FTH1 Pseudogenes in Cancer and Cell Metabolism

 ,
,  and
and
Abstract
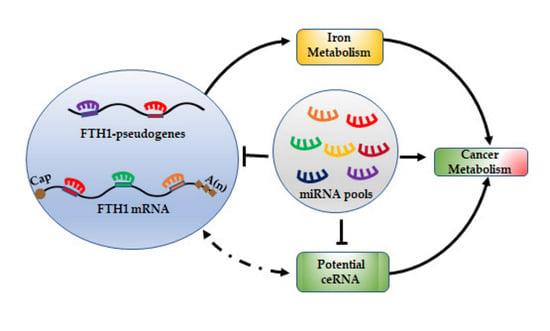
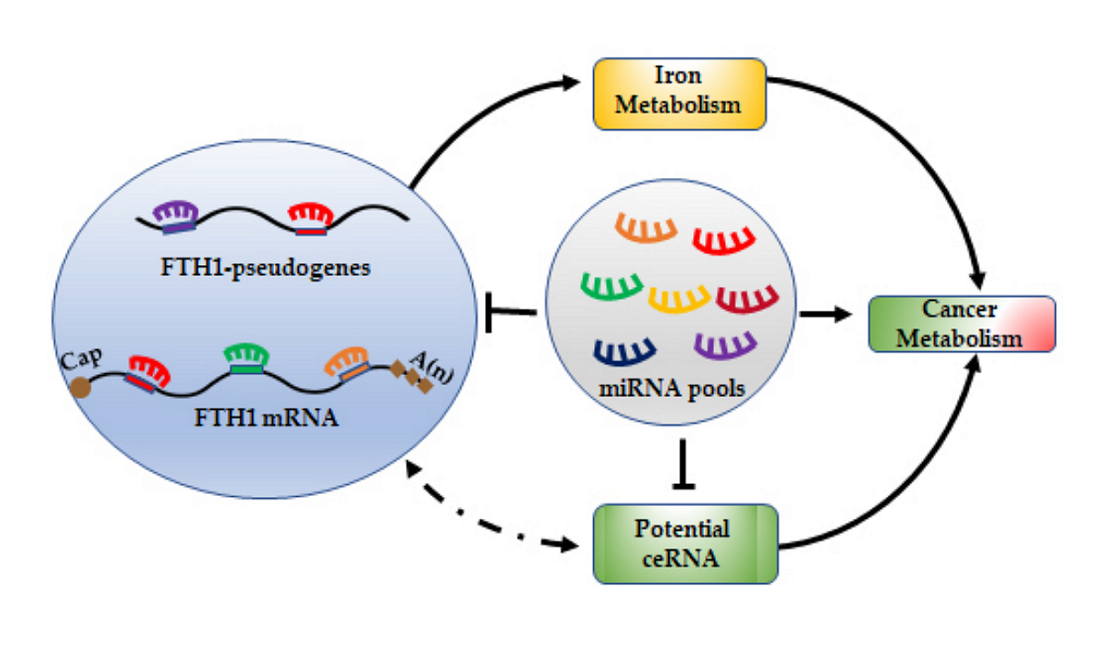
1. Introduction
2. FTH1 and FTL
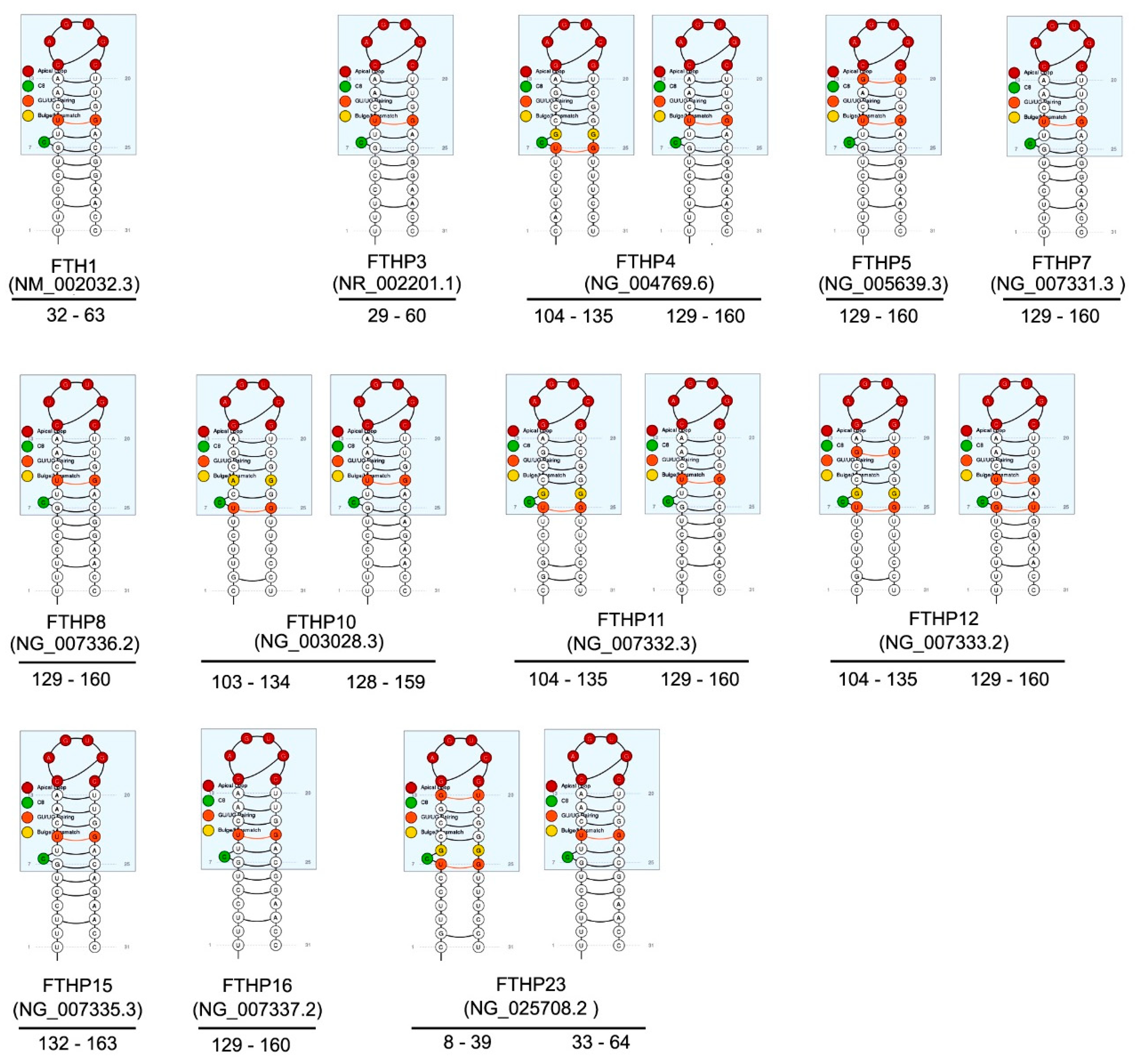
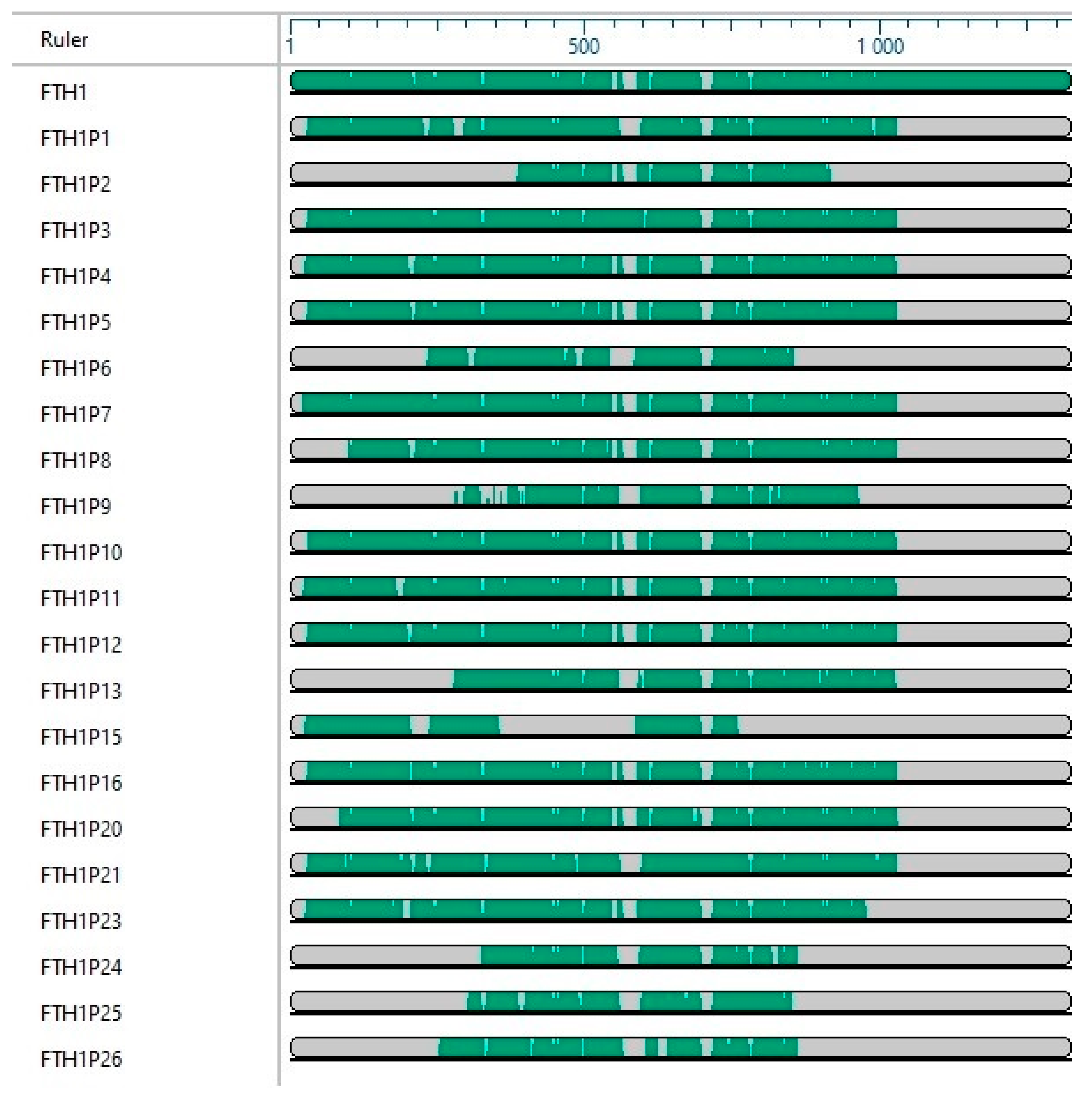
3. FTH1 Pseudogenes
4. Mitochondrial Ferritin (FTMT)
5. FTHL17
6. FTH1P3
7. miRNA/FTH1 Family Network
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheetham, S.W.; Faulkner, G.J.; Dinger, M.E. Overcoming challenges and dogmas to understand the functions of pseudogenes. Nat. Rev. Genet. 2020, 21, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, T.F.; Patrushev, L.I. Pseudogenes as functionally significant elements of the genome. Biochemistry (Moscow) 2018, 83, 1332–1349. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, I.; Darby, A.C. Taking the pseudo out of pseudogenes. Curr. Opin. Microbiol. 2015, 23, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Pink, R.C.; Wicks, K.; Caley, D.P.; Punch, E.K.; Jacobs, L.; Carter, D.R.F. Pseudogenes: Pseudo-functional or key regulators in health and diseasě. RNA 2011, 17, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Maestre, J.; Heidmann, T. Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 2000, 24, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Harrow, J. Gencode pseudogenes. Methods Mol. Biol. 2014, 1167, 129–155. [Google Scholar] [CrossRef] [PubMed]
- Pei, B.; Sisu, C.; Frankish, A.; Howald, C.; Habegger, L.; Mu, X.J.; Harte, R.; Balasubramanian, S.; Tanzer, A.; Diekhans, M.; et al. The GENCODE pseudogene resource. Genome Biol. 2012, 13. [Google Scholar] [CrossRef]
- Sisu, C.; Pei, B.; Leng, J.; Frankish, A.; Zhang, Y.; Balasubramanian, S.; Harte, R.; Wang, D.; Rutenberg-Schoenberg, M.; Clark, W.; et al. Comparative analysis of pseudogenes across three phyla. Proc. Natl. Acad. Sci. USA 2014, 111, 13361–13366. [Google Scholar] [CrossRef]
- Chen, X.; Wan, L.; Wang, W.; Xi, W.J.; Yang, A.G.; Wang, T. Re-recognition of pseudogenes: From molecular to clinical applications. Theranostics 2020, 10, 1479–1499. [Google Scholar] [CrossRef]
- Fiddes, I.T.; Lodewijk, G.A.; Mooring, M.; Bosworth, C.M.; Ewing, A.D.; Mantalas, G.L.; Novak, A.M.; van den Bout, A.; Bishara, A.; Rosenkrantz, J.L.; et al. Human-Specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell 2018, 173, 1356–1369.e22. [Google Scholar] [CrossRef]
- Suzuki, I.K.; Gacquer, D.; Van Heurck, R.; Kumar, D.; Wojno, M.; Bilheu, A.; Herpoel, A.; Lambert, N.; Cheron, J.; Polleux, F.; et al. Human-Specific NOTCH2NL genes expand cortical neurogenesis through delta/notch regulation. Cell 2018, 173, 1370–1384.e16. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.L.; Chang, J.G. Pseudogene-derived endogenous sirnas and their function. Methods Mol. Biol. 2014, 1167, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. Emerging roles for natural microRNA sponges. Curr. Biol. 2010, 20, R858. [Google Scholar] [CrossRef] [PubMed]
- Bak, R.O.; Mikkelsen, J.G. miRNA sponges: Soaking up miRNAs for regulation of gene expression. Wiley Interdiscip. Rev. RNA 2014, 5, 317–333. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef]
- Lam, H.Y.K.; Khurana, E.; Fang, G.; Cayting, P.; Carriero, N.; Cheung, K.H.; Gerstein, M.B. Pseudofam: The pseudogene families database. Nucleic Acids Res. 2009, 37, 738–743. [Google Scholar] [CrossRef]
- Mackenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A Human Mitochondrial Ferritin Encoded by an Intronless Gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef]
- Ruzzenenti, P.; Asperti, M.; Mitola, S.; Crescini, E.; Maccarinelli, F.; Gryzik, M.; Regoni, M.; Finazzi, D.; Arosio, P.; Poli, M. The Ferritin-Heavy-Polypeptide-Like-17 (FTHL17) gene encodes a ferritin with low stability and no ferroxidase activity and with a partial nuclear localization. Biochim. et Biophys. Acta Gen. Subj. 2015, 1850, 1267–1273. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Bjork, S.T.; Levi, S.; Santambrogio, P.; Parsons, P.J.; Kruger, P.C.; Yang, K.X.; Feustel, P.J.; et al. The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef]
- Rodolfo, G.; Mutskova, R.; Schwartz, C. Enrichment and characterization of ferritin. Nanotechnology 2018, 176, 1570–1573. [Google Scholar] [CrossRef]
- Bradley, J.M.; Le Brun, N.E.; Moore, G.R. Ferritins: Furnishing proteins with iron Topical Issue in Honor of R.J.P. Williams. J. Biol. Inorg. Chem. 2016, 21, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Surguladze, N.; Patton, S.; Cozzi, A.; Fried, M.G.; Connor, J.R. Characterization of nuclear ferritin and mechanism of translocation. Biochem. J. 2005, 388, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Alkhateeb, A.A.; Connor, J.R. The significance of ferritin in cancer: Anti-oxidation, inflammation and tumorigenesis. Biochim. et Biophys. Acta Rev. Cancer 2013, 1836, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, S.; Paubelle, E.; Bérard, E.; Saland, E.; Thomas, X.; Tavitian, S.; Larcher, M.V.; Vergez, F.; Delabesse, E.; Sarry, A.; et al. Ferritin heavy/light chain (FTH1/FTL) expression, serum ferritin levels, and their functional as well as prognostic roles in acute myeloid leukemia. Eur. J. Haematol. 2019, 102, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Levi, S. Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 2002, 33, 457–463. [Google Scholar] [CrossRef]
- Theil, E.C. Metal-binding proteins and trace element metabolism ferritin: At the crossroads of iron and oxygen metabolism. J. Nutr. 2003, 133, 1549–1553. [Google Scholar] [CrossRef]
- Ferreira, C.; Santambrogio, P.; Martin, M.E.; Andrieu, V.; Feldmann, G.; Hénin, D.; Beaumont, C. H ferritin knockout mice: A model of hyperferritinemia in the absence of iron overload. Blood 2001, 98, 525–532. [Google Scholar] [CrossRef]
- Ferreira, C.; Bucchini, D.; Martin, M.E.; Levi, S.; Arosio, P.; Grandchamp, B.; Beaumont, C. Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem. 2000, 275, 3021–3024. [Google Scholar] [CrossRef]
- Thompson, K.J.; Fried, M.G.; Ye, Z.; Boyer, P.; Connor, J.R. Thompson 2002 Regulation, mechanisms and proposed function of ferritin translocation to cell nuclei. J. Cell Sci. 2002, 115, 1265–1277. [Google Scholar]
- Hintze, K.J.; Theil, E.C. DNA and mRNA elements with complementary responses to hemin, antioxidant inducers, and iron control ferritin-L expression. Proc. Natl. Acad. Sci. USA 2005, 102, 15048–15052. [Google Scholar] [CrossRef] [PubMed]
