Deletion of Perilipin 5 Protects against Hepatic Injury in Nonalcoholic Fatty Liver Disease via Missing Inflammasome Activation

 , , , ,
, , , , 

Abstract
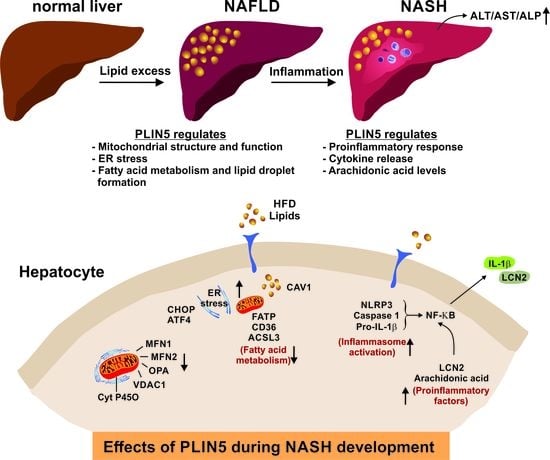
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Generating Plin5-Deficient Mice
2.2. Animals
2.3. Liver Histology and NAFLD/NASH Scoring
2.4. Blood Analysis
2.5. Isolation, Culture and Stimulation of Primary Hepatocytes
2.6. RNA Expression Analysis and Statistical Analyses
2.7. Western Blot Analysis
2.8. Electron Microscopy for Hepatic Mitochondria Imaging
2.9. Proteomics
2.10. Lipidomics
3. Results
3.1. Lack of Plin5 Alleviates Fatty Liver Injury
3.2. Plin5 Regulates Mitochondrial State and ER-Stress
3.3. The Absence of Plin5 Mitigates the Pro-Inflammatory Response in Steatotic Livers
3.4. Lack of Plin5 Alleviates LPS-Induced Inflammasome Activation in Primary Hepatocytes by Suppressing NF-κB Signaling
3.5. Lipid Profile Content of Plin5−/− Livers Reveals Reduced Levels of the Inflammatory Mediator Arachidonic Acid
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef]
- Drescher, H.K.; Weiskirchen, S.; Weiskirchen, R. Current status in testing for nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH). Cells 2019, 8, 845. [Google Scholar] [CrossRef]
- Levene, A.P.; Goldin, R.D. The epidemiology, pathogenesis and histopathology of fatty liver disease. Histopathology 2012, 61, 141–152. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Matsushita, S.; Motojima, K.; Hirose, F.; Osumi, T. MLDP, a novel PAT family protein localized to lipid droplets and enriched in the heart, is regulated by peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 2006, 281, 14232–14240. [Google Scholar] [CrossRef] [PubMed]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Tzekov, A.; Croce, M.A.; Gropler, M.C.; Varma, V.; Yao-Borengasser, A.; Rasouli, N.; Kern, P.A.; et al. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 2006, 55, 3418–3428. [Google Scholar] [CrossRef] [PubMed]
- Dalen, K.T.; Dahl, T.; Holter, E.; Arntsen, B.; Londos, C.; Sztalryd, C.; Nebb, H.I. LSDP5 is a PAT protein specifically expressed in fatty acid oxidizing tissues. Biochim. Biophys. Acta 2007, 1771, 210–227. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Pesce, C.; Canesi, L.; Fugassa, E.; Gallo, G.; Vergani, L. PAT protein mRNA expression in primary rat hepatocytes: Effects of exposure to fatty acids. Int. J. Mol. Med. 2010, 25, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef]
- Langhi, C.; Marquart, T.J.; Allen, R.M.; Baldán, A. Perilipin-5 is regulated by statins and controls triglyceride contents in the hepatocyte. J. Hepatol. 2014, 61, 358–365. [Google Scholar] [CrossRef]
- Trevino, M.B.; Mazur-Hart, D.; Machida, Y.; King, T.; Nadler, J.; Galkina, E.V.; Poddar, A.; Dulta, S.; Imai, Y. Liver Perilipin 5 expression worsens hepatosteatosis but not insulin resistance in high fat-fed mice. Mol. Endocrinol. 2015, 29, 1414–1425. [Google Scholar] [CrossRef]
- Keenan, S.N.; Meex, R.C.; Lo, J.C.; Ryan, A.; Nie, S.; Montgomery, M.K.; Watt, M.J. Perilipin 5 deletion in hepatocytes remodels lipid metabolism and causes hepatic insulin resistance in mice. Diabetes 2019, 68, 543–555. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Vucur, M.; Luedde, T.; Schneiders, S.; Kalampoka, S.; Weiss, T.S.; Weiskirchen, R. Perilipin 5 and Lipocalin 2 Expression in Hepatocellular Carcinoma. Cancers 2019, 11, 385. [Google Scholar] [CrossRef]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef]
