The Proteasomal Deubiquitinating Enzyme PSMD14 Regulates Macroautophagy by Controlling Golgi-to-ER Retrograde Transport

 ,
,  , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Reagents
2.2. Antibodies
2.3. Cell Culture
2.4. High Content siRNA Transfection and Imaging
2.5. siRNA Transfection for the siRNA Screening Validation Stage
2.6. RNA isolation and RT-qPCR Analysis
2.7. Preparation of Protein Extracts, Electrophoresis, SDS-PAGE and Western Blot Analysis
2.8. In vitro Proteasomal Activity Assay
2.9. Immunofluorescence
2.10. Fluorescence Microscopy
2.11. D Golgi Reconstruction and Golgi Volume and Area Measurements
2.12. Densitometric Quantification and Statistical Analysis
3. Results
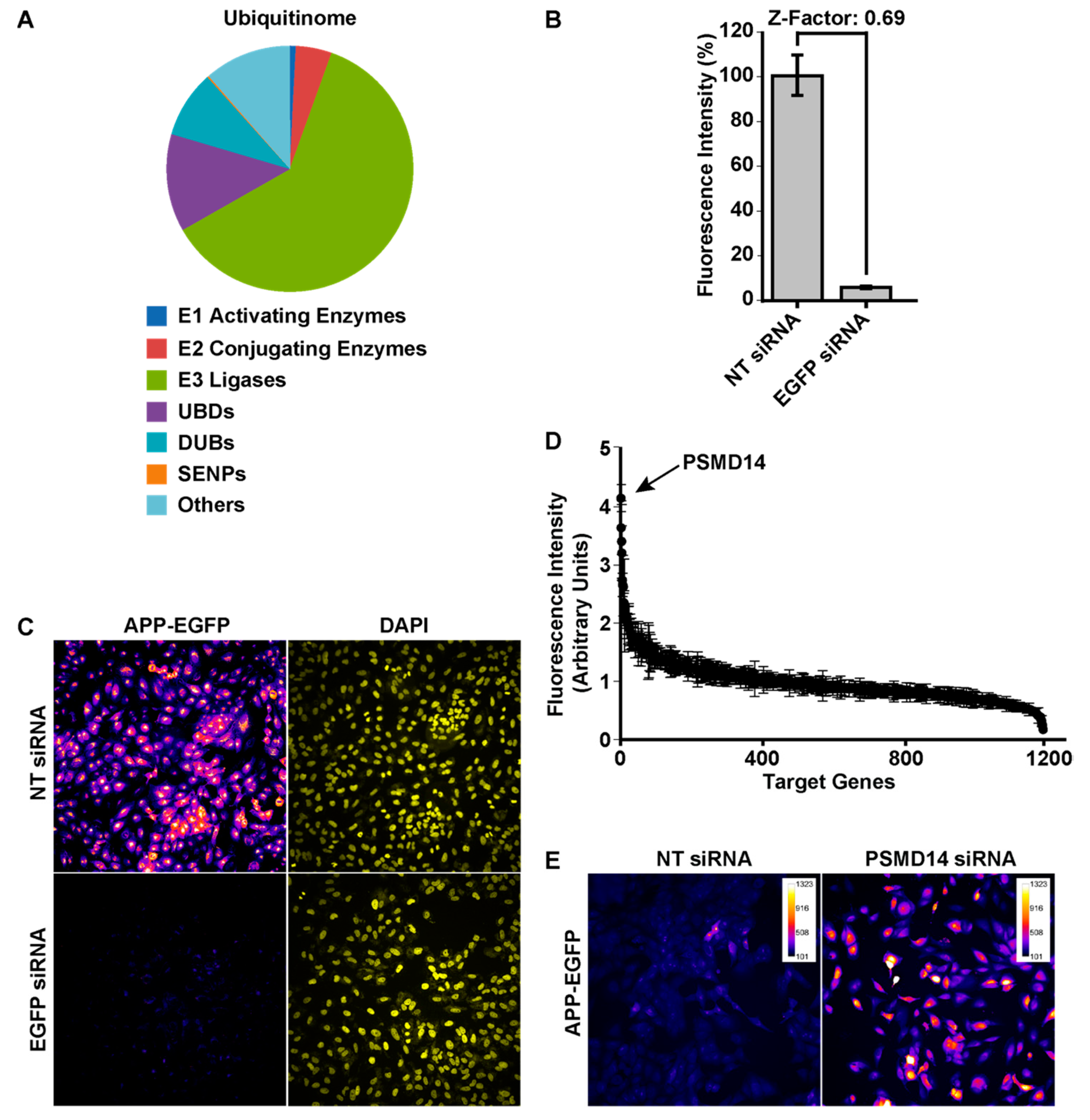
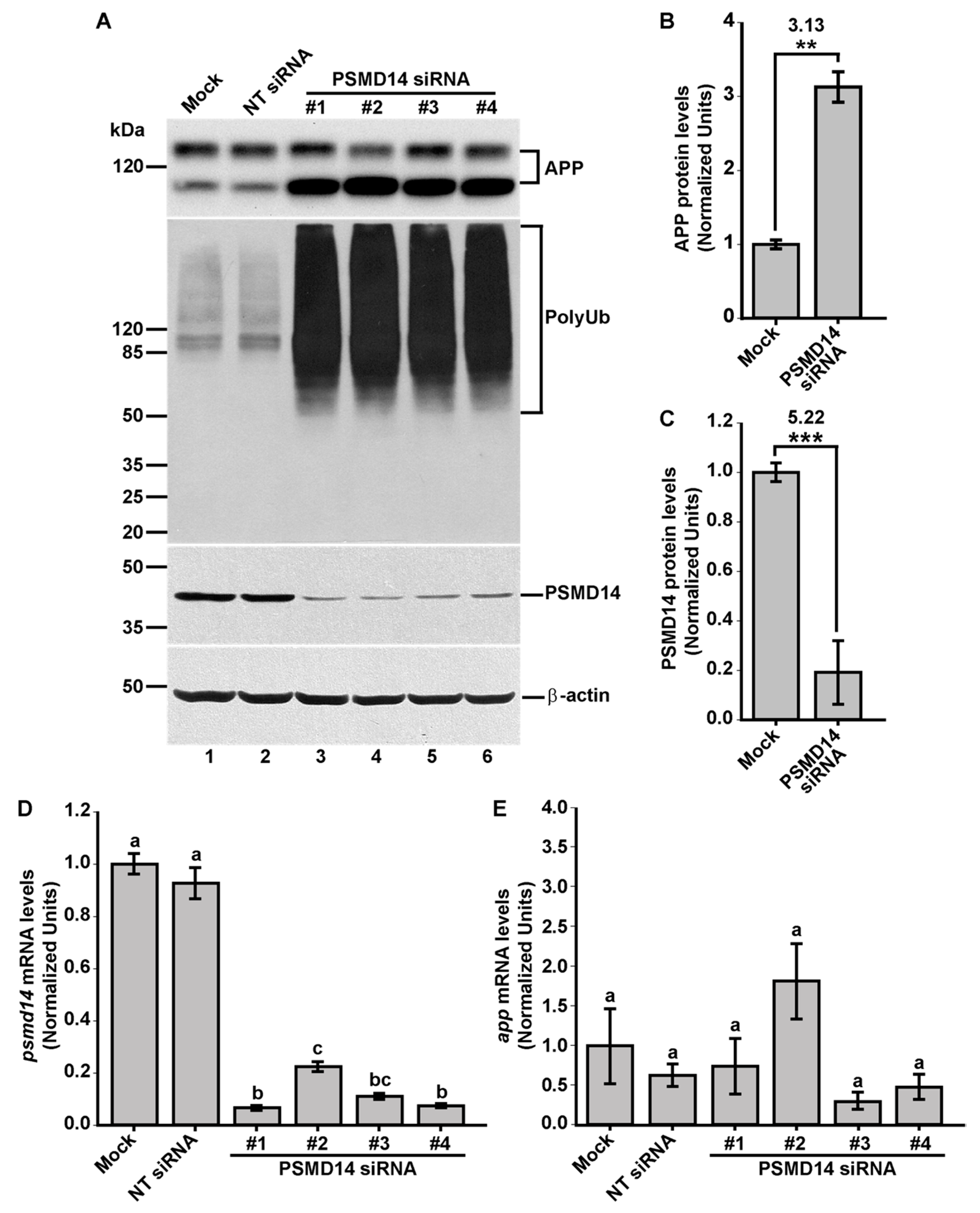
3.1. High-Content siRNA Screening Revealed PSMD14 Deubiquitinating Enzyme as a Novel Regulator of Protein Trafficking
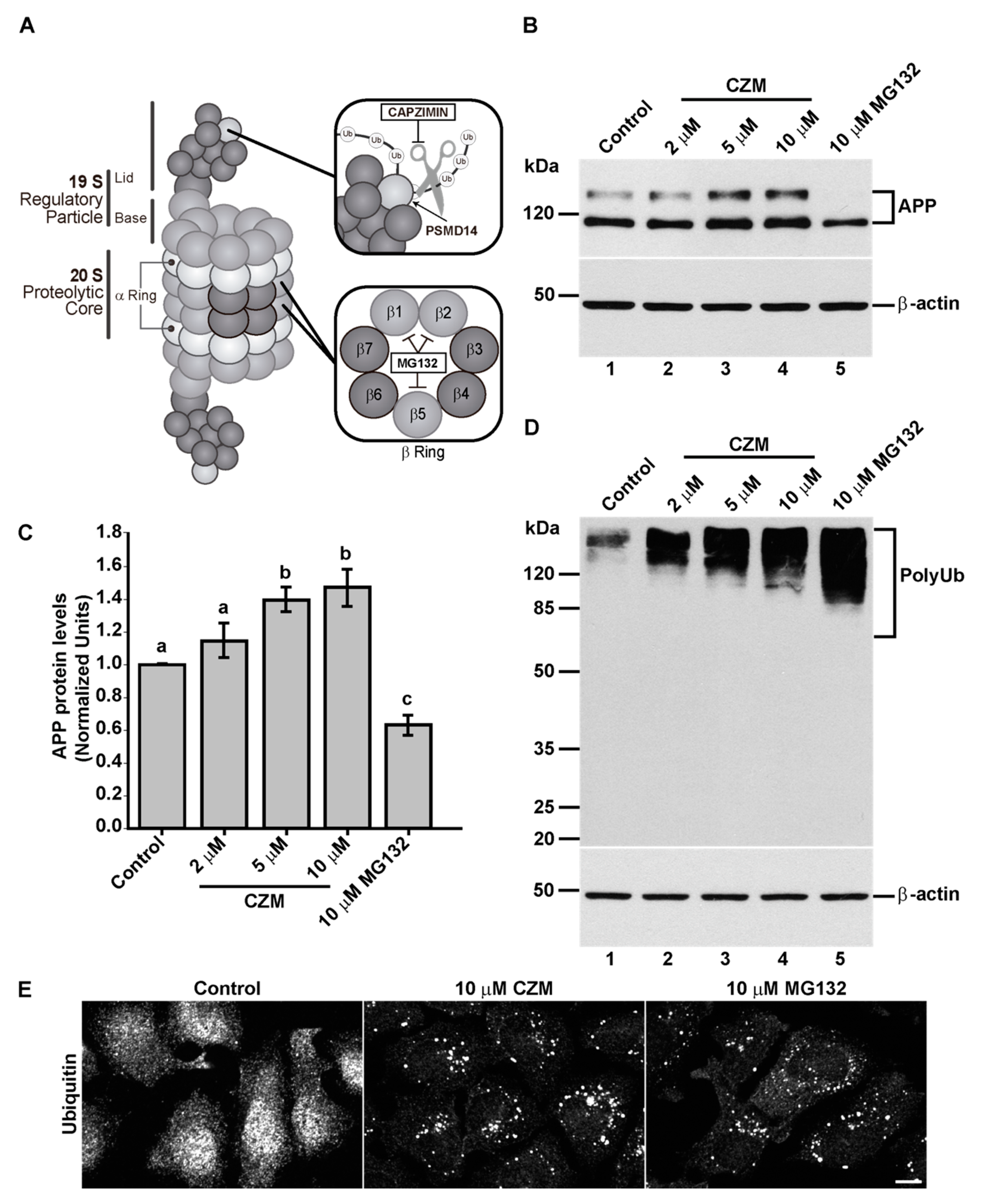
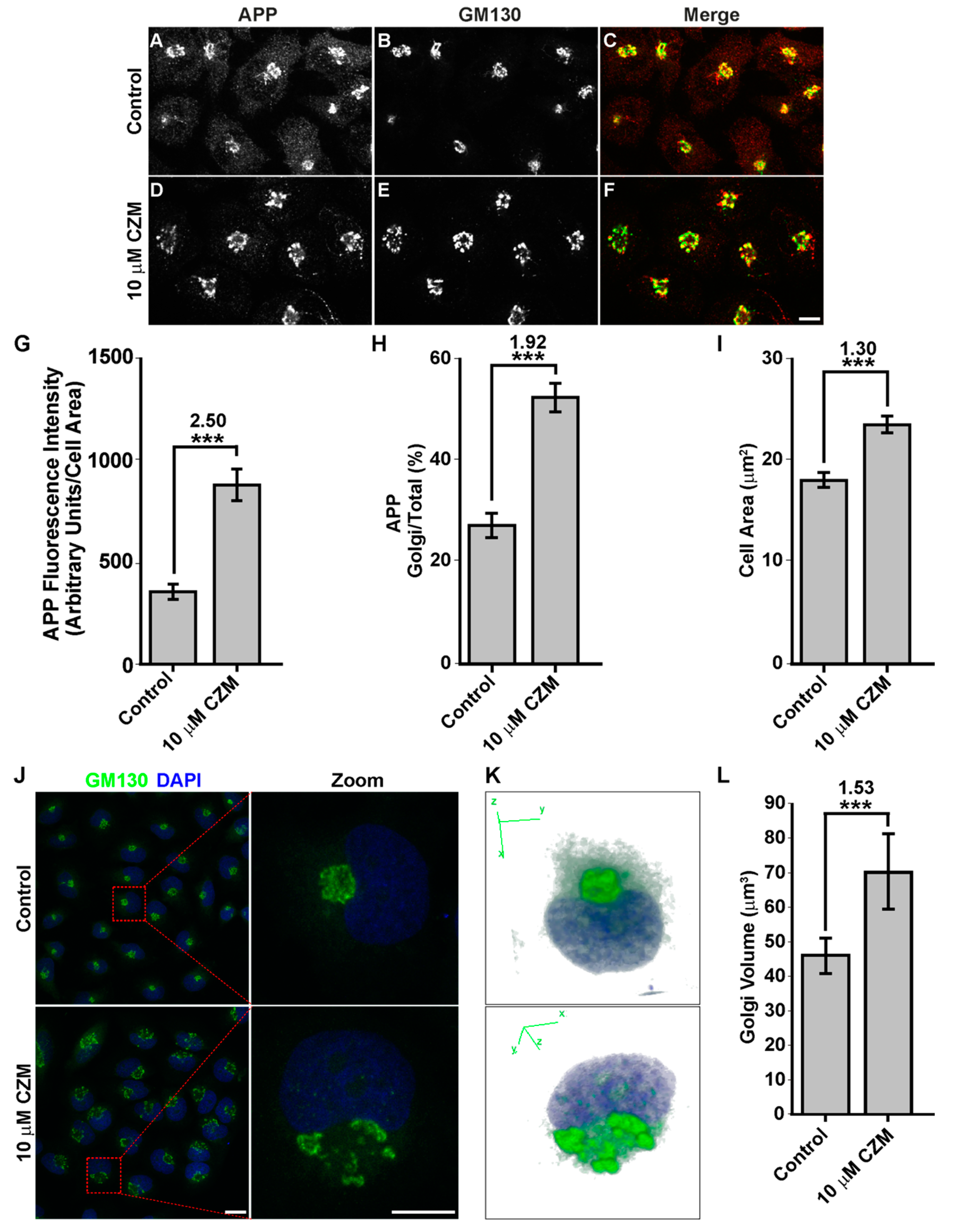
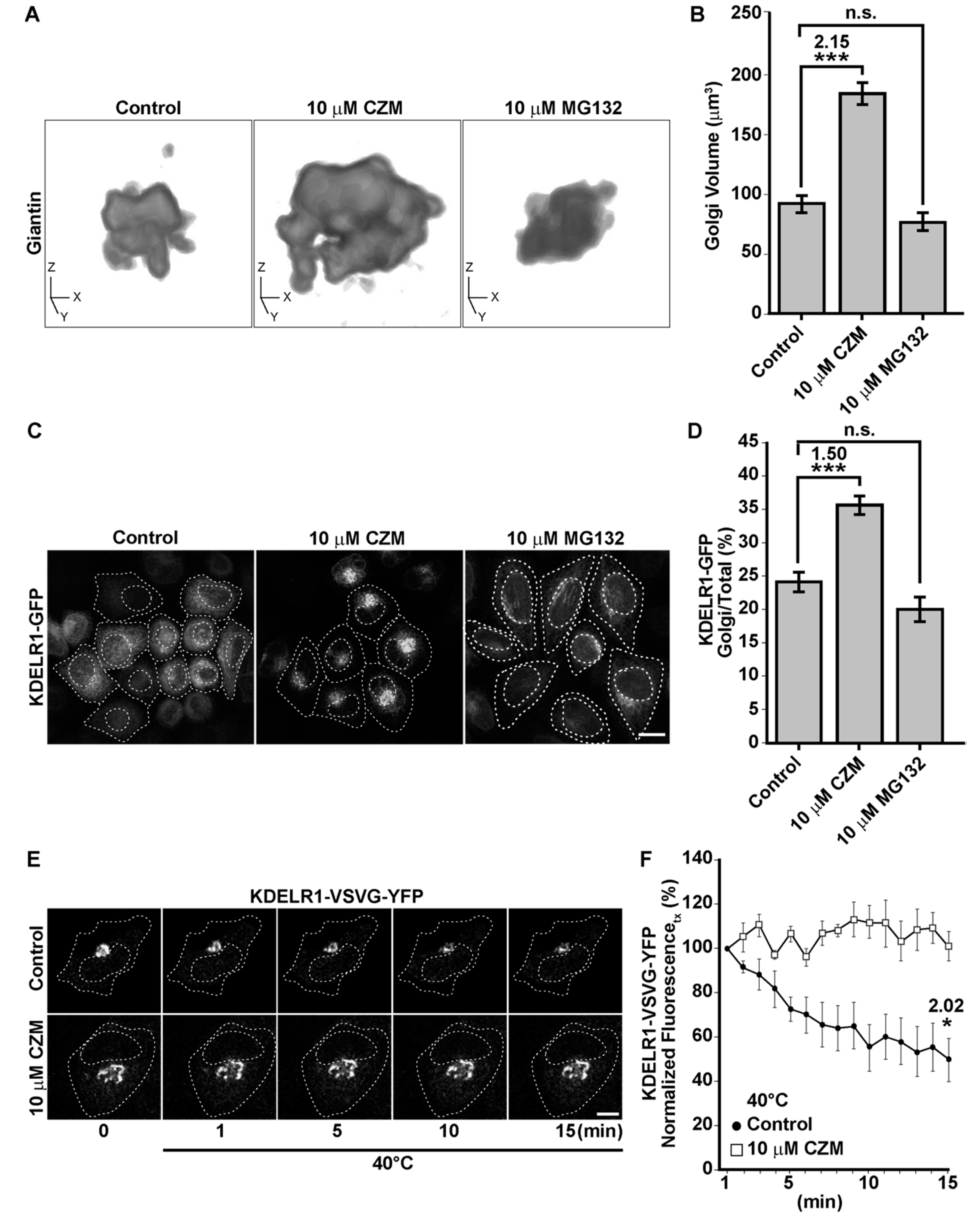
3.2. Acute Inhibition of the Deubiquitinating Enzyme PSMD14 of the 19S RP Accumulates APP in a Swollen Golgi Apparatus
3.3. Acute Inhibition of the Deubiquitinating Enzyme PSMD14 Perturbs Golgi-to-ER Retrograde Transport
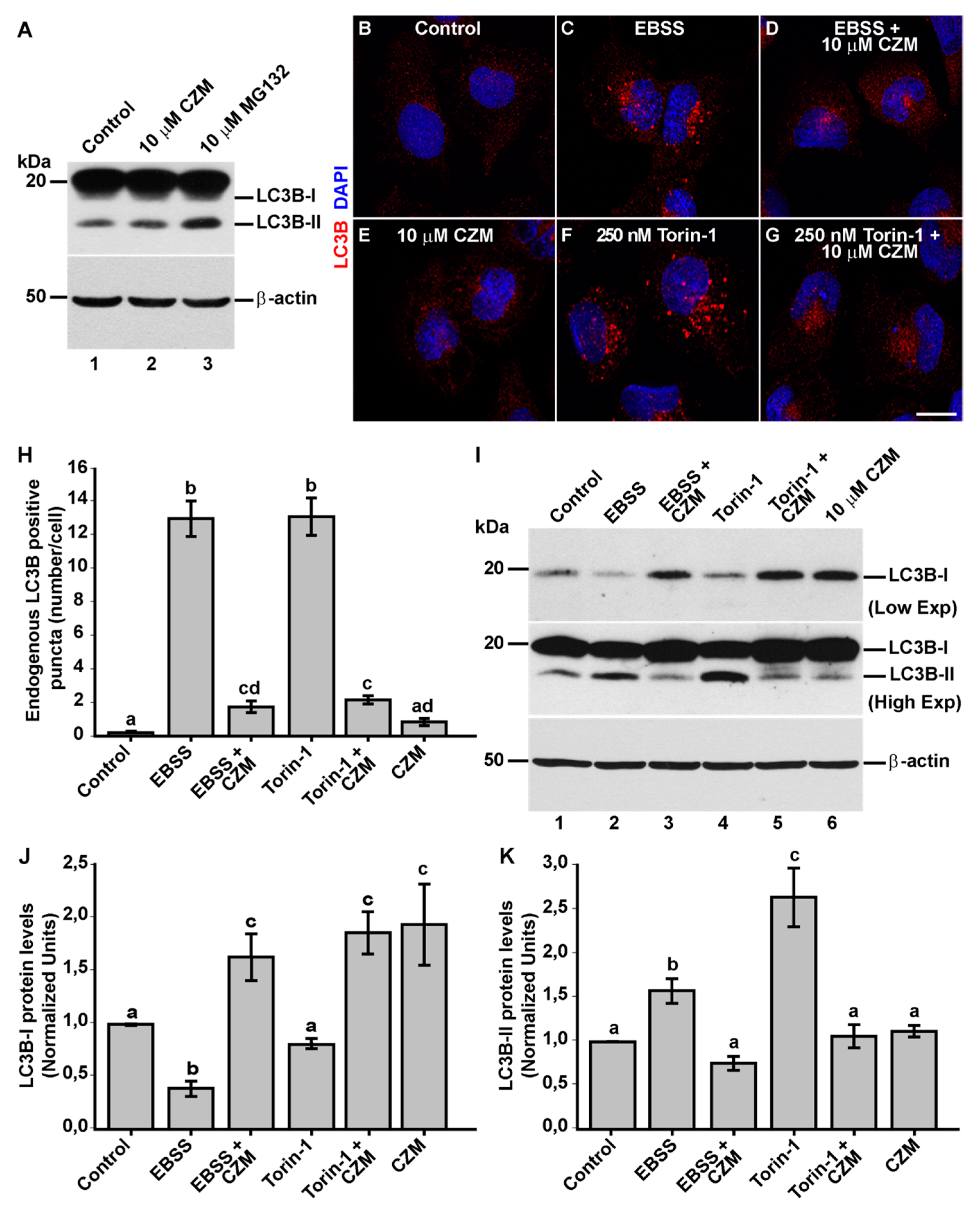
3.4. Inhibition of Golgi-to-ER Retrograde Transport by CZM Has a Negative Impact on Macroautophagy
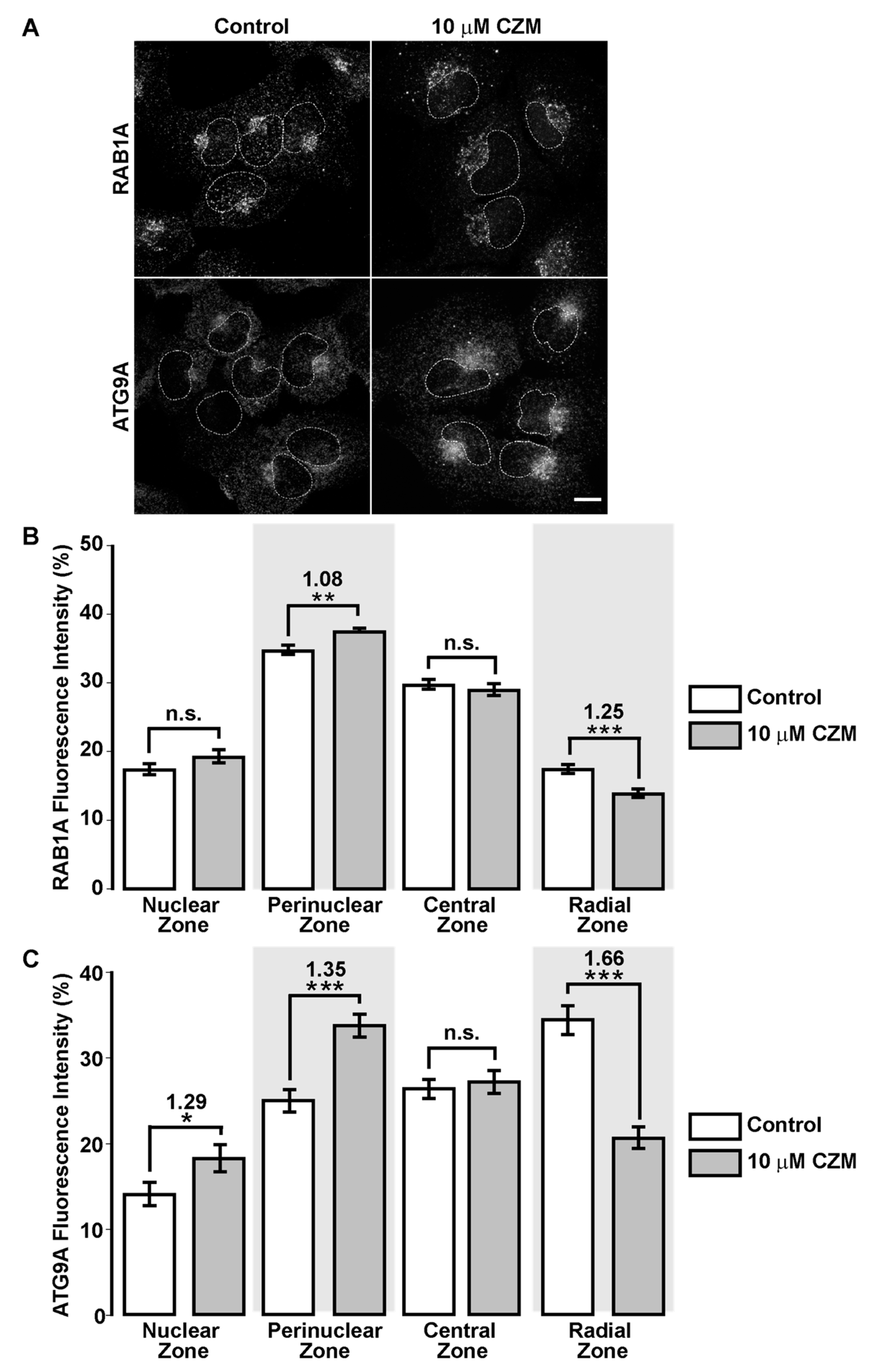
3.5. Inhibition of Golgi-to-ER Retrograde Transport by CZM Accumulates RAB1A and ATG9A at the Golgi Apparatus
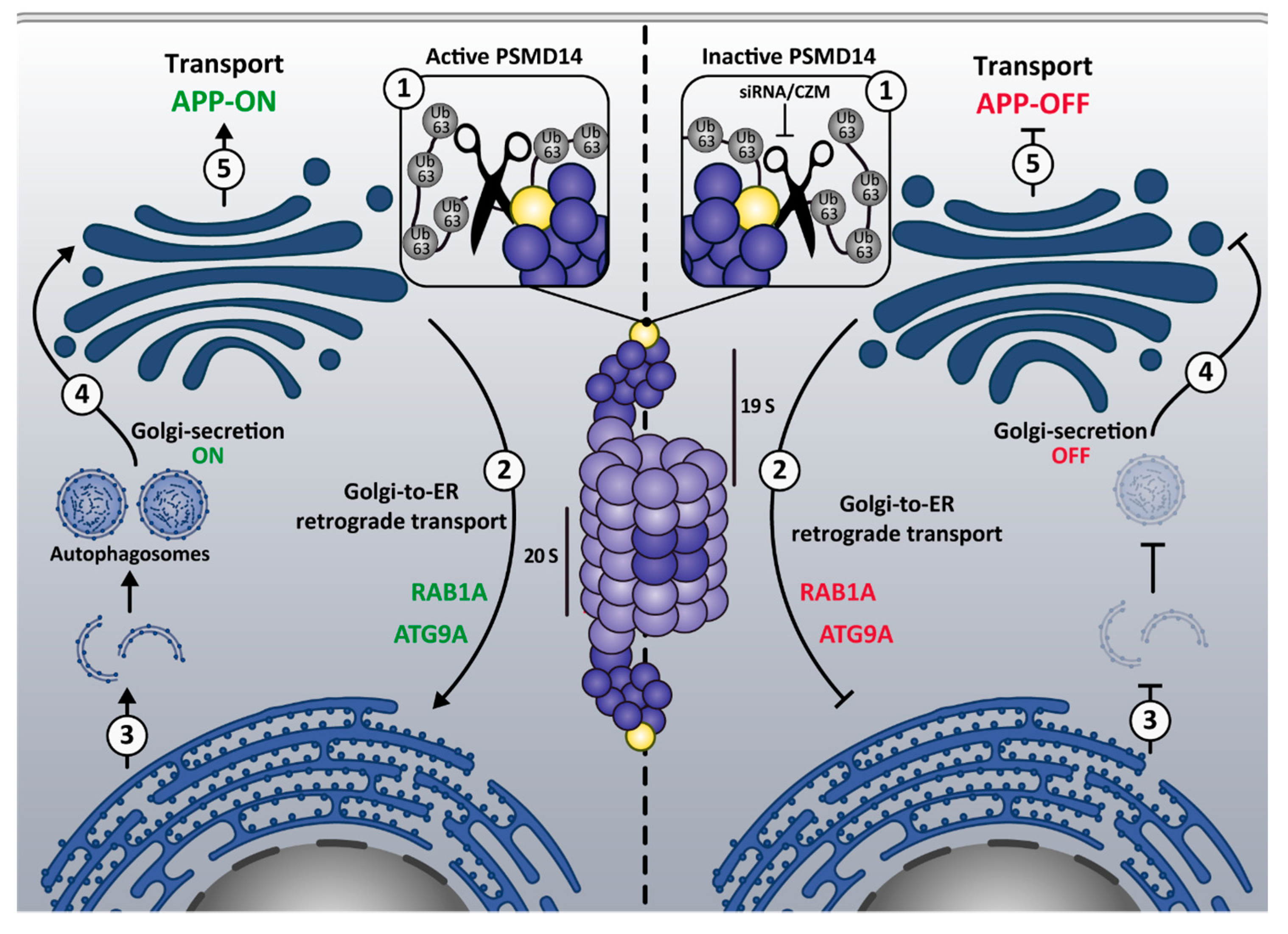
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shinde, S.R.; Maddika, S. Post translational modifications of Rab GTPases. Small GTPases 2017, 9, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.M.; Boyce, M. Directing Traffic: Regulation of COPI Transport by Post-translational Modifications. Front. Cell Dev. Boil. 2019, 7, 190. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- Versus Polyubiquitination: Differential Control of p53 Fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Foot, N.; Henshall, T.; Kumar, S. Ubiquitination and the Regulation of Membrane Proteins. Physiol. Rev. 2017, 97, 253–281. [Google Scholar] [CrossRef]
- Pelham, H.R. Membrane Traffic: GGAs Sort Ubiquitin. Curr. Boil. 2004, 14, R357–R359. [Google Scholar] [CrossRef]
- Yang, B.; Kumar, S. Nedd4 and Nedd4-2: Closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 2010, 17, 68–77. [Google Scholar] [CrossRef]