- Faniello, M.C.; Chirico, G.; Quaresima, B.; Cuda, G.; Allevato, G.; Bevilacqua, M.A.; Baudi, F.; Colantuoni, V.; Cimino, F.; Venuta, S.; et al. An alternative model of H ferritin promoter transactivation by c-Jun. Biochem. J. 2002, 363, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, M.A.; Faniello, M.C.; Quaresima, B.; Tiano, M.T.; Giuliano, P.; Feliciello, A.; Avvedimento, V.E.; Cimino, F.; Costanzo, F. A common mechanism underlying the E1A repression and the cAMP stimulation of the H ferritin transcription. J. Biol. Chem. 1997, 272, 20736–20741. [Google Scholar] [CrossRef]
- Faniello, M.C.; Di Sanzo, M.; Quaresima, B.; Baudi, F.; Di Caro, V.; Cuda, G.; Morrone, G.; Del Sal, G.; Spinelli, G.; Venuta, S.; et al. p53-Mediated downregulation of H ferritin promoter transcriptional efficiency via NF-Y. Int. J. Biochem. Cell Biol. 2008, 40, 2110–2119. [Google Scholar] [CrossRef]
- Faniello, M.C.; Di Sanzo, M.; Quaresima, B.; Nisticò, A.; Fregola, A.; Grosso, M.; Cuda, G.; Costanzo, F. Bilateral cataract in a subject carrying a C to A transition in the L ferritin promoter region. Clin. Biochem. 2009, 42, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Klausner, R.D.; Rouault, T.A.; Harford, J.B. Regulating the fate of mRNA: The control of cellular iron metabolism. Cell 1993, 72, 19–28. [Google Scholar] [CrossRef]
- Sammarco, M.C.; Ditch, S.; Banerjee, A.; Grabczyk, E. Ferritin L and H subunits are differentially regulated on a post-transcriptional level. J. Biol. Chem. 2008, 283, 4578–4587. [Google Scholar] [CrossRef]
- MacKenzie, E.L.; Tsuji, Y. Elevated intracellular calcium increases ferritin H expression through an NFAT-independent post-transcriptional mechanism involving mRNA stabilization. Biochem. J. 2008, 411, 107–113. [Google Scholar] [CrossRef][Green Version]
- Reinheckel, T.; Sitte, N.; Ullrich, O.; Kuckelkorn, U.; Davies, K.J.A.; Grune, T. Comparative resistance of the 20 S and 26 S proteasome to oxidative stress. Biochem. J. 1998, 335, 637–642. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D. The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef]
- Gryzik, M.; Srivastava, A.; Longhi, G.; Bertuzzi, M.; Gianoncelli, A.; Carmona, F.; Poli, M.; Arosio, P. Expression and characterization of the ferritin binding domain of Nuclear Receptor Coactivator-4 (NCOA4). Biochim. et Biophys. Acta Gen. Subj. 2017, 1861, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Coffman, L.G.; Brown, J.C.; Johnson, D.A.; Parthasarathy, N.; D’Agostino, R.B.; Lively, M.O.; Hua, X.; Tilley, S.L.; Muller-Esterl, W.; Willingham, M.C.; et al. Cleavage of high-molecular-weight kininogen by elastase and tryptase is inhibited by ferritin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, L.; Huhn, A.J.; Hatcher, H.; Torti, F.M.; Torti, S.V. Ferritin blocks inhibitory effects of two-chain high molecular weight kininogen (HKa) on adhesion and survival signaling in endothelial cells. PLoS ONE 2012, 7, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Storr, H.L.; Kind, B.; Parfitt, D.A.; Chapple, J.P.; Lorenz, M.; Koehler, K.; Huebner, A.; Clark, A.J.L. Deficiency of ferritin heavy-chain nuclear import in triple A syndrome implies nuclear oxidative damage as the primary disease mechanism. Mol. Endocrinol. 2009, 23, 2086–2094. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Luo, C.; Mines, M.; Zhang, J.; Fan, G.H. Chemokine CXCL12 induces binding of ferritin heavy chain to the chemokine receptor CXCR4, alters CXCR4 signaling, and induces phosphorylation and nuclear translocation of ferritin heavy chain. J. Biol. Chem. 2006, 281, 37616–37627. [Google Scholar] [CrossRef]
