Hyperthrombotic Milieu in COVID-19 Patients

{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Overview
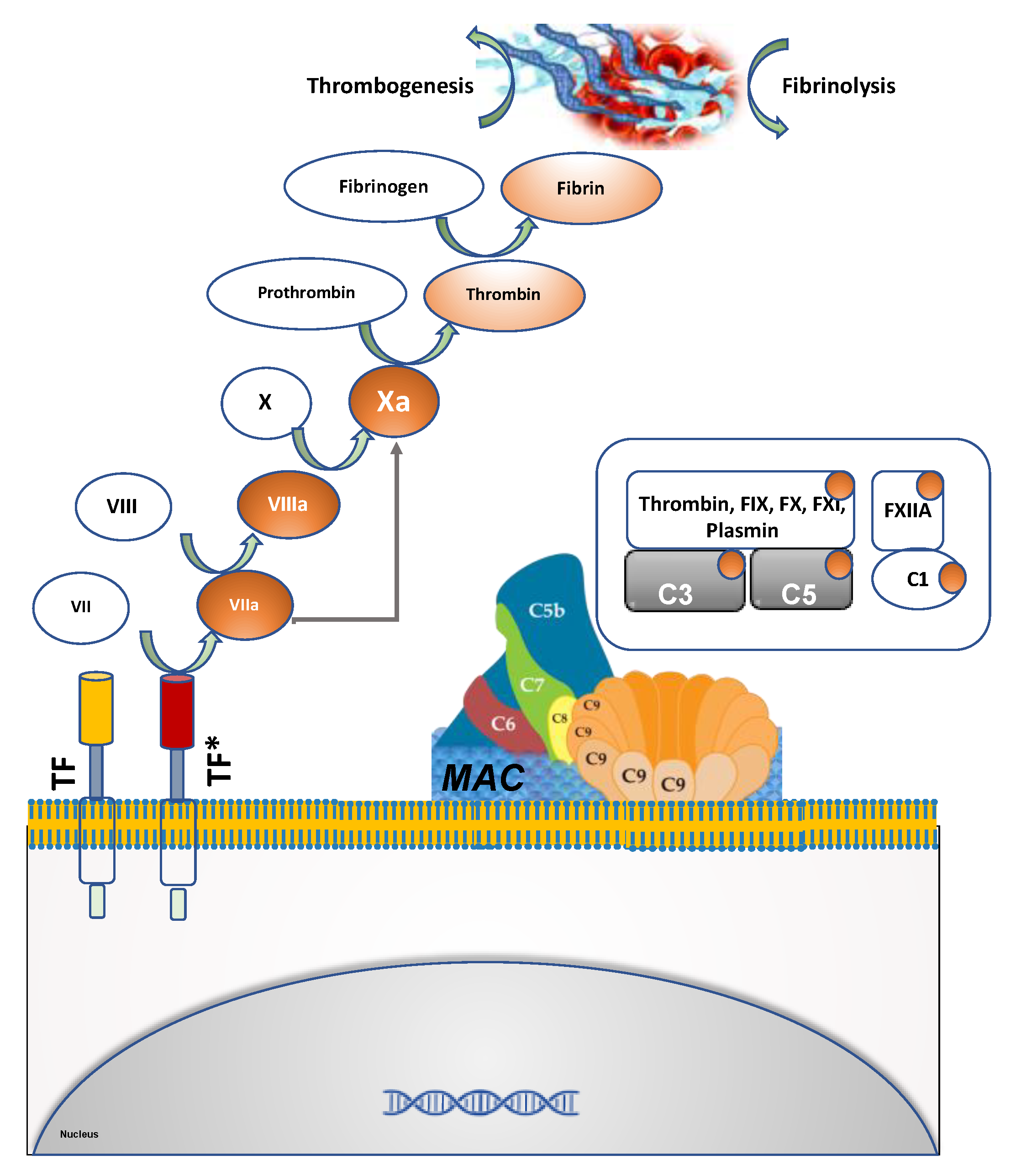
1.2. Components of Hemostasis and Thrombosis
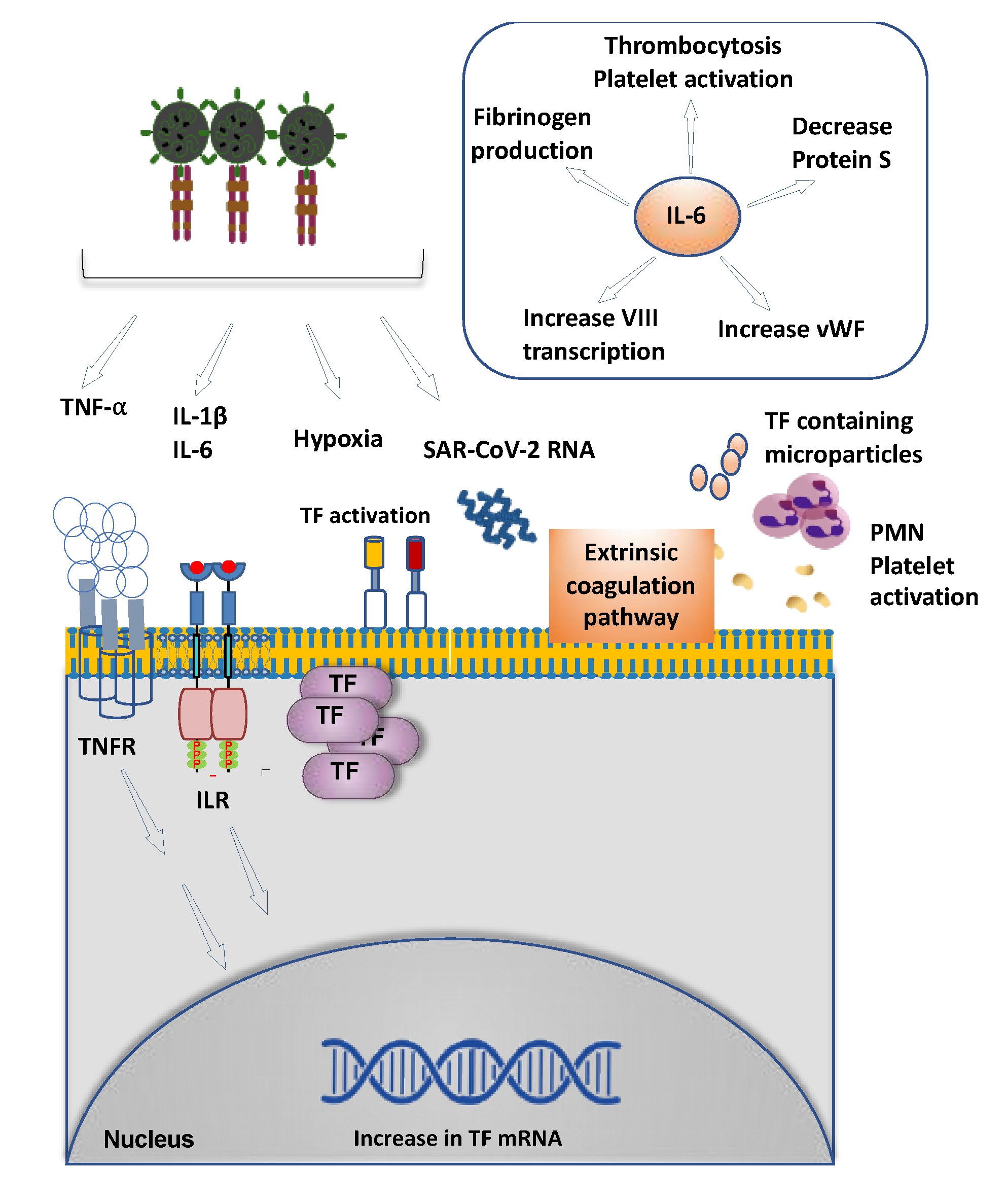
1.3. Viral Infection, Inflammation and Thrombosis
2. Arterial Thrombosis
3. Venous Thrombosis and Pulmonary Embolus
4. Microthrombi in Vascular Beds
5. COVID-19-Induced Thrombosis
5.1. Characteristics of Coagulopathy in COVID-19 Patients
5.2. SARS-CoV-2 and Coagulopathy
5.3. Complement Cascade and Thrombosis
5.4. Antiphospholipid Antibodies
5.5. Platelets and WBCs
6. Therapeutic Considerations
6.1. VTE Prophylaxis
6.2. Prophylactic Anticoagulation in COVID-19 Patients
6.3. Choice of Anticoagulant Agents
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mahase, E. China coronavirus: WHO declares international emergency as death toll exceeds 200. BMJ 2020, 368, m408. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.C.; Zhang, J.X.; Zhang, S.Y.; Wang, P.; Fan, X.H.; Li, L.F.; Li, G.; Dong, B.Q.; Liu, W.; Cheung, C.L.; et al. Prevalence and genetic diversity of coronaviruses in bats from China. J. Virol. 2006, 80, 7481–7490. [Google Scholar] [CrossRef]
- Al-Omari, A.; Rabaan, A.A.; Salih, S.; Al-Tawfiq, J.A.; Memish, Z.A. MERS coronavirus outbreak: Implications for emerging viral infections. Diagn. Microbiol. Infect. Dis. 2019, 93, 265–285. [Google Scholar] [CrossRef]
- Cherry, J.D.; Krogstad, P. SARS: The first pandemic of the 21st century. Pediatr. Res. 2004, 56, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Vijaykrishna, D.; Smith, G.J.; Zhang, J.X.; Peiris, J.S.; Chen, H.; Guan, Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007, 81, 4012–4020. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Simonetti, B.; Antón-Plágaro, C.; Williamson, M.K.; Shoemark, D.K.; Simón-Gracia, L.; Klein, K.; Bauer, M.; Hollandi, R.; Greber, U.F.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Amraie, R.; Napoleon, M.A.; Yin, W.; Berrigan, J.; Suder, E.; Zhao, G.; Olejnik, J.; Gummuluru, S.; Muhlberger, E.; Chitalia, V.; et al. Vipul Chitalia CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2 and are differentially expressed in lung and kidney epithelial and endothelial cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wu, J.T.; Leung, K.; Bushman, M.; Kishore, N.; Niehus, R.; de Salazar, P.M.; Cowling, B.J.; Lipsitch, M.; Leung, G.M. Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China. Nat. Med. 2020, 26, 506–510. [Google Scholar] [CrossRef]
