Tumor Necrosis Factor-Like Weak Inducer of Apoptosis (TWEAK)/Fibroblast Growth Factor-Inducible 14 (Fn14) Axis in Cardiovascular Diseases: Progress and Challenges



Abstract
1. Introduction
2. TWEAK and Fn14: Two Members Belonging to the TNF Superfamily
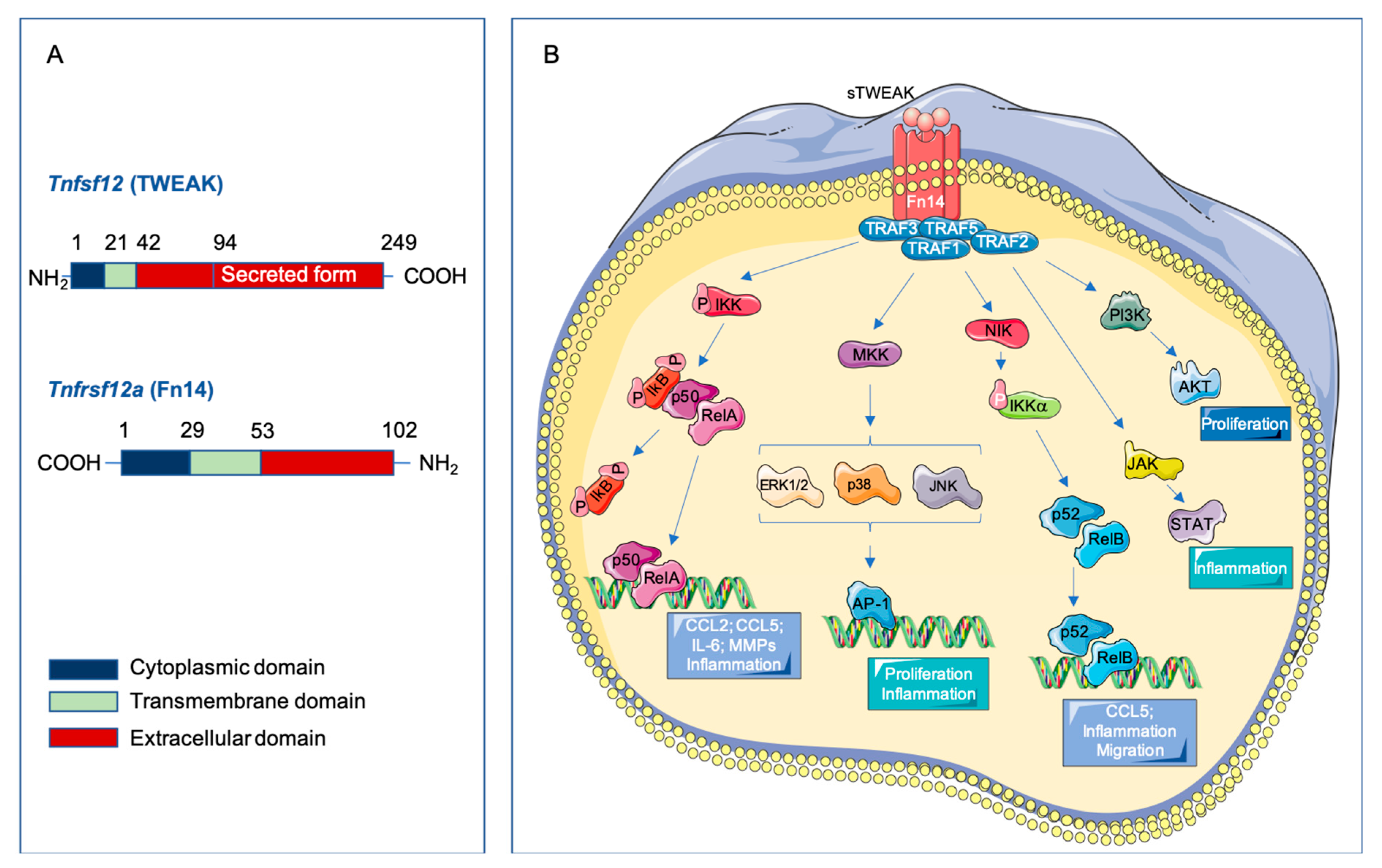
2.1. Structure of TWEAK and Fn14
2.2. Expression of TWEAK and Fn14
2.3. Signaling Pathways Activated by TWEAK-Fn14 Interaction
3. TWEAK and Atherosclerosis
3.1. Plaque Initiation
3.2. Lesion Progression
3.3. Plaque Stability
3.4. Plaque Rupture
4. TWEAK and Restenosis
5. TWEAK and Abdominal Aortic Aneurysm
6. TWEAK and Heart Failure
7. TWEAK and Stroke
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moran, A.E.; Forouzanfar, M.H.; Roth, G.A.; Mensah, G.A.; Ezzati, M.; Murray, C.J.; Naghavi, M. Temporal trends in ischemic heart disease mortality in 21 world regions, 1980 to 2010: The Global Burden of Disease 2010 study. Circulation 2014, 129, 1483–1492. [Google Scholar] [CrossRef]
- Dzau, V.J.; Braun-Dullaeus, R.C.; Sedding, D.G. Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat. Med. 2002, 8, 1249–1256. [Google Scholar] [CrossRef]
- Heusch, G.; Libby, P.; Gersh, B.; Yellon, D.; Böhm, M.; Lopaschuk, G.; Opie, L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 2014, 383, 1933–1943. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Smith, C.A.; Farrah, T.; Goodwin, R.G. The TNF receptor superfamily of cellular and viral proteins: Activation, costimulation, and death. Cell 1994, 76, 959–962. [Google Scholar] [CrossRef]
- Winkles, J.A. The TWEAK-Fn14 cytokine-receptor axis: Discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 2008, 7, 411–425. [Google Scholar] [CrossRef]
- Chicheportiche, Y.; Bourdon, P.R.; Xu, H.; Hsu, Y.M.; Scott, H.; Hession, C.; Garcia, I.; Browing, J.L. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J. Biol. Chem. 1997, 272, 32401–32410. [Google Scholar] [CrossRef]
- Wiley, S.R.; Winkles, J.A. TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev. 2003, 14, 241–249. [Google Scholar] [CrossRef]
- Campbell, S.; Michaelson, J.; Burkly, L.; Putterman, C. The role of TWEAK/Fn14 in the pathogenesis of inflammation and systemic autoimmunity. Front. Biosci. 2004, 9, 2273–2284. [Google Scholar] [CrossRef]
- Marsters, S.A.; Sheridan, J.P.; Pitti, R.M.; Brush, J.; Goddard, A.; Ashekenazi, A. Identification of a ligand for the death-domain containing receptor Apo3. Curr. Biol. 1998, 8, 525–528. [Google Scholar] [CrossRef]
- Wiley, S.R.; Cassiano, L.; Lofton, T.; Davis-Smith, T.; Winkles, J.A.; Lindner, V.; Liu, H.; Daniel, T.O.; Smith, C.A.; Fanslow, W.C. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity 2001, 15, 837–846. [Google Scholar] [CrossRef]
- Meighan-Mantha, R.L.; Hsu, D.K.; Guo, Y.; Brown, S.A.; Feng, S.L.; Peifley, K.A.; Alberts, G.F.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; et al. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J. Biol. Chem. 1999, 274, 33166–33176. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Hanscom, H.N.; Vu, H.; Brew, S.A.; Winkles, J.A. TWEAK binding to the Fn14 cysteine-rich domain depends on charged residues located in both the A1 and D2 modules. Biochem. J. 2006, 397, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Richards, C.M.; Hanscom, H.N.; Feng, S.L.; Winkles, J.A. The Fn14 cytoplasmatic tail binds tumour-necrosis-factor-receptor-associated factors 1, 2, 3 and 5 and mediates nuclear factor-kappa B activation. Biochem. J. 2003, 371, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.L.; McDonough, W.S.; Savitch, B.A.; Sawyer, T.F.; Winkles, J.A.; Berens, M.E. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NF-kappaB pathway activation and BCLXL/BCL-W expression. J. Biol. Chem. 2005, 280, 3483–3492. [Google Scholar] [CrossRef] [PubMed]