- Lee, J.H.; Jang, H.; Cho, E.J.; Youn, H.D. Ferritin binds and activates p53 under oxidative stress. Biochem. Biophys. Res. Commun. 2009, 389, 399–404. [Google Scholar] [CrossRef]
- Biamonte, F.; Battaglia, A.M.; Zolea, F.; Oliveira, D.M.; Aversa, I.; Santamaria, G.; Giovannone, E.D.; Rocco, G.; Viglietto, G.; Costanzo, F. Ferritin heavy subunit enhances apoptosis of non-small cell lung cancer cells through modulation of miR-125b/p53 axis. Cell Death Dis. 2018, 9, 1174. [Google Scholar] [CrossRef]
- Aversa, I.; Zolea, F.; Ieranò, C.; Bulotta, S.; Trotta, A.M.; Faniello, M.C.; De Marco, C.; Malanga, D.; Biamonte, F.; Viglietto, G.; et al. Epithelial-to-mesenchymal transition in FHC-silenced cells: The role of CXCR4/CXCL12 axis. J. Exp. Clin. Cancer Res. 2017, 36, 1–15. [Google Scholar] [CrossRef]
- Lobello, N.; Biamonte, F.; Pisanu, M.E.; Faniello, M.C.; Jakopin, Ž.; Chiarella, E.; Giovannone, E.D.; Mancini, R.; Ciliberto, G.; Cuda, G.; et al. Ferritin heavy chain is a negative regulator of ovarian cancer stem cell expansion and epithelial to mesenchymal transition. Oncotarget 2016, 7, 62019–62033. [Google Scholar] [CrossRef]
- Di Sanzo, M.; Gaspari, M.; Misaggi, R.; Romeo, F.; Falbo, L.; De Marco, C.; Agosti, V.; Quaresima, B.; Barni, T.; Viglietto, G.; et al. H ferritin gene silencing in a human metastatic melanoma cell line: A proteomic analysis. J. Proteome Res. 2011, 10, 5444–5453. [Google Scholar] [CrossRef]
- Biamonte, F.; Zolea, F.; Bisognin, A.; Di Sanzo, M.; Saccoman, C.; Scumaci, D.; Aversa, I.; Panebianco, M.; Faniello, M.C.; Bortoluzzi, S.; et al. H-ferritin-regulated microRNAs modulate gene expression in K562 cells. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Di Sanzo, M.; Chirillo, R.; Aversa, I.; Biamonte, F.; Santamaria, G.; Giovannone, E.D.; Faniello, M.C.; Cuda, G.; Costanzo, F. shRNA targeting of ferritin heavy chain activates H19/miR-675 axis in K562 cells. Gene 2018, 657, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Zolea, F.; Biamonte, F.; Candeloro, P.; Di Sanzo, M.; Cozzi, A.; Di Vito, A.; Quaresima, B.; Lobello, N.; Trecroci, F.; Di Fabrizio, E.; et al. H ferritin silencing induces protein misfolding in K562 cells: A Raman analysis. Free Radic. Biol. Med. 2015, 89, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Aversa, I.; Chirillo, R.; Chiarella, E.; Zolea, F.; Di Sanzo, M.; Biamonte, F.; Palmieri, C.; Costanzo, F. Chemoresistance in H-ferritin silenced cells: The role of NF-κB. Int. J. Mol. Sci. 2018, 19, 2969. [Google Scholar] [CrossRef]
- Shpyleva, S.I.; Tryndyak, V.P.; Kovalchuk, O.; Starlard-Davenport, A.; Chekhun, V.F.; Beland, F.A.; Pogribny, I.P. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res. Treat. 2011, 126, 63–71. [Google Scholar] [CrossRef]
- Chan, J.J.; Kwok, Z.H.; Chew, X.H.; Zhang, B.; Liu, C.; Soong, T.W.; Yang, H.; Tay, Y. A FTH1 gene:pseudogene:microRNA network regulates tumorigenesis in prostate cancer. Nucleic Acids Res. 2018, 46, 1998–2011. [Google Scholar] [CrossRef]