- Tan, J.; Evin, G. β-Site APP-cleaving enzyme 1 trafficking and Alzheimer’s disease pathogenesis. J. Neurochem. 2012, 120, 869–880. [Google Scholar] [CrossRef]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Ren, X.; Hurley, J.H. VHS domains of ESCRT-0 cooperate in high-avidity binding to polyubiquitinated cargo. EMBO J. 2010, 29, 1045–1054. [Google Scholar] [CrossRef]
- Clague, M.J.; Liu, H.; Urbe, S. Governance of Endocytic Trafficking and Signaling by Reversible Ubiquitylation. Dev. Cell 2012, 23, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem. Sci. 2003, 28, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbe, S. Ubiquitin: Same Molecule, Different Degradation Pathways. Cell 2010, 143, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A.J. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbe, S. Endocytosis: The DUB version. Trends Cell Boil. 2006, 16, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Boil. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Mevissen, T.E.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Bienko, M.; Green, C.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R.; et al. Ubiquitin-Binding Domains in Y-Family Polymerases Regulate Translesion Synthesis. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef]
- Bienko, M.; Green, C.; Sabbioneda, S.; Crosetto, N.; Matic, I.; Hibbert, R.G.; Begovic, T.; Niimi, A.; Mann, M.; Lehmann, A.R.; et al. Regulation of Translesion Synthesis DNA Polymerase η by Monoubiquitination. Mol. Cell 2010, 37, 396–407. [Google Scholar] [CrossRef]
- Husnjak, K.; Dikic, I. Ubiquitin-Binding Proteins: Decoders of Ubiquitin-Mediated Cellular Functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Sisodia, S.S.; Trowbridge, I.S. Characterization of sorting signals in the beta-amyloid precursor protein cytoplasmic domain. J. Boil. Chem. 1995, 270, 3565–3573. [Google Scholar] [CrossRef]
- Perez, R.G.; Soriano, S.; Hayes, J.D.; Ostaszewski, B.; Xia, W.; Selkoe, D.J.; Chen, X.; Stokin, G.B.; Koo, E.H. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J. Boil. Chem. 1999, 274, 18851–18856. [Google Scholar] [CrossRef] [PubMed]
- Burgos, P.V.; Mardones, G.A.; Rojas, A.L.; DaSilva, L.L.; Prabhu, Y.; Hurley, J.H.; Bonifacino, J.S. Sorting of the Alzheimer’s Disease Amyloid Precursor Protein Mediated by the AP-4 Complex. Dev. Cell 2010, 18, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Hikichi, Y.; Willuweit, A.; Shintani, Y.; Horiguchi, T. FBL2 Regulates Amyloid Precursor Protein (APP) Metabolism by Promoting Ubiquitination-Dependent APP Degradation and Inhibition of APP Endocytosis. J. Neurosci. 2012, 32, 3352–3365. [Google Scholar] [CrossRef] [PubMed]