- Nakagata, N. Cryopreservation of mouse spermatozoa. Mamm. Genome 2000, 11, 572–576. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Borkham-Kamphorst, E.; Henning, M.; Yagmur, E.; Gassler, N.; Liedtke, C.; Berger, T.; Mak, T.W.; Weiskirchen, R. Lipocalin-2 (LCN2) regulates PLIN5 expressionand intracellular lipid droplet formation in the liver. Biochim. Biophys. Acta 2014, 1842, 1513–1524. [Google Scholar] [CrossRef]
- Seglen, P.O. Preparation of isolated rat liver cells. Methods Cell Biol. 1976, 13, 29–83. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Fülöp, A.; Borkham-Kamphorst, E.; de Leur, E.V.; Gassler, N.; Berger, T.; Beine, B.; Meyer, H.E.; Mak, T.W.; Hopf, C.; et al. Altered mitochondrial and peroxisomal integrity in lipocalin-2-deficient mice with hepatic steatosis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2093–2110. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Engel, K.M.; Sampels, S.; Dzyuba, B.; Podhorec, P.; Policar, T.; Dannenberger, D.; Schiller, J. Swimming at different temperatures: The lipid composition of sperm from three freshwater fish species determined by mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Phys. Lipids 2019, 221, 65–72. [Google Scholar] [CrossRef]
- Engel, K.M.; Schiller, J.; Müller, K.; Dannenberger, D.; Jakop, U. The phospholipid composition of Kangaroo spermatozoa verified by mass spectrometric lipid analysis. Lipids 2017, 52, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Yang, X.F.; Liu, H.L.; Fu, N.; Ouyang, Y.; Qing, K. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: An update. World J. Gastroenterol. 2015, 21, 3492–3498. [Google Scholar] [CrossRef] [PubMed]
- Enooku, K.; Tsutsumi, T.; Kondo, M.; Fujiwara, N.; Sasako, T.; Shibahara, J.; Kado, A.; Okushin, K.; Fujinaga, H.; Nakagomi, R.; et al. Hepatic FATP5 expression is associated with histological progression and loss of hepatic fat in NAFLD patients. J. Gastroenterol. 2020, 55, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, P.; Zhao, L.; Chen, Y.; Zhang, X.; Zeng, S.; Wei, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway. J. Lipid Res. 2019, 60, 844–855. [Google Scholar] [CrossRef]
- Li, M.; Chen, D.; Huang, H.; Wang, J.; Wan, X.; Xu, C.; Li, C.; Ma, H.; Yu, C.; Li, Y. Caveolin1 protects against diet induced hepatic lipid accumulation in mice. PLoS ONE 2017, 12, e0178748. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Caldwell, S.; Ikura, Y.; Dias, D.; Isomoto, K.; Yabu, A.; Moskaluk, C.; Pramoonjago, P.; Simmons, W.; Scruggs, H.; Rosenbaum, N.; et al. Hepatocellular ballooning in NASH. J. Hepatol. 2010, 53, 719–723. [Google Scholar] [CrossRef]
- Machado, M.V.; Cortez-Pinto, H. Cell death and nonalcoholic steatohepatitis: Where is ballooning relevant? Expert Rev. Gastroenterol. Hepatol. 2011, 5, 213–222. [Google Scholar] [CrossRef]