- Zaim, S.; Chong, J.H.; Sankaranarayanan, V.; Harky, A. COVID-19 and Multiorgan Response. Curr. Probl. Cardiol. 2020, 45, 100618. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Davie, E.W.; Ratnoff, O.D. Waterfall Sequence for Intrinsic Blood Clotting. Science 1964, 145, 1310–1312. [Google Scholar] [CrossRef]
- Macfarlane, R.G. An Enzyme Cascade in the Blood Clotting Mechanism, and Its Function as a Biochemical Amplifier. Nature 1964, 202, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arter. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Steffel, J.; Luscher, T.F.; Tanner, F.C. Tissue factor in cardiovascular diseases: Molecular mechanisms and clinical implications. Circulation 2006, 113, 722–731. [Google Scholar] [CrossRef]
- Butenas, S.; Orfeo, T.; Mann, K.G. Tissue factor activity and function in blood coagulation. Thromb. Res. 2008, 122, S42–S46. [Google Scholar] [CrossRef]
- Kamal, A.H.; Tefferi, A.; Pruthi, R.K. How to interpret and pursue an abnormal prothrombin time, activated partial thromboplastin time, and bleeding time in adults. Mayo. Clin. Proc. 2007, 82, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Bruckner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Nagasawa, K.; Horiuchi, T.; Tsuru, T.; Nishizaka, H.; Niho, Y. C5a induces tissue factor activity on endothelial cells. Thromb. Haemost. 1997, 77, 394–398. [Google Scholar] [CrossRef]
- Ritis, K.; Doumas, M.; Mastellos, D.; Micheli, A.; Giaglis, S.; Magotti, P.; Rafail, S.; Kartalis, G.; Sideras, P.; Lambris, J.D. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J. Immunol. 2006, 177, 4794–4802. [Google Scholar] [CrossRef] [PubMed]
- Wojta, J.; Kaun, C.; Zorn, G.; Ghannadan, M.; Hauswirth, A.W.; Sperr, W.R.; Fritsch, G.; Printz, D.; Binder, B.R.; Schatzl, G.; et al. C5a stimulates production of plasminogen activator inhibitor-1 in human mast cells and basophils. Blood 2002, 100, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Wojta, J.; Huber, K.; Valent, P. New aspects in thrombotic research: Complement induced switch in mast cells from a profibrinolytic to a prothrombotic phenotype. Pathophysiol. Haemost. Thromb. 2003, 33, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.K.; Paredes, N. The coagulation system in humans. Methods Mol. Biol. 2013, 992, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M. Interactions between coagulation and inflammation. Scand. J. Infect. Dis. 2003, 35, 545–554. [Google Scholar] [CrossRef] [PubMed]
