Acute Induction of Translocon-Mediated Ca2+ Leak Protects Cardiomyocytes Against Ischemia/Reperfusion Injury

 , ,
, ,  ,
,  and
and
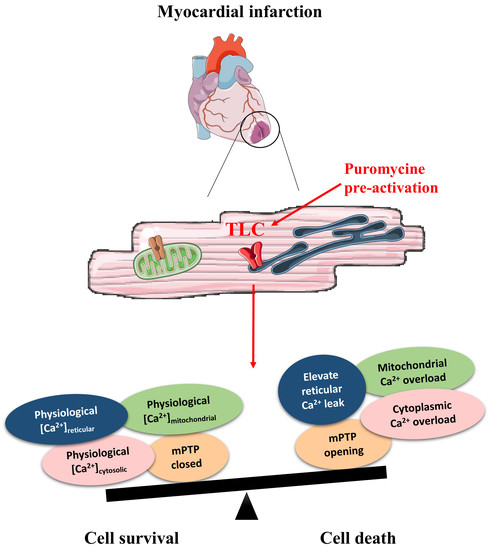
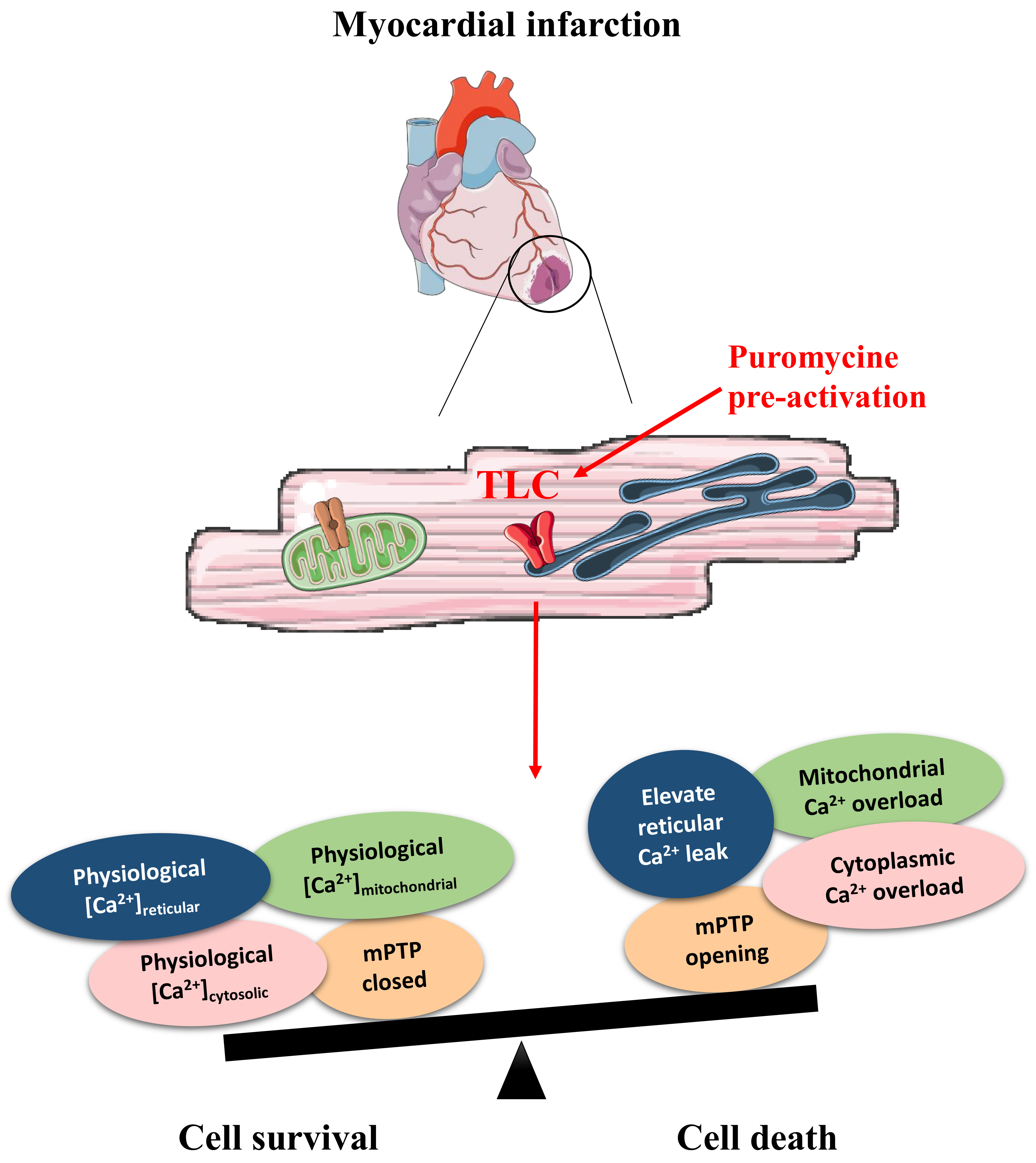
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. CM Isolation
2.3. Adenovirus Injection for Reticular and Mitochondrial Ca2+ Measurements
2.4. Ca2+ Measurements
2.5. Ca2+ Transients
2.6. Calcein Cobalt Protocol
2.7. In Vivo Model of Acute myocardial I/R Injury
2.8. CM Mortality
2.9. Statistical Analysis
3. Results and Discussion
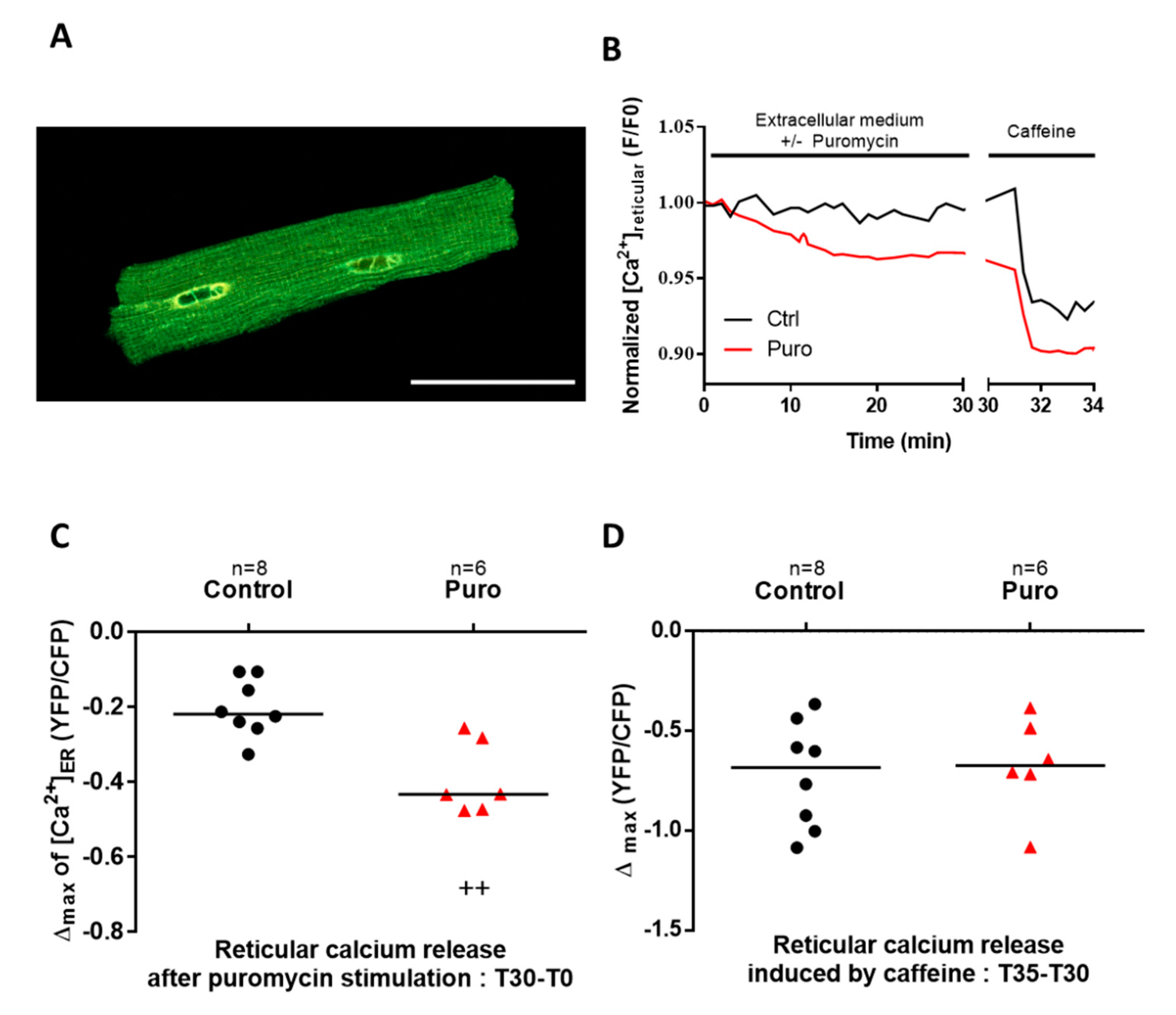
3.1. TLC Is a Functional Reticular Ca2+ Leak Channel in Isolated Mouse CM: Its Activation Mobilized a RyR-Independent Ca2+ Reticular Pool and Did Not Affect Excitation–Contraction (E-C) Coupling
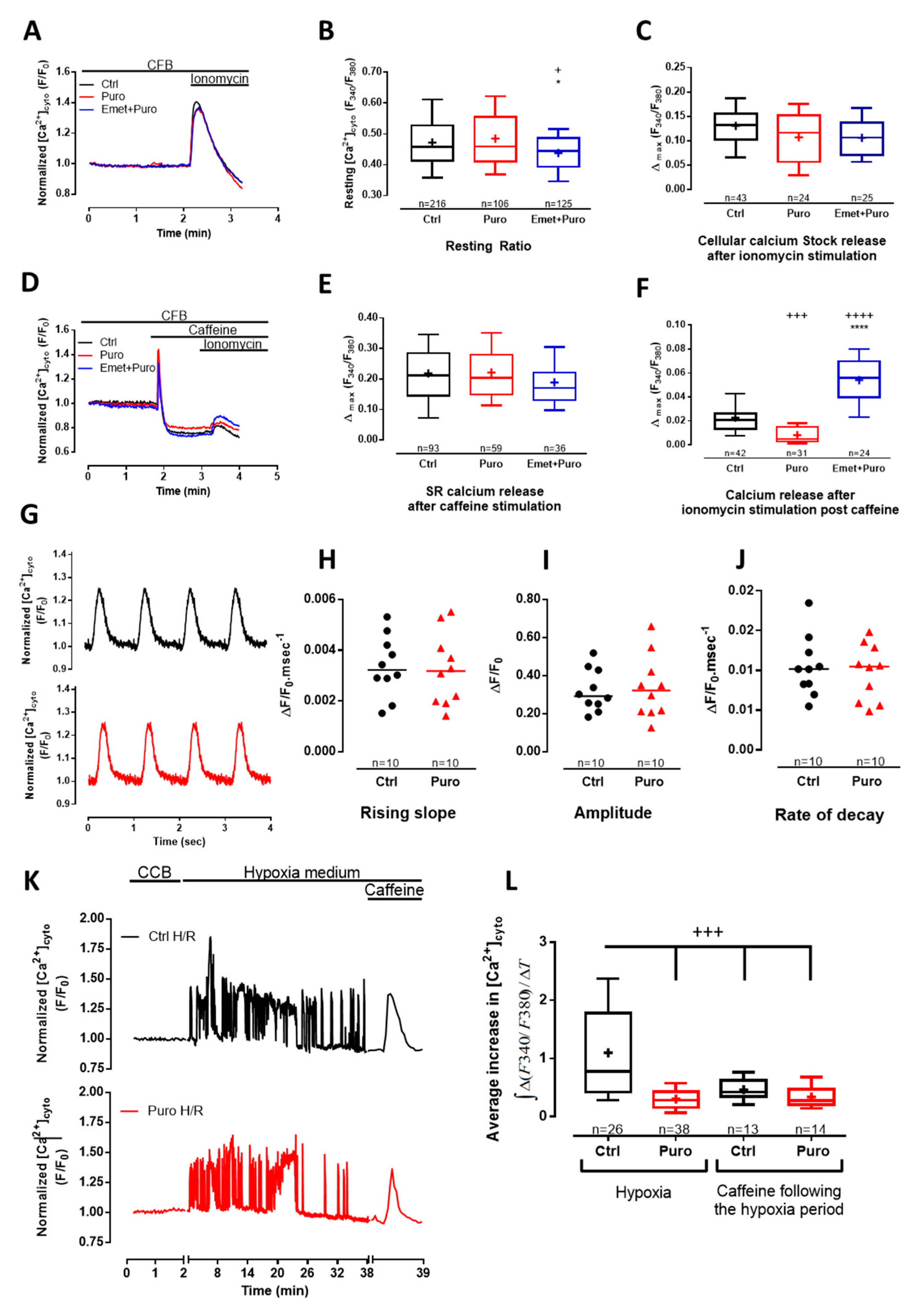
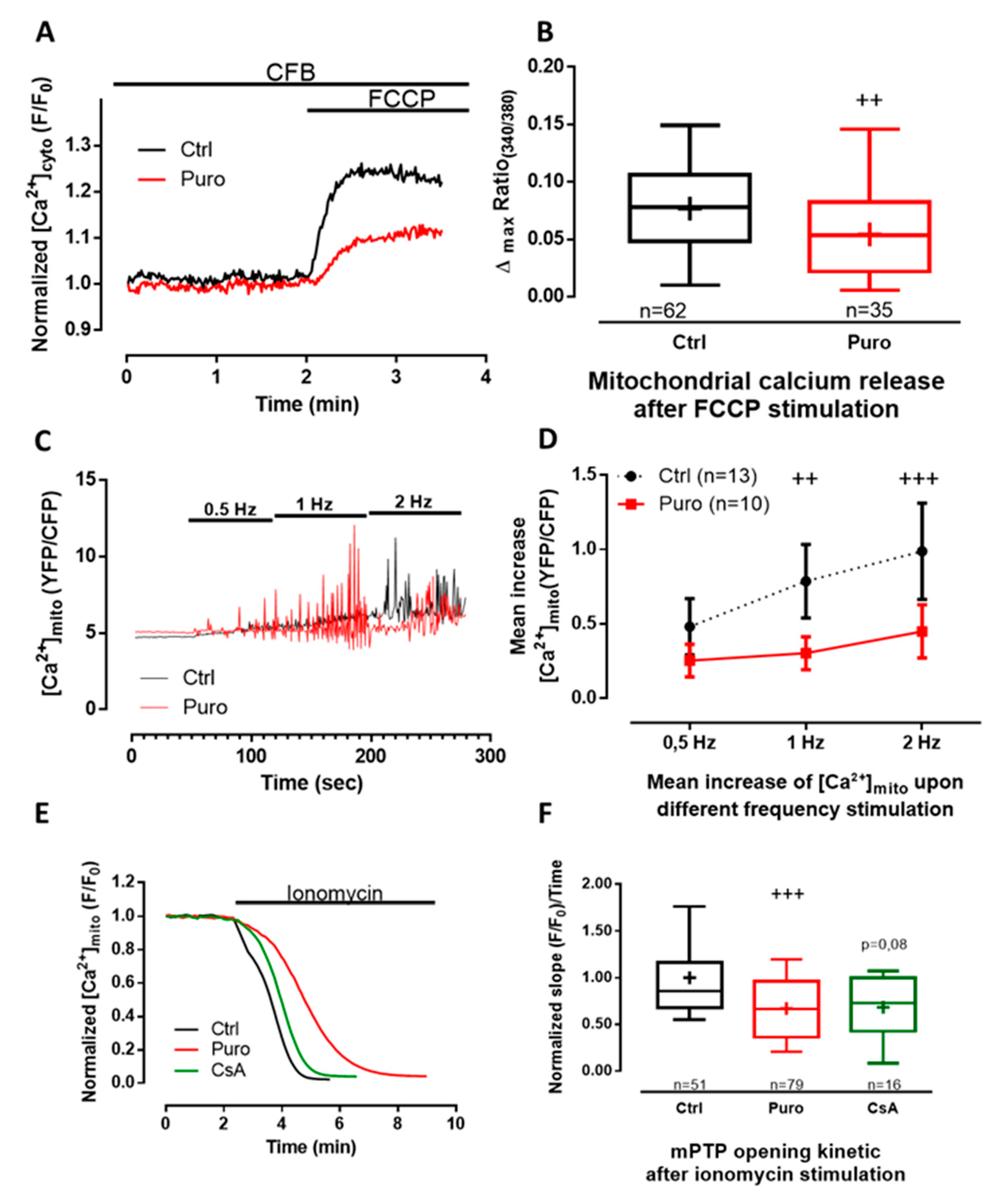
3.2. Pharmacological Modulation of TLC with Puromycin Pretreatment Affected Mitochondrial Ca2+ Content in Beating (But Not in Resting) CM and Slowed down the mPTP Opening
3.3. Pharmacological Modulation of TLC with Puromycin Pretreatment Protected CM after In Vitro H/R, and Reduced Infarct Size in Mice Submitted to In Vivo I/R
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sztark, F.; Payen, J.F.; Piriou, V.; Rigoulet, M.; Ventura-Clapier, R.; Mazat, J.P.; Leverve, X.; Janvier, G. Cellular energy metabolism: Physiologic and pathologic aspects. Ann. Fr. D’anesthesie Reanim. 1999, 18, 261–269. [Google Scholar] [CrossRef]
- Nabel, E.G.; Braunwald, E. A tale of coronary artery disease and myocardial infarction. N. Engl. J. Med. 2012, 366, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.K.; Dhalla, N.S. Defective calcium handling in cardiomyocytes isolated from hearts subjected to ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2260–H2270. [Google Scholar] [CrossRef] [PubMed]