- Bossen, C.; Ingold, K.; Tardivel, A.; Bodmer, J.L.; Gaide, O.; Hertig, S.; Ambrose, C.; Tschopp, J.; Schneider, P. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J. Biol. Chem. 2006, 281, 13946–13971. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.S.; Burkly, L.C. No end in site: TWEAK/Fn14 activation and autoimmunity associated end-organ pathologies. J. Leukoc. Biol. 2008, 84, 338–347. [Google Scholar] [CrossRef]
- Muñoz-García, B.; Martín-Ventura, J.L.; Martínez, E.; Sánchez, S.; Hernández, G.; Ortega, L.; Ortiz, A.; Egido, J.; Blanco-Colio, L.M. Fn14 is upregulated in cytokine-stimulated vascular smooth muscle cells and is expressed in human carotid atherosclerotic plaques: Modulation by atorvastatin. Stroke 2006, 37, 2044–2053. [Google Scholar] [CrossRef]
- Tirnitz-Parker, J.E.; Viebahn, C.S.; Jakubowski, A.; Klopcic, B.R.; Olynyk, J.K.; Yeoh, G.C.; Knight, B. Tumor necrosis factor-like weak inducer of apoptosis is a mitogen for liver progenitor cells. Hepatology 2010, 52, 291–302. [Google Scholar] [CrossRef]
- Mustonen, E.; Säkkinen, H.; Tokola, H.; Isopoussu, E.; Aro, J.; Leskinen, H.; Ruskoaho, H.; Rysä, J. Tumour necrosis factor-like weak inducer of apoptosis (TWEAK) and its receptor Fn14 during cardiac remodelling in rats. Acta Physiol. (Oxf.) 2010, 199, 11–22. [Google Scholar] [CrossRef]
- Méndez-Barbero, N.; Gutierrez-Muñoz, C.; Madrigal-Matute, J.; Mínguez, P.; Egido, J.; Michel, J.B.; Martín-Ventura, J.L.; Esteban, V.; Blanco-Colio, L.M. A major role of TWEAK/Fn14 axis as a therapeutic target for post-angioplasty restenosis. EBioMedicine 2019, 46, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Justo, P.; Sanz, A.B.; Sanchez-Niño, M.D.; Winkles, J.A.; Lorz, C.; Egido, J.; Ortiz, A. Cytokine cooperation in renal tubular cell injury: The role of TWEAK. Kidney Int. 2006, 70, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Jakubowski, A.; Cui, L.; Shi, J.; Su, L.; Bauer, M.; Guan, J.; Lim, C.C.; Naito, Y.; Thompson, J.S.; et al. A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation 2009, 119, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Desplat-Jégo, S.; Varriale, S.; Creidy, R.; Terra, R.; Bernard, D.; Khrestchatisky, M.; Izui, S.; Chicheportiche, Y.; Boucraut, J. TWEAK is expressed by glial cells, induces astrocyte proliferation and increases EAE severity. J. Neuroimmunol. 2002, 133, 116–123. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Lewis, E.E.; Shelden, E.A.; Somers, E.; Pavlic, R.; McCune, W.J.; Richardson, B.C. The apoptotic ligands TRAIL, TWEAK, and Fas ligand mediate monocyte death induced by autologous lupus T cells. J. Immunol. 2002, 169, 6020–6029. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Kayagaki, N.; Yamaguchi, N.; Okumura, K.; Yagita, H. Involvement of TWEAK in interferon gamma-stimulated monocyte cytotoxicity. J. Exp. Med. 2000, 192, 1373–1380. [Google Scholar] [CrossRef]
- Maecker, H.; Varfolomeev, E.; Kischkel, F.; Lawrence, D.; LeBlanc, H.; Lee, W.; Hurst, S.; Danilenko, D.; Li, J.; Filvaroff, E.; et al. TWEAK attenuates the transition from innate to adaptive immunity. Cell 2005, 123, 931–944. [Google Scholar] [CrossRef]
- Mendez-Barbero, N.; Yuste-Montalvo, A.; Nuñez-Borque, E.; Jensen, B.M.; Gutiérrez-Muñoz, C.; Tome-Amat, J.; Garrido-Arandia, M.; Díaz-Perales, A.; Ballesteros-Martinez, C.; Laguna, J.J.; et al. The TNF-like weak inducer of the apoptosis/fibroblast growth factor-inducible molecule 14 axis mediates histamine and platelet-activating factor-induced subcutaneous vascular leakage and anaphylactic shock. J. Allergy Clin. Immunol. 2019, 145, 583–596. [Google Scholar] [CrossRef]
- Liu, Q.; Xiao, S.; Xia, Y. TWEAK/Fn14 Activation Participates in Skin Inflammation. Mediat. Inflamm. 2017, 2017. [Google Scholar] [CrossRef]
- Chorianopoulos, E.; Heger, T.; Lutz, M.; Frank, D.; Bea, F.; Katus, H.A.; Frey, N. FGF-inducible 14-kDa protein (Fn14) is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and mediates nuclear factor-kappaB activation by TWEAK. Basic Res. Cardiol. 2010, 105, 301–313. [Google Scholar] [CrossRef]