- Cragg, S.J.; Drysdale, J.; Worwood, M. Genes for the “H” subunit of human ferritin are present on a number of human chromosomes. Hum. Genet. 1985, 71, 108–112. [Google Scholar] [CrossRef]
- Dugast, I.J.; Papadopoulos, P.; Zappone, E.; Jones, C.; Theriault, K.; Handelman, G.J.; Benarous, R.; Drysdale, J.W. Identification of two human ferritin H genes on the short arm of chromosome 6. Genomics 1990, 6, 204–211. [Google Scholar] [CrossRef]
- Quaresima, B.; Tiano, M.T.; Porcellini, A.; D’Agostino, P.; Faniello, M.C.; Bevilacqua, M.A.; Cimino, F.; Costanzo, F. PCR analysis of the H ferritin multigene family reveals the existence of two classes of processed pseudogenes. Pcr Methods Appl. 1994, 4, 85–88. [Google Scholar] [CrossRef]
- Liu, X.Q.; Tufman, A.; Kiefl, R.; Li, G.F.; Ma, Q.L.; Huber, R.M. Identification of lung adenocarcinoma-specific exosome RNAs in peripheral blood by RNA-Seq analysis. Eur. Rev. Med Pharmacol. Sci. 2020, 24, 1877–1886. [Google Scholar] [CrossRef]
- Lv, P.; Yang, S.; Liu, W.; Qin, H.; Tang, X.; Wu, F.; Liu, Z.; Gao, H.; Liu, X. Circulating plasma lncRNAs as novel markers of EGFR mutation status and monitors of epidermal growth factor receptor-tyrosine kinase inhibitor therapy. Thorac. Cancer 2020, 11, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Bhavsar, D.; Dugast, I.; Zappone, E.; Drysdale, J. Conserved mutations in human ferritin H pseudogenes: A second functional sequence or an evolutionary quirk? Biochim. et Biophys. Acta Gene Struct. Expr. 1997, 1351, 150–156. [Google Scholar] [CrossRef]
- Costanzo, F.; Colombo, M.; Staempfli, S.; Santoro, C.; Marone, M.; Frank, R.; Delius, H.; Cortese, R. Structure of gene and pseudogenes of human apoferritin H. Nucleic Acids Res. 1986, 14, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, P.; Bhavsar, D.; Zappone, E.; David, V.; Jones, C.; Worwood, M.; Drysdale, J. A second human ferritin H locus on chromosome 11. Cytogenet. Cell Genet. 1992, 61, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Bhavsar, D.; Volz, A.; Ziegler, A.; Drysdale, J. Exclusion of ferritins and iron-responsive element (IRE)-binding proteins as candidates for the hemochromatosis gene. Hum. Genet. 1994, 94, 159–164. [Google Scholar] [CrossRef]
- Muzny, D.M.; Scherer, S.E.; Kaul, R.; Wang, J.; Yu, J.; Sudbrak, R.; Buhay, C.J.; Chen, R.; Cree, A.; Ding, Y.; et al. The DNA sequence, annotation and analysis of human chromosome 3. Nature 2006, 440, 1194–1198. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; Fitzhugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Campillos, M.; Cases, I.; Hentze, M.W.; Sanchez, M. SIREs: Searching for iron-responsive elements. Nucleic Acids Res. 2010, 38, W360. [Google Scholar] [CrossRef]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. Methods Mol. Biol. 2000, 132, 71–91. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Corsi, B.; Cozzi, A.; Arosio, P.; Drysdale, J.; Santambrogio, P.; Campanella, A.; Biasiotto, G.; Albertini, A.; Levi, S. Human mitochondrial ferritin expressed in HeLa cells incorporates iron and affects cellular iron metabolism. J. Biol. Chem. 2002, 277, 22430–22437. [Google Scholar] [CrossRef] [PubMed]