- El Ayadi, A.; Stieren, E.S.; Barral, J.M.; Boehning, D. Ubiquilin-1 regulates amyloid precursor protein maturation and degradation by stimulating K63-linked polyubiquitination of lysine Proc. Natl. Acad. Sci. USA 2012, 109, 13416–13421. [Google Scholar] [CrossRef]
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Du Jour, K.S.P.; McCabe, B.D.; et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 2013, 4, 2250. [Google Scholar] [CrossRef]
- Williamson, R.L.; Laulagnier, K.; Miranda, A.M.; Fernandez, M.A.; Wolfe, M.S.; Sadoul, R.; Di Paolo, G. Disruption of amyloid precursor protein ubiquitination selectively increases amyloid β (Aβ) 40 levels via presenilin 2-mediated cleavage. J. Boil. Chem. 2017, 292, 19873–19889. [Google Scholar] [CrossRef]
- Cooper, E.M.; Cutcliffe, C.; Kristiansen, T.Z.; Pandey, A.; Pickart, C.M.; Cohen, R.E. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009, 28, 621–631. [Google Scholar] [CrossRef]
- Prabhu, Y.; Burgos, P.V.; Schindler, C.; Farías, G.G.; Magadár, J.G.; Bonifacino, J.S. Adaptor protein 2–mediated endocytosis of the β-secretase BACE1 is dispensable for amyloid precursor protein processing. Mol. Boil. Cell 2012, 23, 2339–2351. [Google Scholar] [CrossRef]
- Bustamante, H.A.; Rivera-Dictter, A.; Cavieres, V.A.; Muñoz, V.C.; González, A.; Lin, Y.; Mardones, G.A.; Burgos, P.V. Turnover of C99 is Controlled by a Crosstalk between ERAD and Ubiquitin-Independent Lysosomal Degradation in Human Neuroglioma Cells. PLoS ONE 2013, 8, e83096. [Google Scholar] [CrossRef] [PubMed]
- Cancino, J.; Capalbo, A.; Di Campli, A.; Giannotta, M.; Rizzo, R.; Jung, J.E.; Di Martino, R.; Persico, M.; Heinklein, P.; Sallese, M.; et al. Control Systems of Membrane Transport at the Interface between the Endoplasmic Reticulum and the Golgi. Dev. Cell 2014, 30, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Bett, J.S.; Ibrahim, A.F.M.; Garg, A.K.; Rocha, S.; Hay, R. siRNA Screening to Identify Ubiquitin and Ubiquitin-like System Regulators of Biological Pathways in Cultured Mammalian Cells. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Mackay, C.; Carroll, E.; Ibrahim, A.F.; Garg, A.; Inman, G.; Hay, R.; Alpi, A. E3 ubiquitin ligase HOIP attenuates apoptotic cell death induced by cisplatin. Cancer Res. 2014, 74, 2246–2257. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, 45. [Google Scholar] [CrossRef]