- Novoyatleva, T.; Diehl, F.; van Amerongen, M.J.; Patra, C.; Ferrazzi, F.; Bellazzi, R.; Engel, F.B. TWEAK is a positive regulator of cardiomyocyte proliferation. Cardiovasc. Res. 2010, 85, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.N.; Wang, D.J.; Ren, M.Y.; Wang, Q.L.; Sui, S.J. TWEAK/Fn14 promotes the proliferation and collagen synthesis of rat cardiac fibroblasts via the NF-кB pathway. Mol. Biol. Rep. 2012, 39, 8231–8241. [Google Scholar] [CrossRef] [PubMed]
- Donohue, P.J.; Richards, C.M.; Brown, S.A.; Hanscom, H.N.; Buschman, J.; Thangada, S.; Hla, T.; Williams, M.S.; Winkles, J.A. TWEAK is an endothelial cell growth and chemotactic factor that also potentiates FGF-2 and VEGF-A mitogenic activity. Arter. Thromb. Vasc. Biol. 2003, 23, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Burkly, L.C.; Michaelson, J.S.; Hahm, K.; Jakubowski, A.; Zheng, T.S. TWEAKing tissue remodeling by a multifunctional cytokine: Role of TWEAK/Fn14 pathway in health and disease. Cytokine 2007, 40, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Yoon, K.; Lee, K.; Kim, K.; Jang, H.; Lee, N.K.; Hwang, K.; Young Lee, S. TNF-related weak inducer of apoptosis receptor, a TNF receptor superfamily member, activates NF-kappa B through TNF receptor-associated factors. Biochem. Biophys. Res. Commun. 2003, 305, 789–796. [Google Scholar] [CrossRef]
- Saitoh, T.; Nakayama, M.; Nakano, H.; Yagita, H.; Yamamoto, N.; Yamaoka, S. TWEAK induces NF-kappaB2 p100 processing and long lasting NF-kappaB activation. J. Biol. Chem. 2003, 278, 36005–36012. [Google Scholar] [CrossRef]
- Ando, T.; Ichikawa, J.; Wako, M.; Hatsushika, K.; Watanabe, Y.; Sakuma, M.; Tasaka, K.; Ogawa, H.; Hamada, Y.; Yagita, H.; et al. TWEAK/Fn14 interaction regulates RANTES production, BMP-2-induced differentiation, and RANKL expression in mouse osteoblastic MC3T3-E1 cells. Arthritis Res. Ther. 2006, 8, R146. [Google Scholar] [CrossRef]
- Moreno, J.A.; Sastre, C.; Madrigal-Matute, J.; Muñoz-García, B.; Ortega, L.; Burkly, L.C.; Egido, J.; Martín-Ventura, J.L.; Blanco-Colio, L.M. HMGB1 expression and secretion are increased via TWEAK-Fn14 interaction in atherosclerotic plaques and cultured monocytes. Arter. Thromb. Vasc. Biol. 2013, 33, 612–620. [Google Scholar] [CrossRef]
- Fernández-Laso, V.; Sastre, C.; Méndez-Barbero, N.; Egido, J.; Martín-Ventura, J.L.; Gómez-Guerrero, C.; Blanco-Colio, L.M. TWEAK blockade decreases atherosclerotic lesion size and progression through suppression of STAT1 signaling in diabetic mice. Sci. Rep. 2017, 7, 46679. [Google Scholar] [CrossRef]
- Wang, A.; Zhang, F.; Xu, H.; Xu, M.; Cao, Y.; Wang, C.; Xu, Y.; Su, M.; Zhang, M.; Zhuge, Y. TWEAK/Fn14 promotes pro-inflammatory cytokine secretion in hepatic stellate cells via NF-κB/STAT3 pathways. Mol. Immunol. 2017, 87, 67–75. [Google Scholar] [CrossRef]
- Kumar, M.; Makonchuk, D.Y.; Li, H.; Mittal, A.; Kumar, A. TNF-like weak inducer of apoptosis (TWEAK) activates proinflammatory signaling pathways and gene expression through the activation of TGF-beta-activated kinase 1. J. Immunol. 2009, 182, 2439–2448. [Google Scholar] [CrossRef]
- Brown, S.A.; Ghosh, A.; Winkles, J.A. Full-length, membrane-anchored TWEAK can function as a juxtacrine signaling molecule and activate the NF-kappa B pathway. J. Biol. Chem. 2010, 285, 17432–17441. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Colio, L.M. TWEAK/Fn14 Axis: A Promising Target for the Treatment of Cardiovascular Diseases. Front. Immunol. 2014, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.N.; Wang, Y.C.; Lund, J.K.; Chen, Y.W.; Leal, J.A.; Wiley, S.R. TWEAK induces angiogenesis and proliferation of endothelial cells. J. Biol. Chem. 1999, 274, 8455–8849. [Google Scholar] [CrossRef] [PubMed]
- Perper, S.J.; Browning, B.; Burkly, L.C.; Weng, S.; Gao, C.; Giza, K.; Su, L.; Tarilonte, L.; Crowell, T.; Rajman, L.; et al. TWEAK is a novel arthritogenic mediator. J. Immunol. 2006, 177, 2610–2620. [Google Scholar] [CrossRef]