- Buja, L.M. Myocardial ischemia and reperfusion injury. Cardiovasc. Pathol. 2005, 14, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Jennings, R.B.; Sommers, H.M.; Smyth, G.A.; Flack, H.A.; Linn, H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch. Pathol. 1960, 70, 68–78. [Google Scholar]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef]
- Paillard, M.; Tubbs, E.; Thiebaut, P.-A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef]
- Gomez, L.; Thiebaut, P.-A.; Paillard, M.; Ducreux, S.; Abrial, M.; Crola Da Silva, C.; Durand, A.; Alam, M.R.; Van Coppenolle, F.; Sheu, S.-S.; et al. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ. 2016, 23, 313–322. [Google Scholar] [CrossRef]
- Schwaller, B. The Regulation of a Cell’s Ca2+ Signaling Toolkit: The Ca2+ Homeostasome. In Calcium Signaling; Islam, M.d.S., Ed.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2012; Volume 740, pp. 1–25. ISBN 978-94-007-2888-2. [Google Scholar]
- Sammels, E.; Parys, J.B.; Missiaen, L.; De Smedt, H.; Bultynck, G. Intracellular Ca2+ storage in health and disease: A dynamic equilibrium. Cell Calcium 2010, 47, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, C.J.; Roderick, H.L.; Bootman, M.D. Calcium Signaling in Cardiac Myocytes. Cold Spring Harb. Perspect. Biol. 2011, 3, a004242. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Barrabes, J.A.; Bøtker, H.E.; Davidson, S.M.; Di Lisa, F.; Downey, J.; Engstrom, T.; Ferdinandy, P.; Carbrera-Fuentes, H.A.; Heusch, G.; et al. Ischaemic conditioning and targeting reperfusion injury: A 30 year voyage of discovery. Basic Res. Cardiol. 2016, 111, 70. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, G.; Chiari, P.; Fauconnier, J.; Abrial, M.; Couture-Lepetit, E.; Harisseh, R.; Pillot, B.; Lacampagne, A.; Tourneur, Y.; Gharib, A.; et al. Involvement of Cyclophilin D and Calcium in Isoflurane-induced Preconditioning. Anesthesiology 2015, 123, 1374–1384. [Google Scholar] [CrossRef]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn, G.W.; Yellon, D.M.; Hausenloy, D.J. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016, 7, e2238. [Google Scholar] [CrossRef]
- Cung, T.-T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Lang, S.; Erdmann, F.; Jung, M.; Wagner, R.; Cavalie, A.; Zimmermann, R. Sec61 complexes form ubiquitous ER Ca2+ leak channels. Channels 2011, 5, 228–235. [Google Scholar] [CrossRef]
- Tyedmers, J.; Lerner, M.; Bies, C.; Dudek, J.; Skowronek, M.H.; Haas, I.G.; Heim, N.; Nastainczyk, W.; Volkmer, J.; Zimmermann, R. Homologs of the yeast Sec complex subunits Sec62p and Sec63p are abundant proteins in dog pancreas microsomes. Proc. Natl. Acad. Sci. USA 2000, 97, 7214–7219. [Google Scholar] [CrossRef]
- Hamman, B.D.; Chen, J.C.; Johnson, E.E.; Johnson, A.E. The aqueous pore through the translocon has a diameter of 40-60 A during cotranslational protein translocation at the ER membrane. Cell 1997, 89, 535–544. [Google Scholar] [CrossRef]
- Tinker, A.; Williams, A.J. Probing the structure of the conduction pathway of the sheep cardiac sarcoplasmic reticulum calcium-release channel with permeant and impermeant organic cations. J. Gen. Physiol. 1993, 102, 1107–1129. [Google Scholar] [CrossRef]
- Mead, F.; Williams, A.J. Block of the ryanodine receptor channel by neomycin is relieved at high holding potentials. Biophys. J. 2002, 82, 1953–1963. [Google Scholar] [CrossRef][Green Version]