- Mlynarczuk-Bialy, I.; Doeppner, T.R.; Golab, J.; Nowis, D.; Wilczyński, G.; Parobczak, K.; Wigand, M.E.; Hajdamowicz, M.; Bialy, L.; Aniolek, O.; et al. Biodistribution and Efficacy Studies of the Proteasome Inhibitor BSc2118 in a Mouse Melanoma Model. Transl. Oncol. 2014, 7, 570–579. [Google Scholar] [CrossRef]
- González, A.E.; Muñoz, V.C.; Cavieres, V.A.; Bustamante, H.A.; Cornejo, V.-H.; Januário, Y.C.; González, I.; Hetz, C.; DaSilva, L.L.; Rojas-Fernández, A.; et al. Autophagosomes cooperate in the degradation of intracellular C-terminal fragments of the amyloid precursor protein via the MVB/lysosomal pathway. FASEB J. 2017, 31, 2446–2459. [Google Scholar] [CrossRef]
- Moffat, J.; Reiling, J.H.; Sabatini, D.M. Off-target effects associated with long dsRNAs in Drosophila RNAi screens. Trends Pharmacol. Sci. 2007, 28, 149–151. [Google Scholar] [CrossRef]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R.; Koonin, E.V.; Deshaies, R. Role of Rpn11 Metalloprotease in Deubiquitination and Degradation by the 26S Proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef]
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yakushi, T.; Parlati, F.; Mackinnon, A.L.; Perez, C.; Ma, Y.; Carter, K.P.; Colayco, S.; Magnuson, G.; Brown, B.; et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase RpnNat. Chem. Biol. 2017, 13, 486–493. [Google Scholar]
- Tsubuki, S.; Saito, Y.; Tomioka, M.; Ito, H.; Kawashima, S. Differential Inhibition of Calpain and Proteasome Activities by Peptidyl Aldehydes of Di-Leucine and Tri-Leucine. J. Biochem. 1996, 119, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Boil. 2001, 8, 739–758. [Google Scholar] [CrossRef]
- Wieland, F.T.; Gleason, M.L.; Serafini, T.A.; Rothman, J.E. The rate of bulk flow from the endoplasmic reticulum to the cell surface. Cell 1987, 50, 289–300. [Google Scholar] [CrossRef]
- Martínez-Menárguez, J.A.; Geuze, H.J.; Slot, J.W.; Klumperman, J. Vesicular Tubular Clusters between the ER and Golgi Mediate Concentration of Soluble Secretory Proteins by Exclusion from COPI-Coated Vesicles. Cell 1999, 98, 81–90. [Google Scholar] [CrossRef]
- Klumperman, J. Transport between ER and Golgi. Curr. Opin. Cell Boil. 2000, 12, 445–449. [Google Scholar] [CrossRef]
- Thor, F.; Gautschi, M.; Geiger, R.; Helenius, A. Bulk Flow Revisited: Transport of a Soluble Protein in the Secretory Pathway. Traffic 2009, 10, 1819–1830. [Google Scholar] [CrossRef]
- Tenorio, M.J.; Luchsinger, C.; Mardones, G.A. Protein Kinase A Activity Is Necessary for Fission and Fusion of Golgi to Endoplasmic Reticulum Retrograde Tubules. PLoS ONE 2015, 10, e0135260. [Google Scholar] [CrossRef][Green Version]