- Harada, N.; Nakayama, M.; Nakano, H.; Fukuchi, Y.; Yagita, H.; Okumura, K. Pro-inflammatory effect of TWEAK/Fn14 interaction on human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2002, 299, 488–493. [Google Scholar] [CrossRef]
- Yang, B.; Yan, P.; Gong, H.; Zuo, L.; Shi, Y.; Guo, J.; Guo, R.; Xie, J.; Li, B. TWEAK protects cardiomyocyte against apoptosis in a PI3K/AKT pathway dependent manner. Am. J. Transl. Res. 2016, 8, 3848–3860. [Google Scholar]
- Schapira, K.; Burkly, L.C.; Zheng, T.S.; Wu, P.; Groeneweg, M.; Rousch, M.; Kockx, M.M.; Daemen, M.J.; Heeneman, S. Fn14-Fc fusion protein regulates atherosclerosis in ApoE-/- mice and inhibits macrophage lipid uptake in vitro. Arter. Thromb. Vasc. Biol. 2009, 29, 2021–2027. [Google Scholar] [CrossRef]
- Hénaut, L.; Sanz, A.B.; Martin-Sanchez, D.; Carrasco, S.; Villa-Bellosta, R.; Aldamiz-Echevarria, G.; Massy, Z.A.; Sanchez-Nino, M.D.; Ortiz, A. TWEAK favors phosphate-induced calcification of vascular smooth muscle cells through canonical and non-canonical activation of NFκB. Cell Death Dis. 2016, 7, e2305. [Google Scholar] [CrossRef]
- Muñoz-García, B.; Madrigal-Matute, J.; Moreno, J.A.; Martin-Ventura, J.L.; López-Franco, O.; Sastre, C.; Ortega, L.; Burkly, L.C.; Egido, J.; Blanco-Colio, L.M. TWEAK-Fn14 interaction enhances plasminogen activator inhibitor 1 and tissue factor expression in atherosclerotic plaques and in cultured vascular smooth muscle cells. Cardiovasc. Res. 2011, 89, 225–233. [Google Scholar] [CrossRef]
- Novoyatleva, T.; Janssen, W.; Wietelmann, A.; Schermuly, R.T.; Engel, F.B. TWEAK/Fn14 axis is a positive regulator of cardiac hypertrophy. Cytokine 2013, 64, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Bover, L.C.; Cardó-Vila, M.; Kuniyasu, A.; Sun, J.; Rangel, R.; Takeya, M.; Aggarwal, B.B.; Arap, W.; Pasqualini, R. A previously unrecognized protein-protein interaction between TWEAK and CD163: Potential biological implications. J. Immunol. 2007, 178, 8183–8194. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Muñoz-García, B.; Martín-Ventura, J.L.; Madrigal-Matute, J.; Orbe, J.; Páramo, J.A.; Ortega, L.; Egido, J.; Blanco-Colio, L.M. The CD163-expressing macrophages recognize and internalize TWEAK: Potential consequences in atherosclerosis. Atherosclerosis 2009, 207, 103–110. [Google Scholar] [CrossRef]
- Akahori, H.; Karmali, V.; Polavarapu, R.; Lyle, A.N.; Weiss, D.; Shin, E.; Husain, A.; Naqvi, N.; Van Dam, R.; Habib, A.; et al. CD163 interacts with TWEAK to regulate tissue regeneration after ischaemic injury. Nat. Commun. 2015, 6, 7792. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Gallio, A.; Aboyans, V.; Diehm, C.; Cosentino, F.; Stricker, H.; Falk, E.; Schouten, O.; Lekakis, J.; Amann-Vesti, B.; Siclari, F.; et al. European Society of Cardiology Working Group on Peripheral Circulation. Non-coronary atherosclerosis. Eur. Heart J. 2014, 35, 1112–1119. [Google Scholar]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. the road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef]
- Blanco-Colio, L.M.; Martín-Ventura, J.L.; Munoz-Garcia, B.; Moreno, J.A.; Meilhac, O.; Ortiz, A.; Egido, J. TWEAK and Fn14. New players in the pathogenesis of atherosclerosis. Front. Biosci. 2007, 12, 3648–3655. [Google Scholar] [CrossRef]
- Nus, M.; Mallat, Z. Immune-mediated mechanisms of atherosclerosis and implications for the clinic. Expert Rev. Clin. Immunol. 2016, 12, 1217–1237. [Google Scholar] [CrossRef]
- Brand, K.; Page, S.; Rogler, G.; Bartsch, A.; Brandl, R.; Knuechel, R.; Page, M.; Kaltschmidt, C.; Baeuerle, P.A.; Neumeier, D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J. Clin. Investig. 1996, 97, 1715–1722. [Google Scholar] [CrossRef]