- Lindsay, A.R.; Manning, S.D.; Williams, A.J. Monovalent cation conductance in the ryanodine receptor-channel of sheep cardiac muscle sarcoplasmic reticulum. J. Physiol. 1991, 439, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Alder, N.N.; Shen, Y.; Brodsky, J.L.; Hendershot, L.M.; Johnson, A.E. The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J. Cell Biol. 2005, 168, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Hammadi, M.; Oulidi, A.; Gackière, F.; Katsogiannou, M.; Slomianny, C.; Roudbaraki, M.; Dewailly, E.; Delcourt, P.; Lepage, G.; Lotteau, S.; et al. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: Involvement of GRP78. FASEB J. 2013, 27, 1600–1609. [Google Scholar] [CrossRef]
- Ménétret, J.-F.; Hegde, R.S.; Aguiar, M.; Gygi, S.P.; Park, E.; Rapoport, T.A.; Akey, C.W. Single copies of Sec61 and TRAP associate with a non-translating mammalian ribosome. Structure 2008, 16, 1126–1137. [Google Scholar] [CrossRef]
- Becker, T.; Bhushan, S.; Jarasch, A.; Armache, J.-P.; Funes, S.; Jossinet, F.; Gumbart, J.; Mielke, T.; Berninghausen, O.; Schulten, K.; et al. Structure of Monomeric Yeast and Mammalian Sec61 Complexes Interacting with the Translating Ribosome. Science 2009, 326, 1369–1373. [Google Scholar] [CrossRef]
- Roy, A.; Wonderlin, W.F. The Permeability of the Endoplasmic Reticulum Is Dynamically Coupled to Protein Synthesis. J. Biol. Chem. 2003, 278, 4397–4403. [Google Scholar] [CrossRef]
- Lang, S.; Benedix, J.; Fedeles, S.V.; Schorr, S.; Schirra, C.; Schäuble, N.; Jalal, C.; Greiner, M.; Haßdenteufel, S.; Tatzelt, J.; et al. Different effects of Sec61α, Sec62 and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 2012, 125, 1958–1969. [Google Scholar] [CrossRef]