- Hsu, V.W.; Shah, N.; Klausner, R.D. A brefeldin A-like phenotype is induced by the overexpression of a human ERD-2-like protein, ELP-1. Cell 1992, 69, 625–635. [Google Scholar] [CrossRef]
- Cole, N.B.; Ellenberg, J.; Song, J.; DiEuliis, D.; Lippincott-Schwartz, J. Retrograde Transport of Golgi-localized Proteins to the ER. J. Cell Boil. 1998, 140, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.J.; Rayner, J.C.; Pelham, H.R.B. A novel SNARE complex implicated in vesicle fusion with the endoplasmic reticulum. EMBO J. 1997, 16, 3017–3024. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Ni, D.; Ma, B.; Lee, J.-H.; Zhang, T.; Ghozalli, I.; Pirooz, S.D.; Zhao, Z.; Bharatham, N.; Li, B.; et al. PtdIns(3)P-bound UVRAG coordinates Golgi–ER retrograde and Atg9 transport by differential interactions with the ER tether and the beclin 1 complex. Nature 2013, 15, 1206–1219. [Google Scholar] [CrossRef] [PubMed]
- Lemus, L.; Ribas, J.L.; Sikorska, N.; Goder, V. An ER-Localized SNARE Protein Is Exported in Specific COPII Vesicles for Autophagosome Biogenesis. Cell Rep. 2016, 14, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xiao, Y.; Chai, P.; Zheng, P.; Teng, J.; Chen, J. ATL3 Is a Tubular ER-Phagy Receptor for GABARAP-Mediated Selective Autophagy. Curr. Boil. 2019, 29, 846–855.e6. [Google Scholar] [CrossRef]
- Hao, R.; Nanduri, P.; Rao, Y.; Panichelli, R.S.; Ito, A.; Yoshida, M.; Yao, T.-P. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 2013, 51, 819–828. [Google Scholar] [CrossRef]
- Nanduri, P.; Hao, R.; Fitzpatrick, T.; Yao, T.-P. Chaperone-mediated 26S Proteasome Remodeling Facilitates Free K63 Ubiquitin Chain Production and Aggresome Clearance*. J. Boil. Chem. 2015, 290, 9455–9464. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Munafó, D.B.; Colombo, M.I. A novel assay to study autophagy: Regulation of autophagosome vacuole size by amino acid deprivation. J. Cell Sci. 2001, 114, 3619–3629. [Google Scholar]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORCJ. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.-W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; A Peden, A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Boil. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.; Ferraiuolo, L.; Myszczynska, M.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Boil. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014, 127, 4984. [Google Scholar] [CrossRef]