- Martín-Ventura, J.L.; Blanco-Colio, L.M.; Muñoz-García, B.; Gómez-Hernández, A.; Arribas, A.; Ortega, L.; Tuñón, J.; Egido, J. NF-kappaB activation and Fas ligand overexpression in blood and plaques of patients with carotid atherosclerosis: Potential implication in plaque instability. Stroke 2004, 35, 458–463. [Google Scholar] [CrossRef]
- Muñoz-García, B.; Moreno, J.A.; López-Franco, O.; Sanz, A.B.; Martín-Ventura, J.L.; Blanco, J.; Jakubowski, A.; Burkly, L.C.; Ortiz, A.; Egido, J.; et al. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances vascular and renal damage induced by hyperlipidemic diet in ApoE-knockout mice. Arter. Thromb. Vasc. Biol. 2009, 29, 2061–2068. [Google Scholar] [CrossRef]
- Sastre, C.; Fernández-Laso, V.; Madrigal-Matute, J.; Muñoz-García, B.; Moreno, J.A.; Pastor-Vargas, C.; Llamas-Granda, P.; Burkly, L.C.; Egido, J.; Martín-Ventura, J.L.; et al. Genetic deletion or TWEAK blocking antibody administration reduce atherosclerosis and enhance plaque stability in mice. J. Cell Mol. Med. 2014, 18, 721–734. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, Y.J.; Kim, W.J.; Woo, D.K.; Lee, Y.; Kim, D.I.; Park, Y.B.; Kwon, B.S.; Park, J.E.; Lee, W.H. TWEAK can induce pro-inflammatory cytokines and matrix metalloproteinase-9 in macrophages. Circ. J. 2004, 68, 396–399. [Google Scholar] [CrossRef]
- Kalinina, N.; Agrotis, A.; Antropova, Y.; DiVitto, G.; Kanellakis, P.; Kostolias, G.; Ilyinskaya, O.; Tararak, E.; Bobik, A. Increased expression of the DNA-binding cytokine HMGB1 in human atherosclerotic lesions: Role of activated macrophages and cytokines. Arter. Thromb. Vasc. Biol. 2004, 24, 2320–2325. [Google Scholar] [CrossRef]
- Martin-Ventura, J.L.; Rodrigues-Diez, R.; Martinez-Lopez, D.; Salaices, M.; Blanco-Colio, L.M.; Briones, A.M. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 2315. [Google Scholar] [CrossRef]
- Lassègue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arter. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef]
- Lu, Y.; Wahl, L.M. Oxidative stress augments the production of matrix metalloproteinase-1, cyclooxygenase-2, and prostaglandin E2 through enhancement of NF-kappa B activity in lipopolysaccharide-activated human primary monocytes. J. Immunol. 2005, 175, 5423–5429. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Fernandez-Laso, V.; Sastre, C.; Llamas-Granda, P.; Egido, J.; Martin-Ventura, J.L.; Zalba, G.; Blanco-Colio, L.M. TWEAK/Fn14 interaction promotes oxidative stress through NADPH oxidase activation in macrophages. Cardiovasc. Res. 2015, 108, 139–147. [Google Scholar] [CrossRef]
- Ketelhuth, D.F.; Bäck, M. The role of matrix metalloproteinases in atherothrombosis. Curr. Atheroscler. Rep. 2011, 13, 162–169. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, W.H.; Kwon, B.S.; Oh, G.T.; Choi, Y.H.; Park, J.E. Tumor necrosis factor receptor superfamily 12 may destabilize atherosclerotic plaques by inducing matrix metalloproteinases. Jpn. Circ. J. 2001, 65, 136–138. [Google Scholar] [CrossRef][Green Version]
- Huang, H.; Virmani, R.; Younis, H.; Burke, A.P.; Kamm, R.D.; Lee, R.T. The impact of calcification on the biomechanical stability of atherosclerotic plaques. Circulation 2001, 103, 1051–1056. [Google Scholar] [CrossRef]
- Agirbasli, M. Pivotal role of plasminogen-activator inhibitor-1 in vascular disease. Int. J. Clin. Pract. 2005, 59, 102–106. [Google Scholar] [CrossRef]
- Jukema, J.W.; Ahmed, T.A.; Verschuren, J.J.; Quax, P.H. Restenosis after PCI. Part 2: Prevention and therapy. Nat. Rev. Cardiol. 2011, 9, 79–90. [Google Scholar] [CrossRef]
- Schwartz, S.M. Perspectives series: Cell adhesion in vascular biology. Smooth muscle migration in atherosclerosis and restenosis. J. Clin. Investig. 1997, 99, 2814–2816. [Google Scholar] [CrossRef]
- Chaabane, C.; Otsuka, F.; Virmani, R.; Bochaton-Piallat, M.-L. Biological responses in tented arteries. Cardiovasc. Res. 2013, 99, 353–363. [Google Scholar] [CrossRef]