- Guaraldo, M.; Santambrogio, P.; Rovelli, E.; Di Savino, A.; Saglio, G.; Cittaro, D.; Roetto, A.; Levi, S. Characterization of human mitochondrial ferritin promoter: Identification of transcription factors and evidences of epigenetic control. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, F.; Hentze, M.W. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 2004, 5, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Invernizzi, R.; Bergamaschi, G.; Levi, S.; Corsi, B.; Travaglino, E.; Rolandi, V.; Biasiotto, G.; Drysdale, J.; Arosio, P. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood 2003, 101, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Guan, H.; Yang, M.; Liu, Z.; Takeuchi, S.; Yanagisawa, D.; Vincent, S.R.; Zhao, S.; Tooyama, I. Upregulation of Mitochondrial Ferritin by Proinflammatory Cytokines: Implications for a Role in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.M.; Wang, X.; Patton, S.M.; Arosio, P.; Levi, S.; Earley, C.J.; Allen, R.P.; Connor, J.R. Mitochondrial ferritin in the substantia nigra in restless legs syndrome. J. Neuropathol. Exp. Neurol. 2009, 68, 1193–1199. [Google Scholar] [CrossRef]
- Shi, Z.H.; Shi, F.F.; Wang, Y.Q.; Sheftel, A.D.; Nie, G.; Zhao, Y.S.; You, L.H.; Gou, Y.J.; Duan, X.L.; Zhao, B.L.; et al. Mitochondrial ferritin, a new target for inhibiting neuronal tumor cell proliferation. Cell. Mol. Life Sci. 2015, 72, 983–997. [Google Scholar] [CrossRef]
- Wang, P.J.; McCarrey, J.R.; Yang, F.; Page, D.C. An abundance of X-linked genes expressed in spermatogonia. Nat. Genet. 2001, 27, 422–426. [Google Scholar] [CrossRef]
- Loriot, A.; Boon, T.; De Smet, C. Five new human cancer-germline genes identified among 12 genes expressed in spermatogonia. Int. J. Cancer 2003, 105, 371–376. [Google Scholar] [CrossRef]
- Aoki, N.; Mochizuki, K.; Matsui, Y. DNA methylation of the Fthl17 5’-upstream region regulates differential Fthl17 expression in lung cancer cells and germline stem cells. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Di Sanzo, M.; Aversa, I.; Santamaria, G.; Gagliardi, M.; Panebianco, M.; Biamonte, F.; Zolea, F.; Faniello, M.C.; Cuda, G.; Costanzo, F. FTH1P3, a novel H-ferritin pseudogene transcriptionally active, is ubiquitously expressed and regulated during cell differentiation. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Chen, W.; Li, W.; Ding, Y.; Tu, P. E2F1-induced ferritin heavy chain 1 pseudogene 3 (FTH1P3) accelerates non-small cell lung cancer gefitinib resistance. Biochem. Biophys. Res. Commun. 2020, 530, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Tang, H.; Zhao, X.; Sun, Y.; Jiang, Y.; Liu, Y. Long non-coding RNA FTH1P3 facilitates uveal melanoma cell growth and invasion through miR-224-5p. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Zhang, C.Z. Long non-coding RNA FTH1P3 facilitates oral squamous cell carcinoma progression by acting as a molecular sponge of miR-224-5p to modulate fizzled 5 expression. Gene 2017, 607, 47–55. [Google Scholar] [CrossRef]
- Yang, L.; Sun, K.; Chu, J.; Qu, Y.; Zhao, X.; Yin, H.; Ming, L.; Wan, J.; He, F. Long non-coding RNA FTH1P3 regulated metastasis and invasion of esophageal squamous cell carcinoma through SP1/NF-kB pathway. Biomed. Pharmacother. 2018, 106, 1570–1577. [Google Scholar] [CrossRef]