- van der Poll, T.; Levi, M. Crosstalk between inflammation and coagulation: The lessons of sepsis. Curr. Vasc. Pharm. 2012, 10, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; van der Poll, T.; Buller, H.R. Bidirectional relation between inflammation and coagulation. Circulation 2004, 109, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Muta, T.; Iwanaga, S. Clotting and immune defense in Limulidae. Prog. Mol. Subcell. Biol. 1996, 15, 154–189. [Google Scholar] [CrossRef]
- Esmon, C.T.; Xu, J.; Lupu, F. Innate immunity and coagulation. J. Thromb. Haemost. 2011, 9, 182–188. [Google Scholar] [CrossRef]
- Antoniak, S. The coagulation system in host defense. Res Pr. Thromb Haemost. 2018, 2, 549–557. [Google Scholar] [CrossRef]
- Burzynski, L.C.; Humphry, M.; Pyrillou, K.; Wiggins, K.A.; Chan, J.N.E.; Figg, N.; Kitt, L.L.; Summers, C.; Tatham, K.C.; Martin, P.B.; et al. The Coagulation and Immune Systems Are Directly Linked through the Activation of Interleukin-1alpha by Thrombin. Immunity 2019, 50, 1033–1042.e1036. [Google Scholar] [CrossRef]
- Basler, C.F. Molecular pathogenesis of viral hemorrhagic fever. Semin. Immunopathol. 2017, 39, 551–561. [Google Scholar] [CrossRef]
- Matsuo, K.; Uozumi, Y.; Miyamoto, H.; Tatsumi, S.; Kohmura, E. Varicella-zoster vasculitis presenting with cerebellar hemorrhage. J. Stroke Cereb. Dis. 2015, 24, e153–e155. [Google Scholar] [CrossRef]
- Zhang, W.; Ruan, Q.L.; Yan, F.; Hu, Y.K. Fatal hemorrhagic varicella in a patient with abdominal pain: A case report. BMC Infect. Dis. 2020, 20, 54. [Google Scholar] [CrossRef]
- Maness, D.L.; Rogers, D.Y. Hemorrhagic complications of varicella. Am. Fam. Phys. 1987, 35, 151–155. [Google Scholar]
- Fonkoua, L.K.; Zhang, S.; Canty, E.; Fairfull, A.; Benich, S.; Knab, A.; Polimera, H.; Songdej, N. Purpura fulminans from reduced protein S following cytomegalovirus and varicella infection. Am. J. Hematol. 2019, 94, 491–495. [Google Scholar] [CrossRef]