- Waseda, K.; Miyazawa, A.; Ako, J.; Hasegawa, T.; Tsujino, I.; Sakurai, R.; Yock, P.G.; Honda, Y.; Kandzari, D.E.; Leon, M.B.; et al. ENDEAVOR IV Trial Investigators. Intravascular ultrasound results from the ENDEAVOR IV trial: Randomized comparison between zotarolimus- and paclitaxel-eluting stents in patients with coronary artery disease. JACC Cardiovasc. Interv. 2009, 2, 779–784. [Google Scholar] [CrossRef][Green Version]
- Simone, T.M.; Higgins, S.P.; Archambeault, J.; Higgins, C.E.; Ginnan, R.G.; Singer, H.; Higgins, P.J. A small molecule PAI-1 functional inhibitor attenuates neointimal hyperplasia and vascular smooth muscle cell survival by promoting PAI-1 cleavage. Cell Signal. 2015, 27, 923–933. [Google Scholar] [CrossRef]
- Golledge, J.; Muller, J.; Daugherty, A.; Norman, P. Abdominal aortic aneurysm: Pathogenesis and implications for management. Arter. Thromb. Vasc. Biol. 2006, 26, 2605–2613. [Google Scholar] [CrossRef]
- Powell, J.T.; Brady, A.R. Detection, management, and prospects for the medical treatment of small abdominal aortic aneurysms. Arter. Thromb. Vasc. Biol. 2004, 24, 241–245. [Google Scholar] [CrossRef]
- Martín-Ventura, J.L.; Lindholt, J.S.; Moreno, J.A.; de Céniga, M.V.; Meilhac, O.; Michel, J.B.; Egido, J.; Blanco-Colio, L.M. Soluble TWEAK plasma levels predict expansion of human abdominal aortic aneurysms. Atherosclerosis 2011, 214, 486–489. [Google Scholar] [CrossRef]
- Tarín, C.; Fernández-Laso, V.; Sastre, C.; Madrigal-Matute, J.; Gómez, M.; Zaragoza, C.; Egido, J.; Burkly, L.C.; Martín-Ventura, J.L.; Blanco-Colio, L.M. Tumor necrosis factor-like weak inducer of apoptosis or Fn14 deficiency reduce elastase perfusion-induced aortic abdominal aneurysm in mice. J. Am. Heart Assoc. 2014, 3, e000723. [Google Scholar] [CrossRef]
- Sakalihasan, N.; Michel, J.B.; Katsargyris, A.; Kuivaniemi, H.; Defraigne, J.O.; Nchimi, A.; Powell, J.T.; Yoshimura, K.; Hultgren, R. Abdominal aortic aneurysms. Nat. Rev. Dis. Primers 2018, 4, 34. [Google Scholar] [CrossRef]
- Houard, X.; Touat, Z.; Ollivier, V.; Louedec, L.; Philippe, M.; Sebbag, U.; Meilhac, O.; Rossignol, P.; Michel, J.B. Mediators of neutrophil recruitment in human abdominal aortic aneurysms. Cardiovasc. Res. 2009, 82, 532–541. [Google Scholar] [CrossRef]
- Middleton, R.K.; Bown, M.J.; Lloyd, G.M.; Jones, J.L.; London, N.J.; Sayers, R.D. Characterisation of Interleukin-8 and monocyte chemoattractant protein-1 expression within the abdominal aortic aneurysm and their association with mural inflammation. Eur. J. Vasc. Endovasc. Surg. 2009, 37, 46–55. [Google Scholar] [CrossRef]
- Middleton, R.K.; Lloyd, G.M.; Bown, M.J.; Cooper, N.J.; London, N.J.; Sayers, R.D. The pro-inflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: A protein array study. J. Vasc. Surg. 2007, 45, 574–580. [Google Scholar] [CrossRef]
- Moehle, C.W.; Bhamidipati, C.M.; Alexander, M.R.; Mehta, G.S.; Irvine, J.N.; Salmon, M.; Upchurch, G.R., Jr.; Kron, I.L.; Owens, G.K.; Ailawadi, G. Bone marrow-derived MCP1 required for experimental aortic aneurysm formation and smooth muscle phenotypic modulation. J. Thorac. Cardiovasc. Surg. 2011, 142, 1567–1574. [Google Scholar] [CrossRef]
- Iida, Y.; Xu, B.; Xuan, H.; Glover, K.J.; Tanaka, H.; Hu, X.; Fujimura, N.; Wang, W.; Schultz, J.R.; Turner, C.R.; et al. Peptide inhibitor of CXCL4-CCL5 heterodimer formation, MKEY, inhibits experimental aortic aneurysm initiation and progression. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 718–726. [Google Scholar] [CrossRef]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, R.; Oo, A.Y.; Xiao, Q. Matrix Metalloproteinase in Abdominal Aortic Aneurysm and Aortic Dissection. Pharmaceuticals (Basel) 2019, 12, E118. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef]
- Novoyatleva, T.; Sajjad, A.; Engel, F.B. TWEAK-Fn14 Cytokine-Receptor Axis: A New Player of Myocardial Remodeling and Cardiac Failure. Front. Immunol. 2014, 5, 50. [Google Scholar] [CrossRef]