- Karanasios, E.; Walker, S.; Okkenhaug, H.; Manifava, M.; Hummel, E.; Zimmermann, H.; Ahmed, Q.; Domart, M.-C.; Collinson, L.M.; Ktistakis, N.T. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat. Commun. 2016, 7, 12420. [Google Scholar] [CrossRef]
- Tapia, D.; Jiménez, T.; Zamora, C.; Espinoza, J.; Rizzo, R.; González-Cárdenas, A.; Fuentes, D.; Hernández, S.; Cavieres, V.A.; Soza, A.; et al. KDEL receptor regulates secretion by lysosome relocation- and autophagy-dependent modulation of lipid-droplet turnover. Nat. Commun. 2019, 10, 735. [Google Scholar] [CrossRef]
- Iwata, A.; Riley, B.E.; Johnston, J.; Kopito, R.R. HDAC6 and Microtubules Are Required for Autophagic Degradation of Aggregated Huntingtin. J. Boil. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, N.B.; Ron, D.; Yin, X.-M. Linking of Autophagy to Ubiquitin-Proteasome System Is Important for the Regulation of Endoplasmic Reticulum Stress and Cell Viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef]
- Lan, D.; Wang, W.; Zhuang, J.; Zhao, Z. Proteasome inhibitor-induced autophagy in PC12 cells overexpressing A53T mutant α-synuclein. Mol. Med. Rep. 2014, 11, 1655–1660. [Google Scholar] [CrossRef]
- Peng, H.; Yang, J.; Li, G.; You, Q.; Han, W.; Li, T.; Gao, D.; Xie, X.; Lee, B.-H.; Du, J.; et al. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res. 2017, 27, 657–674. [Google Scholar] [CrossRef]
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A.L. The Role of ATF4 Stabilization and Autophagy in Resistance of Breast Cancer Cells Treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423. [Google Scholar] [CrossRef]
- Albornoz, N.; Bustamante, H.; Soza, A.; Burgos, P. Cellular Responses to Proteasome Inhibition: Molecular Mechanisms and Beyond. Int. J. Mol. Sci. 2019, 20, 3379. [Google Scholar] [CrossRef]
- Zhou, F.; Van Laar, T.; Huang, H.; Zhang, L. APP and APLP1 are degraded through autophagy in response to proteasome inhibition in neuronal cells. Protein Cell 2011, 2, 377–383. [Google Scholar] [CrossRef]
- Swaminathan, G.; Zhu, W.; Plowey, E.D. BECN1/Beclin 1 sorts cell-surface APP/amyloid β precursor protein for lysosomal degradation. Autophagy 2016, 12, 2404–2419. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-J.; Her, G.M.; Hu, M.-K.; Chen, Y.-W.; Tung, Y.-T.; Wu, P.-Y.; Hsu, W.-M.; Lee, H.; Jin, L.-W.; Hwang, S.-P.L.; et al. ErbB2 regulates autophagic flux to modulate the proteostasis of APP-CTFs in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3129–E3138. [Google Scholar] [CrossRef]
