N.A. Differential regulation of the autophagy and proteasome pathways in skeletal muscles in sepsis. Crit. Care Med. 2017, 45, e971–e979. [Google Scholar] [CrossRef]
- Buras, J.A.; Holzmann, B.; Sitkovsky, M. Animal models of sepsis: Setting the stage. Nat. Rev. Drug Discov. 2005, 4, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G. Parkin overexpression protects from ageing-related loss of muscle mass and strength. J. Physiol. 2019, 597, 1975–1991. [Google Scholar] [CrossRef] [PubMed]
- Moarbes, V.; Mayaki, D.; Huck, L.; Leblanc, P.; Vassilakopoulos, T.; Petrof, B.J.; Hussain, S.N.A. Differential regulation of myofibrillar proteins in skeletal muscles of septic mice. Physiol. Rep. 2019, 7, e14248. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.P.; Picard, M.; St-Jean Pelletier, F.; Sgarioto, N.; Auger, M.J.; Vallee, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef] [Green Version]
- Briguet, A.; Courdier-Fruh, I.; Foster, M.; Meier, T.; Magyar, J.P. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul. Disord. 2004, 14, 675–682. [Google Scholar] [CrossRef]
- Picard, M.; Gentil, B.J.; McManus, M.J.; White, K.; St Louis, K.; Gartside, S.E.; Wallace, D.C.; Turnbull, D.M. Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J. Appl. Physiol. 2013, 115, 1562–1571. [Google Scholar] [CrossRef]
- Picard, M.; White, K.; Turnbull, D.M. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: A quantitative three-dimensional electron microscopy study. J. Appl. Physiol. 2013, 114, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. Pink1 phosphorylates ubiquitin to activate parkin e3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [Green Version]
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of parkin in skeletal muscle contractile and mitochondrial function. J. Physiol. 2018, 596, 2565–2579. [Google Scholar] [CrossRef] [PubMed]
- Peker, N.; Donipadi, V.; Sharma, M.; McFarlane, C.; Kambadur, R. Loss of parkin impairs mitochondrial function and leads to muscle atrophy. Am. J. Physiol. Cell Physiol. 2018, 315, C164–C185. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouspillou, G.; Sgarioto, N.; Norris, B.; Barbat-Artigas, S.; Aubertin-Leheudre, M.; Morais, J.A.; Burelle, Y.; Taivassalo, T.; Hepple, R.T. The relationship between muscle fiber type-specific pgc-1alpha content and mitochondrial content varies between rodent models and humans. PLoS ONE 2014, 9, e103044. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Alvarez, M.I.; Thabit, H.; Burns, N.; Shah, S.; Brema, I.; Hatunic, M.; Finucane, F.; Liesa, M.; Chiellini, C.; Naon, D.; et al. Subjects with early-onset type 2 diabetes show defective activation of the skeletal muscle pgc-1{alpha}/mitofusin-2 regulatory pathway in response to physical activity. Diabetes Care 2010, 33, 645–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, B.J.; Capaldi, R.A.; Marusich, M.F.; Sherwood, S.W. An immunocytochemical approach to detection of mitochondrial disorders. J. Histochem. Cytochem. 2002, 50, 1281–1288. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharm. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [Green Version]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. Drp1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Sun, Y.; Guo, S.; Lu, B. The pink1/parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet. 2011, 20, 3227–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishta, O.A.; Guo, Y.; Mofarrahi, M.; Stana, F.; Lands, L.C.; Hussain, S.N.A. Pulmonary pseudomonas aeruginosa infection induces autophagy and proteasome proteolytic pathways in skeletal muscles: Effects of a pressurized whey protein-based diet in mice. Food Nutr. Res. 2017, 61, 1325309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of bnip3 and bnip3l via their bh3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Gong, G.; Burelle, Y.; Gustafsson, A.B.; Kitsis, R.N.; Matkovich, S.J.; Dorn, G.W., 2nd. Interdependence of parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ. Res. 2015, 117, 346–351. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leduc-Gaudet, J.-P.; Mayaki, D.; Reynaud, O.; Broering, F.E.; Chaffer, T.J.; Hussain, S.N.A.; Gouspillou, G. Parkin Overexpression Attenuates Sepsis-Induced Muscle Wasting. Cells 2020, 9, 1454. https://doi.org/10.3390/cells9061454
Leduc-Gaudet J-P, Mayaki D, Reynaud O, Broering FE, Chaffer TJ, Hussain SNA, Gouspillou G. Parkin Overexpression Attenuates Sepsis-Induced Muscle Wasting. Cells. 2020; 9(6):1454. https://doi.org/10.3390/cells9061454
Chicago/Turabian StyleLeduc-Gaudet, Jean-Philippe, Dominique Mayaki, Olivier Reynaud, Felipe E. Broering, Tomer J. Chaffer, Sabah N. A. Hussain, and Gilles Gouspillou. 2020. "Parkin Overexpression Attenuates Sepsis-Induced Muscle Wasting" Cells 9, no. 6: 1454. https://doi.org/10.3390/cells9061454
APA StyleLeduc-Gaudet, J.-P., Mayaki, D., Reynaud, O., Broering, F. E., Chaffer, T. J., Hussain, S. N. A., & Gouspillou, G. (2020). Parkin Overexpression Attenuates Sepsis-Induced Muscle Wasting. Cells, 9(6), 1454. https://doi.org/10.3390/cells9061454