- Koc, B.; Bircan, H.Y.; Altaner, S.; Cinar, O.; Ozcelik, U.; Yavuz, A.; Kemik, O. Massive Alimentary Tract Bleeding due to Cytomegalovirus Infection in an Elderly Patient. Infect. Dis. Rep. 2014, 6, 5512. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Khamaisi, M.; Leitersdorf, E. Association between cytomegalovirus infection and venous thromboembolism. Am. J. Med. Sci. 2007, 334, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Sabat, S.; Agarwal, A.; Zacharia, T.; Labib, S.; Yousef, J. Epstein-Barr virus encephalitis presenting as cerebellar hemorrhage. Neuroradiol. J. 2015, 28, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Goeijenbier, M.; van Wissen, M.; van de Weg, C.; Jong, E.; Gerdes, V.E.; Meijers, J.C.; Brandjes, D.P.; van Gorp, E.C. Review: Viral infections and mechanisms of thrombosis and bleeding. J. Med. Virol. 2012, 84, 1680–1696. [Google Scholar] [CrossRef]
- Rovery, C.; Granel, B.; Parola, P.; Foucault, C.; Brouqui, P. Acute cytomegalovirus infection complicated by venous thrombosis: A case report. Ann. Clin. Microbiol. Antimicrob. 2005, 4, 11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arav-Boger, R.; Reif, S.; Bujanover, Y. Portal vein thrombosis caused by protein C and protein S deficiency associated with cytomegalovirus infection. J. Pediatr. 1995, 126, 586–588. [Google Scholar] [CrossRef]
- Labarca, J.A.; Rabaggliati, R.M.; Radrigan, F.J.; Rojas, P.P.; Perez, C.M.; Ferres, M.V.; Acuna, G.G.; Bertin, P.A. Antiphospholipid syndrome associated with cytomegalovirus infection: Case report and review. Clin. Infect. Dis. 1997, 24, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Umapathi, T.; Kor, A.C.; Venketasubramanian, N.; Lim, C.C.; Pang, B.C.; Yeo, T.T.; Lee, C.C.; Lim, P.L.; Ponnudurai, K.; Chuah, K.L.; et al. Large artery ischaemic stroke in severe acute respiratory syndrome (SARS). J. Neurol. 2004, 251, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.D.; Sacco, C.; Alexia, B.; et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef]
- Vulliamy, P.; Jacob, S.; Davenport, R.A. Acute aorto-iliac and mesenteric arterial thromboses as presenting features of COVID-19. Br. J. Haematol. 2020, 189, 1053–1054. [Google Scholar] [CrossRef]
- Lia, A.B.; Pacioni, C.; Ponton, S.; Francavilla, S.; Cuzzoli, A. Arterial Mesenteric Thrombosis as a Complication of SARS-CoV-2 Infection. Eur. J. Case Rep. Intern. Med. 2020, 7, 001690. [Google Scholar] [CrossRef]