- Yang, C.; Cai, C.-Z.; Song, J.; Tan, J.-Q.; Durairajan, S.S.K.; Iyaswamy, A.; Wu, M.-Y.; Chen, L.-L.; Yue, Z.; Li, M.; et al. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 2017, 13, 2028–2040. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Norris, A.; Grant, B.D.; Wang, X. A novel requirement for ubiquitin-conjugating enzyme UBC-13 in retrograde recycling of MIG-14/Wntless and Wnt signaling. Mol. Biol. Cell 2018, 29, 2098–2112. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Jiang, C.; Chen, L.; Jin, J.; Wei, J.; Zhao, L.; Chen, M.; Pan, W.; Xu, Y.; Chu, H.; et al. The Ubiquitination of RagA GTPase by RNF152 Negatively Regulates mTORC1 Activation. Mol. Cell 2015, 58, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.H.; Kim, H.Y.; Heo, A.J.; Lee, S.H.; Lee, M.J.; Bin Kim, S.; SrinivasRao, G.; Mun, S.R.; Cha-Molstad, H.; Ciechanover, A.; et al. The N-Degron Pathway Mediates ER-phagy. Mol. Cell 2019, 75, 1058–1072.e9. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.; Ito, A.; Yao, T.-P. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Cohen, M.M.; Stutz, F.; Dargemont, C. Deubiquitination, a New Player in Golgi to Endoplasmic Reticulum Retrograde Transport. J. Boil. Chem. 2003, 278, 51989–51992. [Google Scholar] [CrossRef] [PubMed]
- Millard, S.; Wood, S. Riding the DUBway: Regulation of protein trafficking by deubiquitylating enzymes. J. Cell Boil. 2006, 173, 463–468. [Google Scholar] [CrossRef]
- Cohen, M.M.; Stutz, F.; Belgareh, N.; Haguenauer-Tsapis, R.; Dargemont, C. Ubp3 requires a cofactor, Bre5, to specifically de-ubiquitinate the COPII protein, Sec23. Nature 2003, 5, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Chang, J.C.; Luo, W.; Aryal, S.; Varotsis, D.; Randolph, L.; Netzer, W.J.; Greengard, P.; Flajolet, M. δ-COP modulates Aβ peptide formation via retrograde trafficking of APP. Proc. Natl. Acad. Sci. USA 2016, 113, 5412–5417. [Google Scholar] [CrossRef] [PubMed]
- He, S.; O’Connell, D.; Zhang, X.; Yang, Y.; Liang, C. The intersection of Golgi-ER retrograde and autophagic trafficking. Autophagy 2013, 10, 180–181. [Google Scholar] [CrossRef][Green Version]
- Rinaldi, L.; Donne, R.D.; Catalanotti, B.; Torres-Quesada, O.; Enzler, F.; Moraca, F.; Nisticò, R.; Chiuso, F.; Piccinin, S.; Bachmann, V.; et al. Feedback inhibition of cAMP effector signaling by a chaperone-assisted ubiquitin system. Nat. Commun. 2019, 10, 2572. [Google Scholar] [CrossRef]