- Leri, A.; Kajstura, J.; Anversa, P. Myocyte proliferation and ventricular remodeling. J. Card Fail. 2002, 8, S518–S525. [Google Scholar] [CrossRef]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef]
- Das, N.A.; Carpenter, A.J.; Yoshida, T.; Kumar, S.A.; Gautam, S.; Mostany, R.; Izadpanah, R.; Kumar, A.; Mummidi, S.; Siebenlist, U.; et al. TRAF3IP2 mediates TWEAK/TWEAKR-induced pro-fibrotic responses in cultured cardiac fibroblasts and the heart. J. Mol. Cell Cardiol. 2018, 121, 107–123. [Google Scholar] [CrossRef]
- Shi, J.; Jiang, B.; Qiu, Y.; Guan, J.; Jain, M.; Cao, X.; Bauer, M.; Su, L.; Burkly, L.C.; Leone, T.C.; et al. PGC1α plays a critical role in TWEAK-induced cardiac dysfunction. PLoS ONE 2013, 8, e54054. [Google Scholar] [CrossRef]
- Jarr, K.U.; Eschricht, S.; Burkly, L.C.; Preusch, M.; Katus, H.A.; Frey, N.; Chorianopoulos, E. TNF-like weak inducer of apoptosis aggravates left ventricular dysfunction after myocardial infarction in mice. Mediat. Inflamm. 2014, 2014, 131950. [Google Scholar] [CrossRef]
- Hao, L.; Ren, M.; Rong, B.; Xie, F.; Lin, M.-J.; Zhao, Y.-C.; Yue, X.; Han, W.-Q.; Zhon, J.-Q. TWEAK/Fn14 mediates atrial-derived HL-1 myocytes hypertrophy via JAK2/STAT3 signalling pathway. J. Cell Mol. Med. 2018, 22, 4344–4353. [Google Scholar] [CrossRef]
- Orogo, A.M.; Gustafsson, Å.B. Cell death in the myocardium: My heart won’t go on. IUBMB Life 2013, 65, 651–656. [Google Scholar] [CrossRef]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res. 2014, 103, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, M.W.; Rechner, C.; Freund, C.; Baurand, A.; El Jamali, A.; Dietz, R. Statins inhibit reoxygenation-induced cardiomyocyte apoptosis: Role for glycogen synthase kinase 3beta and transcription factor beta-catenin. J. Mol. Cell Cardiol. 2004, 37, 681–690. [Google Scholar] [CrossRef]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M.; Farbood, Y.; Moghaddam, H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017, 38, 1167–1186. [Google Scholar] [CrossRef]
- Inta, I.; Frauenknecht, K.; Dörr, H.; Kohlhof, P.; Rabsilber, T.; Auffarth, G.U.; Burkly, L.; Mittelbronn, M.; Hahm, K.; Sommer, C.; et al. Induction of the cytokine TWEAK and its receptor Fn14 in ischemic stroke. J. Neurol. Sci. 2008, 275, 117–120. [Google Scholar] [CrossRef]
- Potrovita, I.; Zhang, W.; Burkly, L.; Hahm, K.; Lincecum, J.; Wang, M.Z.; Maurer, M.H.; Rossner, M.; Schneider, A.; Schwaninger, M. Tumor necrosis factor-like weak inducer of apoptosis-induced neurodegeneration. J. Neurosci. 2004, 24, 8237–8244. [Google Scholar] [CrossRef]
- Yepes, M.; Brown, S.A.; Moore, E.G.; Smith, E.P.; Lawrence, D.A.; Winkles, J.A. A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am. J. Pathol. 2005, 166, 511–520. [Google Scholar] [CrossRef]
- Polavarapu, R.; Gongora, M.C.; Winkles, J.A.; Yepes, M. Tumor necrosis factor-like weak inducer of apoptosis increases the permeability of the neurovascular unit through nuclear factor-kappa B pathway activation. J. Neurosci. 2005, 25, 10094–10100. [Google Scholar] [CrossRef]
- Zhang, X.; Winkles, J.A.; Gongora, M.C.; Polavarapu, R.; Michaelson, J.S.; Hahm, K.; Burkly, L.; Friedman, M.; Li, X.J.; Yepes, M. TWEAK-Fn14 pathway inhibition protects the integrity of the neurovascular unit during cerebral ischemia. J. Cereb. Blood Flow Metab. 2007, 27, 534–544. [Google Scholar] [CrossRef]
- Saas, P.; Boucraut, J.; Walker, P.R.; Quiquerez, A.L.; Billot, M.; Desplat-Jego, S.; Chicheportiche, Y.; Dietrich, P.Y. TWEAK stimulation of astrocytes and the proinflammatory consequences. Glia 2000, 32, 102–107. [Google Scholar] [CrossRef]
- Haile, W.B.; Echeverry, R.; Wu, J.; Yepes, M. The interaction between tumor necrosis factor-like weak inducer of apoptosis and its receptor fibroblast growth factor-inducible 14 promotes the recruitment of neutrophils into the ischemic brain. J. Cereb Blood Flow Metab. 2010, 30, 1147–1156. [Google Scholar] [CrossRef]