- Lomax, R.B.; Camello, C.; Van Coppenolle, F.; Petersen, O.H.; Tepikin, A.V. Basal and physiological Ca2+ leak from the endoplasmic reticulum of pancreatic acinar cells. Second messenger-activated channels and translocons. J. Biol. Chem. 2002, 277, 26479–26485. [Google Scholar] [CrossRef]
- Coppenolle, F.V.; Abeele, F.V.; Slomianny, C.; Flourakis, M.; Hesketh, J.; Dewailly, E.; Prevarskaya, N. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J. Cell Sci. 2004, 117, 4135–4142. [Google Scholar] [CrossRef]
- Flourakis, M.; Van Coppenolle, F.; Lehen’kyi, V.; Beck, B.; Skryma, R.; Prevarskaya, N. Passive calcium leak via translocon is a first step for iPLA2-pathway regulated store operated channels activation. FASEB J. 2006, 20, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Paredes, R.M.; Bollo, M.; Holstein, D.; Lechleiter, J.D. Luminal Ca2+ depletion during the unfolded protein response in Xenopus oocytes: Cause and consequence. Cell Calcium 2013, 53, 286–296. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Layhadi, J.A.; Fountain, S.J. Influence of ER leak on resting cytoplasmic Ca2+ and receptor-mediated Ca2+ signalling in human macrophage. Biochem. Biophys. Res. Commun. 2017, 487, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Giunti, R.; Gamberucci, A.; Fulceri, R.; Bánhegyi, G.; Benedetti, A. Both translocon and a cation channel are involved in the passive Ca2+ leak from the endoplasmic reticulum: A mechanistic study on rat liver microsomes. Arch. Biochem. Biophys. 2007, 462, 115–121. [Google Scholar] [CrossRef]
- Erdmann, F.; Schäuble, N.; Lang, S.; Jung, M.; Honigmann, A.; Ahmad, M.; Dudek, J.; Benedix, J.; Harsman, A.; Kopp, A.; et al. Interaction of calmodulin with Sec61α limits Ca2+ leakage from the endoplasmic reticulum. EMBO J. 2011, 30, 17–31. [Google Scholar] [CrossRef]