- Feng, X.; Jia, Y.; Zhang, Y.; Ma, F.; Zhu, Y.; Hong, X.; Zhou, Q.; He, R.; Zhang, H.; Jin, J.; et al. Ubiquitination of UVRAG by SMURF1 promotes autophagosome maturation and inhibits hepatocellular carcinoma growth. Autophagy 2019, 15, 1130–1149. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef]
- Mattera, R.; Park, S.Y.; De Pace, R.; Guardia, C.M.; Bonifacino, J.S. AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc. Natl. Acad. Sci. USA 2017, 114, E10697–E10706. [Google Scholar] [CrossRef]
- Song, Y.; Li, S.; Ray, A.; Das, D.S.; Qi, J.; Samur, M.K.; Tai, Y.-T.; Munshi, N.; Carrasco, R.D.; Chauhan, D.; et al. Blockade of deubiquitylating enzyme Rpn11 triggers apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Oncogene 2017, 36, 5631–5638. [Google Scholar] [CrossRef]
- Wang, C.-H.; Lu, S.-X.; Liu, L.-L.; Li, Y.; Yang, X.; He, Y.-F.; Chen, S.-L.; Cai, S.; Wang, H.; Yun, J. POH1 Knockdown Induces Cancer Cell Apoptosis via p53 and Bim. Neoplasia 2018, 20, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Boil. 2014, 12, 94. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustamante, H.A.; Cereceda, K.; González, A.E.; Valenzuela, G.E.; Cheuquemilla, Y.; Hernández, S.; Arias-Muñoz, E.; Cerda-Troncoso, C.; Bandau, S.; Soza, A.; et al. The Proteasomal Deubiquitinating Enzyme PSMD14 Regulates Macroautophagy by Controlling Golgi-to-ER Retrograde Transport. Cells 2020, 9, 777. https://doi.org/10.3390/cells9030777
Bustamante HA, Cereceda K, González AE, Valenzuela GE, Cheuquemilla Y, Hernández S, Arias-Muñoz E, Cerda-Troncoso C, Bandau S, Soza A, et al. The Proteasomal Deubiquitinating Enzyme PSMD14 Regulates Macroautophagy by Controlling Golgi-to-ER Retrograde Transport. Cells. 2020; 9(3):777. https://doi.org/10.3390/cells9030777
Chicago/Turabian StyleBustamante, Hianara A, Karina Cereceda, Alexis E González, Guillermo E Valenzuela, Yorka Cheuquemilla, Sergio Hernández, Eloisa Arias-Muñoz, Cristóbal Cerda-Troncoso, Susanne Bandau, Andrea Soza, and et al. 2020. "The Proteasomal Deubiquitinating Enzyme PSMD14 Regulates Macroautophagy by Controlling Golgi-to-ER Retrograde Transport" Cells 9, no. 3: 777. https://doi.org/10.3390/cells9030777
APA StyleBustamante, H. A., Cereceda, K., González, A. E., Valenzuela, G. E., Cheuquemilla, Y., Hernández, S., Arias-Muñoz, E., Cerda-Troncoso, C., Bandau, S., Soza, A., Kausel, G., Kerr, B., Mardones, G. A., Cancino, J., Hay, R. T., Rojas-Fernandez, A., & Burgos, P. V. (2020). The Proteasomal Deubiquitinating Enzyme PSMD14 Regulates Macroautophagy by Controlling Golgi-to-ER Retrograde Transport. Cells, 9(3), 777. https://doi.org/10.3390/cells9030777