- Zhang, Y.; Ying, L.I.; Wang, J.; Lei, P. Long non-coding RNA ferritin heavy polypeptide 1 pseudogene 3 controls glioma cell proliferation and apoptosis via regulation of the microRNA-224-5p/tumor protein D52 axis. Mol. Med. Rep. 2018, 18, 4239–4246. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, T.; Yang, Z.; Jiang, C.; Seng, J. Long non-coding RNA FTH1P3 activates paclitaxel resistance in breast cancer through miR-206/ABCB1. J. Cell. Mol. Med. 2018, 22, 4068–4075. [Google Scholar] [CrossRef]
- Lv, R.; Zhang, Q.W. The long noncoding RNA FTH1P3 promotes the proliferation and metastasis of cervical cancer through microRNA-145. Oncol. Rep. 2020, 43, 31–40. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y. Long non-coding RNA FTH1P3 promotes the metastasis and aggressiveness of non-small cell lung carcinoma by inducing epithelial-mesenchymal transition. Int. J. Clin. Exp. Pathol. 2019, 12, 3782–3790. [Google Scholar]
- Zhang, S.; Tian, L.; Ma, P.; Sun, Q.; Zhang, K.; Wang, G.; Liu, H.; Xu, B. Potential role of differentially expressed lncRNAs in the pathogenesis of oral squamous cell carcinoma. Arch. Oral Biol. 2015, 60, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gao, X.; Liu, C.L. Increased expression of lncRNA FTH1P3 promotes oral squamous cell carcinoma cells migration and invasion by enhancing PI3K/Akt/GSK3B/Wnt/β-catenin signaling. Eur. Rev. Med Pharmacol. Sci. 2018, 22, 8306–8314. [Google Scholar] [CrossRef] [PubMed]
- Groen, J.N.; Capraro, D.; Morris, K.V. The emerging role of pseudogene expressed non-coding RNAs in cellular functions. Int. J. Biochem. Cell Biol. 2014, 54, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Lou, W.; Ding, B.; Fu, P. Pseudogene-Derived lncRNAs and their miRNA sponging mechanism in human cancer. Front. Cell Dev. Biol. 2020, 8, 85. [Google Scholar] [CrossRef]
- Sanchez-Mejias, A.; Tay, Y. Competing endogenous RNA networks: Tying the essential knots for cancer biology and therapeutics. J. Hematol. Oncol. 2012. [Google Scholar] [CrossRef]
- Ala, U.; Karreth, F.A.; Bosia, C.; Pagnani, A.; Taulli, R.; Léopold, V.; Tay, Y.; Provero, P.; Zecchina, R.; Pandolfi, P.P. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proc. Natl. Acad. Sci. USA 2013, 110, 7154–7159. [Google Scholar] [CrossRef]
- Sen, R.; Ghosal, S.; Das, S.; Balti, S.; Chakrabarti, J. Competing endogenous RNA: The key to posttranscriptional regulation. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef]
- Ke, Z.; Qiu, Z.; Xiao, T.; Zeng, J.; Zou, L.; Lin, X.; Hu, X.; Lin, S.; Lv, H. Downregulation of miR-224-5p promotes migration and proliferation in human dental pulp stem cells. Biomed Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Ke, T.W.; Hsu, H.L.; Wu, Y.H.; Chen, W.T.L.; Cheng, Y.W.; Cheng, C.W. MicroRNA-224 suppresses colorectal cancer cell migration by targeting Cdc42. Dis. Markers 2014, 2014. [Google Scholar] [CrossRef]
- Li, Q.; Ding, C.; Chen, C.; Zhang, Z.; Xiao, H.; Xie, F.; Lei, L.; Chen, Y.; Mao, B.; Jiang, M.; et al. miR-224 promotion of cell migration and invasion by targeting Homeobox D 10 gene in human hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2014, 29, 835–842. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.; Zhao, Y.; Wang, F.; Chen, Y.; Fei, X. mir-224 enhances invasion and metastasis by targeting HOXD10 in non-small cell lung cancer cells. Oncol. Lett. 2018, 15, 7069–7075. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zhou, L.; Liu, H.; Shan, Y.; Zhang, X. MicroRNA-224 Promotes Pancreatic Cancer Cell Proliferation and Migration by Targeting the TXNIP-Mediated HIF1α Pathway. Cell. Physiol. Biochem. 2018, 48, 1735–1746. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