- Cassel, R.; Ducreux, S.; Alam, M.R.; Dingreville, F.; Berlé, C.; Burda-Jacob, K.; Chauvin, M.A.; Chikh, K.; Païta, L.; Al-Mawla, R.; et al. Protection of Human Pancreatic Islets from Lipotoxicity by Modulation of the Translocon. PLoS ONE 2016, 11, e0148686. [Google Scholar] [CrossRef]
- Linxweiler, M.; Schick, B.; Zimmermann, R. Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target. Ther. 2017, 2, 17002. [Google Scholar] [CrossRef]
- Linxweiler, M.; Schorr, S.; Schäuble, N.; Jung, M.; Linxweiler, J.; Langer, F.; Schäfers, H.-J.; Cavalié, A.; Zimmermann, R.; Greiner, M. Targeting cell migration and the endoplasmic reticulum stress response with calmodulin antagonists: A clinically tested small molecule phenocopy of SEC62 gene silencing in human tumor cells. BMC Cancer 2013, 13, 574. [Google Scholar] [CrossRef]
- Lu, Z.; Zhou, L.; Killela, P.; Rasheed, A.B.; Di, C.; Poe, W.E.; McLendon, R.E.; Bigner, D.D.; Nicchitta, C.; Yan, H. Glioblastoma Proto-Oncogene SEC61γ Is Required for Tumor Cell Survival and Response to Endoplasmic Reticulum Stress. Cancer Res. 2009, 69, 9105–9111. [Google Scholar] [CrossRef]
- Lang, S.; Pfeffer, S.; Lee, P.-H.; Cavalié, A.; Helms, V.; Förster, F.; Zimmermann, R. An Update on Sec61 Channel Functions, Mechanisms, and Related Diseases. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Palmer, A.E.; Tsien, R.Y. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat. Protoc. 2006, 1, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Greensmith, D.J. Ca analysis: An Excel based program for the analysis of intracellular calcium transients including multiple, simultaneous regression analysis. Comput. Methods Programs Biomed. 2014, 113, 241–250. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hom, J.R.; Quintanilla, R.A.; Hoffman, D.L.; de Mesy Bentley, K.L.; Molkentin, J.D.; Sheu, S.-S.; Porter, G.A. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev. Cell 2011, 21, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Miotto, G.; Canton, M.; Brini, M.; Colonna, R.; Bernardi, P.; Di Lisa, F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 1999, 76, 725–734. [Google Scholar] [CrossRef]
- Paccalet, A.; Tessier, N.; Paillard, M.; Païta, L.; Gomez, L.; Gallo-Bona, N.; Chouabe, C.; Léon, C.; Badawi, S.; Harhous, Z.; et al. An innovative sequence of hypoxia-reoxygenation on adult mouse cardiomyocytes in suspension to perform multilabeling analysis by flow cytometry. Am. J. Physiol. Cell Physiol. 2019, 318, C439–C447. [Google Scholar] [CrossRef]
- Amer, M.S.; Li, J.; O’Regan, D.J.; Steele, D.S.; Porter, K.E.; Sivaprasadarao, A.; Beech, D.J. Translocon closure to Ca2+ leak in proliferating vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H910–H916. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Sleiman, N.H.; McFarland, T.P.; Jones, L.R.; Cala, S.E. Transitions of protein traffic from cardiac ER to junctional SR. J. Mol. Cell. Cardiol. 2015, 81, 34–45. [Google Scholar] [CrossRef][Green Version]
- Hajnóczky, G.; Csordás, G.; Madesh, M.; Pacher, P. The machinery of local Ca2+ signalling between sarco-endoplasmic reticulum and mitochondria. J. Physiol. 2000, 529, 69–81. [Google Scholar] [CrossRef]
- Bochaton, T.; Crola-Da-Silva, C.; Pillot, B.; Villedieu, C.; Ferreras, L.; Alam, M.R.; Thibault, H.; Strina, M.; Gharib, A.; Ovize, M.; et al. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. J. Mol. Cell. Cardiol. 2015, 84, 61–69. [Google Scholar] [CrossRef]