- Griffin, D.O.; Jensen, A.; Khan, M.; Chin, J.; Chin, K.; Parnell, R.; Awwad, C.; Patel, D. Arterial thromboembolic complications in COVID-19 in low risk patients despite prophylaxis. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Sathe, P.M.; Patwa, U.D. D Dimer in acute care. Int. J. Crit. Illn. Inj. Sci. 2014, 4, 229–232. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, X.; Yang, P.; Zhang, S. COVID-19 Complicated by Acute Pulmonary Embolism. Radiol. Cardiothorac. Imaging 2020, 2. [Google Scholar] [CrossRef]
- Marone, E.M.; Rinaldi, L.F. Upsurge of deep venous thrombosis in patients affected by COVID-19: Preliminary data and possible explanations. J. Vasc. Surg. Venous. Lymphat. Disord. 2020, 8, 694–695. [Google Scholar] [CrossRef] [PubMed]
- Danzi, G.B.; Loffi, M.; Galeazzi, G.; Gherbesi, E. Acute pulmonary embolism and COVID-19 pneumonia: A random association? Eur. Heart J. 2020, 41, 1858. [Google Scholar] [CrossRef] [PubMed]
- Poggiali, E.; Bastoni, D.; Ioannilli, E.; Vercelli, A.; Magnacavallo, A. Deep Vein Thrombosis and Pulmonary Embolism: Two Complications of COVID-19 Pneumonia? Eur. J. Case Rep. Intern. Med. 2020, 7, 001646. [Google Scholar] [CrossRef] [PubMed]
- Linkins, L.A.; Takach Lapner, S. Review of D-dimer testing: Good, Bad, and Ugly. Int. J. Lab. Hematol. 2017, 39, 98–103. [Google Scholar] [CrossRef]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424. [Google Scholar] [CrossRef]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, D. COVID-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 201, 1299–1300. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S.; et al. Pulmonary Embolism in Patients With COVID-19: Awareness of an Increased Prevalence. Circulation 2020, 142, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, D.; Sperhake, J.-P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann. Intern. Med. 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Dolhnikoff, M.; Duarte-Neto, A.N.; de Almeida Monteiro, R.A.; da Silva, L.F.F.; de Oliveira, E.P.; Saldiva, P.H.N.; Mauad, T.; Negri, E.M. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 2020, 18, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- White, R.H.; Keenan, C.R. Effects of race and ethnicity on the incidence of venous thromboembolism. Thromb. Res. 2009, 123, S11–S17. [Google Scholar] [CrossRef]
- Zakai, N.A.; McClure, L.A.; Judd, S.E.; Safford, M.M.; Folsom, A.R.; Lutsey, P.L.; Cushman, M. Racial and regional differences in venous thromboembolism in the United States in 3 cohorts. Circulation 2014, 129, 1502–1509. [Google Scholar] [CrossRef]