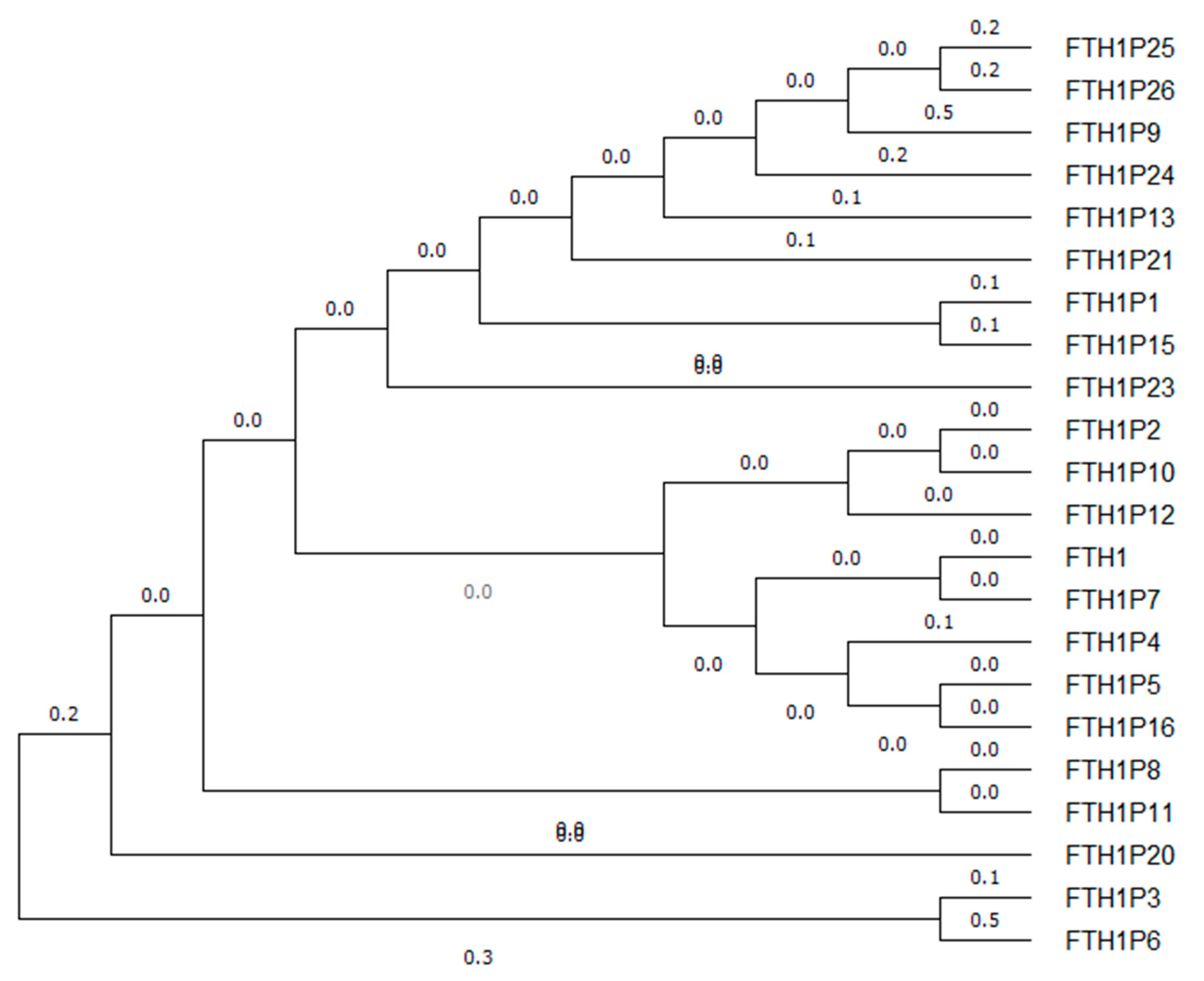{kind=link}
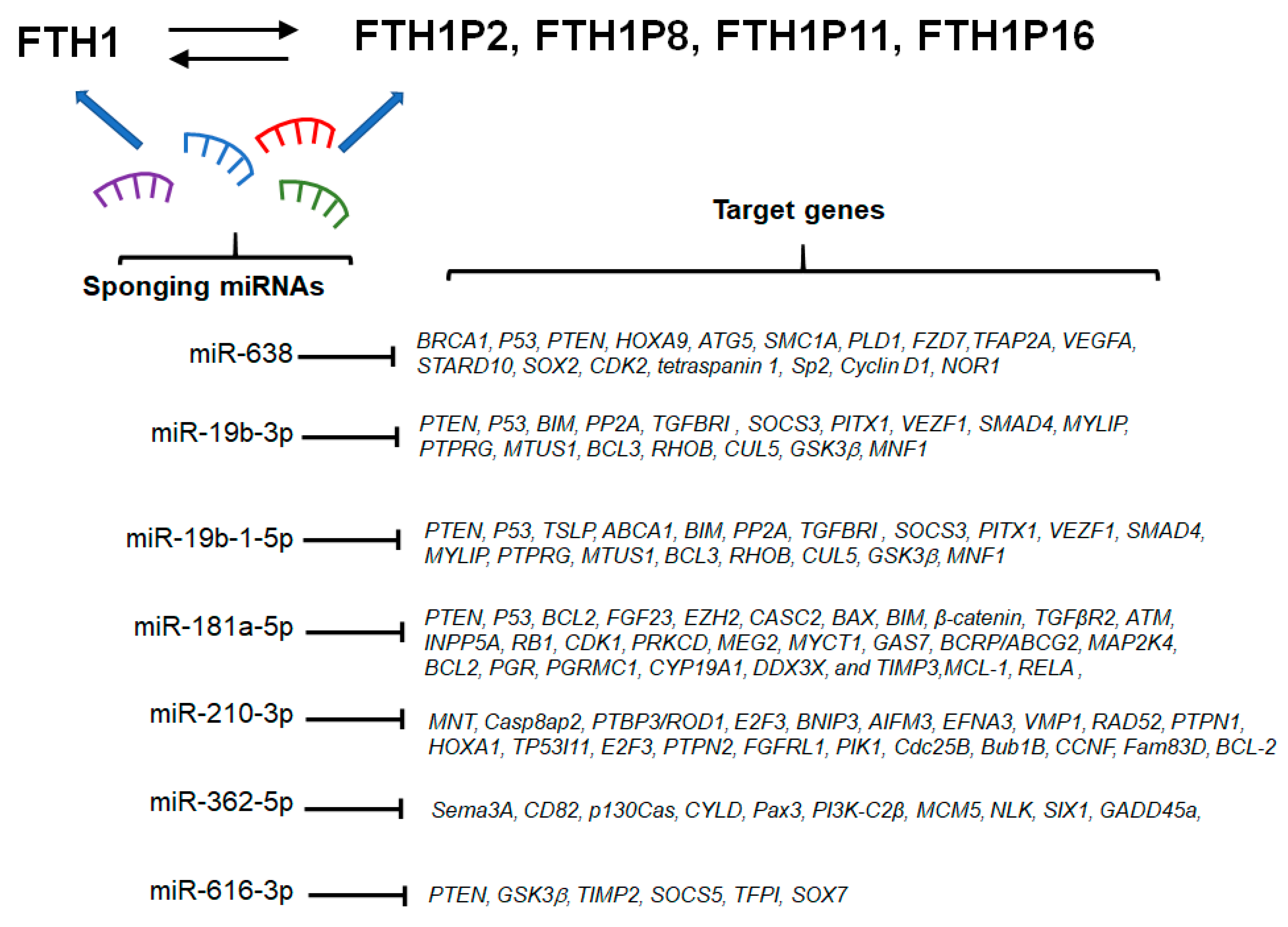{kind=link}
| Name | NCBI Reference | Location | Length (bp) | Evidence | Type | Putative Protein% Identity FTH1 | Reference |
|---|---|---|---|---|---|---|---|
| FTH1P1 | NG_004768.3 | 1p34.3 | 1096 | INFERRED | PROCESSED | 71aa (91.23%) | [57,58] |
| FTH1P2 | NG_007330.2 | 1q42.13 | 663 | VALIDATED | PROCESSED | 113aa (94.69%) | [56,57,59,60] |
| FTH1P3 | NR_002201.1 | 2p23.3 | 954 | VALIDATED | PROCESSED | 103aa (91.78%) | [57] |
| FTH1P4 | NG_004769.6 | 3q21.3 | 1112 | VALIDATED | PROCESSED | 183aa (91.8%) | [57,61] |
| FTH1P5 | NG_005639.3 | 6p12.3 | 1113 | INFERRED | PROCESSED | 79aa (92.41%) | [57] |
| FTH1P6 | NG_043893.1 | 2p16.2 | 336 | INFERRED | PROCESSED | 50aa (0%) | Direct submission |
| FTH1P7 | NG_007331.3 | 13q12.12 | 1122 | INFERRED | PROCESSED | 79aa (94.94%) | [57] |
| FTH1P8 | NG_007336.2 | Xq28 | 1112 | VALIDATED | PROCESSED | 87aa (95.12%) | [56,57,59] |
| FTH1P9 | NG_043625.1 | 5q14.2 | 555 | INFERRED | PROCESSED | 45aa (0%) | Direct submission |
| FTH1P10 | NG_003028.3 | 5p15.1 | 1119 | VALIDATED | PROCESSED | 253aa (95.78%) | [57,61] |
| FTH1P11 | NG_007332.3 | 8q21.13 | 1108 | VALIDATED | PROCESSED | 113aa (96.46%) | [56,57,59,60,61] |
| FTH1P12 | NG_007333.2 | 9p22.3 | 1115 | VALIDATED | PROCESSED | 152aa (91.16%) | [57] |
| FTH1P13 | NG_007334.1 | 14q23.3 | 877 | VALIDATED | PROCESSED | 83aa (79.52%) | [61,62] |
| FTH1P15 | NG_007335.3 | 6p12.1 | 655 | INFERRED | PROCESSED | 95aa (69.14%) | [58] |
| FTH1P16 | NG_007337.2 | 11q14.1 | 1118 | VALIDATED | PROCESSED | 183aa (95.63%) | [56,57,59,63,64] |
| FTH1P20 | NG_023542.2 | 2q31.3 | 1061 | VALIDATED | PROCESSED | 49aa (0%) | [61] |
| FTH1P21 | NG_001121.4 | 4q32.1 | 1115 | INFERRED | PROCESSED | 108aa (0%) | [65,66,67] |
| FTH1P23 | NG_025708.2 | 3p13 | 952 | VALIDATED | PROCESSED | 87aa (91.03%) | [61,65,66,67] |
| FTH1P24 | NG_022082.2 | 4q31.1 | 747 | INFERRED | PROCESSED | 47aa (79.31%) | Direct submission |
| FTH1P25 | NG_022745.1 | 1q25.3 | 671 | INFERRED | PROCESSED | 77aa (57.75%) | Direct submission |
| FTH1P26 | NG_028813.1 | 6q23.2 | 714 | INFERRED | PROCESSED | 61aa (53.57%) | Direct submission |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Sanzo, M.; Quaresima, B.; Biamonte, F.; Palmieri, C.; Faniello, M.C. FTH1 Pseudogenes in Cancer and Cell Metabolism. Cells 2020, 9, 2554. https://doi.org/10.3390/cells9122554
Di Sanzo M, Quaresima B, Biamonte F, Palmieri C, Faniello MC. FTH1 Pseudogenes in Cancer and Cell Metabolism. Cells. 2020; 9(12):2554. https://doi.org/10.3390/cells9122554
Chicago/Turabian StyleDi Sanzo, Maddalena, Barbara Quaresima, Flavia Biamonte, Camillo Palmieri, and Maria Concetta Faniello. 2020. "FTH1 Pseudogenes in Cancer and Cell Metabolism" Cells 9, no. 12: 2554. https://doi.org/10.3390/cells9122554
APA StyleDi Sanzo, M., Quaresima, B., Biamonte, F., Palmieri, C., & Faniello, M. C. (2020). FTH1 Pseudogenes in Cancer and Cell Metabolism. Cells, 9(12), 2554. https://doi.org/10.3390/cells9122554