- Fagan, S.C.; Hess, D.C.; Hohnadel, E.J.; Pollock, D.M.; Ergul, A. Targets for vascular protection after acute ischemic stroke. Stroke 2004, 35, 2220–2225. [Google Scholar] [CrossRef]
- del Zoppo, G.J.; Mabuchi, T. Cerebral microvessel responses to focal ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 879–894. [Google Scholar] [CrossRef]
- Yepes, M. Tweak and FN14 in central nervous system health and disease. Front. Biosci. 2007, 12, 2772–2781. [Google Scholar] [CrossRef]
- Haile, W.B.; Echeverry, R.; Wu, F.; Guzman, J.; An, J.; Wu, J.; Yepes, M. Tumor necrosis factor-like weak inducer of apoptosis and fibroblast growth factor-inducible 14 mediate cerebral ischemia-induced poly(ADP-ribose) polymerase-1 activation and neuronal death. Neuroscience 2010, 171, 1256–1264. [Google Scholar] [CrossRef][Green Version]
- Cheng, E.; Armstrong, C.L.; Galisteo, R.; Winkles, J.A. TWEAK/Fn14 Axis-Targeted Therapeutics: Moving Basic Science Discoveries to the Clinic. Front. Immunol. 2013, 4, 473. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cellular Response | Cell Type(s) | Pathology | References |
|---|---|---|---|
| Proliferation | Human endothelial cells | In vitro | [44] |
| Human smooth muscle cells | In vitro | [44] | |
| Rat endothelial cells | Angiogenesis in rat corneas | [44] | |
| Mice endothelial cells | Arthritis | [45] | |
| Mice smooth muscle cells | Restenosis | [21] | |
| Post-natal rat cardiomyocytes | In vitro | [31] | |
| Rat cardiac fibroblasts | In vitro | [32] | |
| Migration | Human endothelial cells | In vitro | [33,46] |
| Rat aortic smooth muscle cells | In vitro | [35] | |
| Mice smooth muscle cells | In vitro | [21] | |
| Survival | Cardiomyocyte cell line H9C2 | In vitro/rat ischemia/reperfusion model | [47] |
| Differentiation | Mice Smooth muscle cells. (Contractile-Synthetic) | In vitro/In vivo in diabetes- induced atherosclerosis in ApoE-/- mice | [35] |
| Human endothelial cells (Endothelial activation/Expression of Adhesion molecules) | In vitro | [46] | |
| Mice endothelial cells (Endothelial activation/Expression of Adhesion molecules) | In vivo in diabetes induced atherosclerosis in ApoE-/- mice | [35] | |
| Bone marrow-derived macrophages. (Lipid uptake/Foam cells formation) | In vitro | [48] | |
| Human vascular smooth muscle cells (Osteogenic transition/Calcification) | In vitro | [49] | |
| Human aortic vascular smooth muscle cells (Prothrombotic phenotype/Expression of Prothrombotic factors) | In vitro | [50] | |
| Adult rat cardiomyocytes (Hypertrophy) | In vitro | [51] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Méndez-Barbero, N.; Gutiérrez-Muñoz, C.; Blázquez-Serra, R.; Martín-Ventura, J.L.; Blanco-Colio, L.M. Tumor Necrosis Factor-Like Weak Inducer of Apoptosis (TWEAK)/Fibroblast Growth Factor-Inducible 14 (Fn14) Axis in Cardiovascular Diseases: Progress and Challenges. Cells 2020, 9, 405. https://doi.org/10.3390/cells9020405
Méndez-Barbero N, Gutiérrez-Muñoz C, Blázquez-Serra R, Martín-Ventura JL, Blanco-Colio LM. Tumor Necrosis Factor-Like Weak Inducer of Apoptosis (TWEAK)/Fibroblast Growth Factor-Inducible 14 (Fn14) Axis in Cardiovascular Diseases: Progress and Challenges. Cells. 2020; 9(2):405. https://doi.org/10.3390/cells9020405
Chicago/Turabian StyleMéndez-Barbero, Nerea, Carmen Gutiérrez-Muñoz, Rafael Blázquez-Serra, Jose L. Martín-Ventura, and Luis M. Blanco-Colio. 2020. "Tumor Necrosis Factor-Like Weak Inducer of Apoptosis (TWEAK)/Fibroblast Growth Factor-Inducible 14 (Fn14) Axis in Cardiovascular Diseases: Progress and Challenges" Cells 9, no. 2: 405. https://doi.org/10.3390/cells9020405
APA StyleMéndez-Barbero, N., Gutiérrez-Muñoz, C., Blázquez-Serra, R., Martín-Ventura, J. L., & Blanco-Colio, L. M. (2020). Tumor Necrosis Factor-Like Weak Inducer of Apoptosis (TWEAK)/Fibroblast Growth Factor-Inducible 14 (Fn14) Axis in Cardiovascular Diseases: Progress and Challenges. Cells, 9(2), 405. https://doi.org/10.3390/cells9020405