- Petrovski, G.; Das, S.; Juhasz, B.; Kertesz, A.; Tosaki, A.; Das, D.K. Cardioprotection by endoplasmic reticulum stress-induced autophagy. Antioxid. Redox Signal. 2011, 14, 2191–2200. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, Z.; Chen, L. Endoplasmic reticulum stress: A novel mechanism and therapeutic target for cardiovascular diseases. Acta Pharmacol. Sin. 2016, 37, 425–443. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Mawla, R.; Ducrozet, M.; Tessier, N.; Païta, L.; Pillot, B.; Gouriou, Y.; Villedieu, C.; Harhous, Z.; Paccalet, A.; Crola Da Silva, C.; et al. Acute Induction of Translocon-Mediated Ca2+ Leak Protects Cardiomyocytes Against Ischemia/Reperfusion Injury. Cells 2020, 9, 1319. https://doi.org/10.3390/cells9051319
Al-Mawla R, Ducrozet M, Tessier N, Païta L, Pillot B, Gouriou Y, Villedieu C, Harhous Z, Paccalet A, Crola Da Silva C, et al. Acute Induction of Translocon-Mediated Ca2+ Leak Protects Cardiomyocytes Against Ischemia/Reperfusion Injury. Cells. 2020; 9(5):1319. https://doi.org/10.3390/cells9051319
Chicago/Turabian StyleAl-Mawla, Ribal, Mallory Ducrozet, Nolwenn Tessier, Lucille Païta, Bruno Pillot, Yves Gouriou, Camille Villedieu, Zeina Harhous, Alexandre Paccalet, Claire Crola Da Silva, and et al. 2020. "Acute Induction of Translocon-Mediated Ca2+ Leak Protects Cardiomyocytes Against Ischemia/Reperfusion Injury" Cells 9, no. 5: 1319. https://doi.org/10.3390/cells9051319
APA StyleAl-Mawla, R., Ducrozet, M., Tessier, N., Païta, L., Pillot, B., Gouriou, Y., Villedieu, C., Harhous, Z., Paccalet, A., Crola Da Silva, C., Ovize, M., Bidaux, G., Ducreux, S., & Van Coppenolle, F. (2020). Acute Induction of Translocon-Mediated Ca2+ Leak Protects Cardiomyocytes Against Ischemia/Reperfusion Injury. Cells, 9(5), 1319. https://doi.org/10.3390/cells9051319