- Wan, X.; Xu, C.; Yu, C.; Li, Y. Role of NLRP3 inflammasome in the progression of NAFLD to NASH. Can. J. Gastroenterol. Hepatol. 2016, 2016, 6489012. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Petta, S.; Mannisto, V.; Mancina, R.M.; Pipitone, R.; Karja, V.; Maggioni, M.; Kakela, P.; Wiklund, O.; Mozzi, E.; et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J. Hepatol. 2015, 63, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Oseini, A.M.; Sanyal, A.J. Therapies in non-alcoholic steatohepatitis (NASH). Liver Int. 2017, 37 (Suppl. 1), 97–103. [Google Scholar] [CrossRef] [PubMed]
- Laurens, C.; Bourlier, V.; Mairal, A.; Louche, K.; Badin, P.M.; Mouisel, E.; Montagner, A.; Marette, A.; Tremblay, A.; Weisnagel, J.S.; et al. Perilipin 5 fine-tunes lipid oxidation to metabolic demand and protects against lipotoxicity in skeletal muscle. Sci. Rep. 2016, 6, 38310. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Mokhtar, R.; Bayliss, J.; Parkington, H.C.; Suturin, V.M.; Bruce, C.R.; Watt, M.J. Perilipin 5 deletion unmasks an endoplasmic reticulum stress-fibroblast growth factor 21 axis in skeletal muscle. Diabetes 2018, 67, 594–606. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Luan, J.; Ju, D. Inflammasome: A double-edged sword in liver diseases. Front. Immunol. 2018, 9, 2201. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef]
- Boaru, S.G.; Borkham-Kamphorst, E.; Van de Leur, E.; Lehnen, E.; Liedtke, C.; Weiskirchen, R. NLRP3 inflammasome expression is driven by NF-κB in cultured hepatocytes. Biochem. Biophys. Res. Commun. 2015, 458, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska, D.; Ossowski, P.; Drozd, A.; Ryterska, K.; Jamioł-Milc, D.; Banaszczak, M.; Kaczorowska, M.; Sabinicz, A.; Raszeja-Wyszomirska, J.; Stachowska, E. Metabolites of arachidonic acid and linoleic acid in early stages of non-alcoholic fatty liver disease—A pilot study. Prostaglandins Other Lipid Mediat. 2015, 121, 184–189. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asimakopoulou, A.; Engel, K.M.; Gassler, N.; Bracht, T.; Sitek, B.; Buhl, E.M.; Kalampoka, S.; Pinoé-Schmidt, M.; van Helden, J.; Schiller, J.; et al. Deletion of Perilipin 5 Protects against Hepatic Injury in Nonalcoholic Fatty Liver Disease via Missing Inflammasome Activation. Cells 2020, 9, 1346. https://doi.org/10.3390/cells9061346
Asimakopoulou A, Engel KM, Gassler N, Bracht T, Sitek B, Buhl EM, Kalampoka S, Pinoé-Schmidt M, van Helden J, Schiller J, et al. Deletion of Perilipin 5 Protects against Hepatic Injury in Nonalcoholic Fatty Liver Disease via Missing Inflammasome Activation. Cells. 2020; 9(6):1346. https://doi.org/10.3390/cells9061346
Chicago/Turabian StyleAsimakopoulou, Anastasia, Kathrin M. Engel, Nikolaus Gassler, Thilo Bracht, Barbara Sitek, Eva M. Buhl, Stavroula Kalampoka, Manuela Pinoé-Schmidt, Josef van Helden, Jürgen Schiller, and et al. 2020. "Deletion of Perilipin 5 Protects against Hepatic Injury in Nonalcoholic Fatty Liver Disease via Missing Inflammasome Activation" Cells 9, no. 6: 1346. https://doi.org/10.3390/cells9061346
APA StyleAsimakopoulou, A., Engel, K. M., Gassler, N., Bracht, T., Sitek, B., Buhl, E. M., Kalampoka, S., Pinoé-Schmidt, M., van Helden, J., Schiller, J., & Weiskirchen, R. (2020). Deletion of Perilipin 5 Protects against Hepatic Injury in Nonalcoholic Fatty Liver Disease via Missing Inflammasome Activation. Cells, 9(6), 1346. https://doi.org/10.3390/cells9061346