- Yancy, C.W. COVID-19 and African Americans. JAMA 2020, 323, 1891–1892. [Google Scholar] [CrossRef]
- Wright, J.E., II; Merritt, C.C. Social Equity and COVID-19: The Case of African Americans. Public Adm. Rev. 2020. [Google Scholar] [CrossRef]
- Hamidian Jahromi, A.; Hamidianjahromi, A. Why African Americans Are a Potential Target for COVID-19 Infection in the United States. J. Med. Internet. Res. 2020, 22, e19934. [Google Scholar] [CrossRef]
- Frydman, G.H.; Boyer, E.W.; Nazarian, R.M.; Van Cott, E.M.; Piazza, G. Coagulation Status and Venous Thromboembolism Risk in African Americans: A Potential Risk Factor in COVID-19. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620943671. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- McGonagle, D.; Plein, S.; O’Donnell, J.S.; Sharif, K.; Bridgewood, C. Increased cardiovascular mortality in African Americans with COVID-19. Lancet Respir. Med. 2020, 8, 649–651. [Google Scholar] [CrossRef]
- Leisman, D.E.; Deutschman, C.S.; Legrand, M. Facing COVID-19 in the ICU: Vascular dysfunction, thrombosis, and dysregulated inflammation. Intensive Care Med. 2020, 46, 1105–1108. [Google Scholar] [CrossRef] [PubMed]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Deigendesch, H.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; Bassetti, S.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Brown, J.Q.; Vander Heide, R.S. Pulmonary and Cardiac Pathology in Covid-19: The First Autopsy Series from New Orleans. medRxiv 2020. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020. [Google Scholar] [CrossRef]
- Escher, R.; Breakey, N.; Lammle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Zhong, N.S. Clinical Characteristics of Covid-19 in China. Reply. N. Engl. J. Med. 2020, 382, 1861–1862. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Lippi, G.; Plebani, M.; Henry, B.M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin. Chim. Acta 2020, 506, 145–148. [Google Scholar] [CrossRef]
- Di Micco, P.; Russo, V.; Carannante, N.; Imparato, M.; Rodolfi, S.; Cardillo, G.; Lodigiani, C. Clotting Factors in COVID-19: Epidemiological Association and Prognostic Values in Different Clinical Presentations in an Italian Cohort. J. Clin. Med. 2020, 9, 1371. [Google Scholar] [CrossRef]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- O’Sullivan, J.M.; Gonagle, D.M.; Ward, S.E.; Preston, R.J.S.; O’Donnell, J.S. Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol. 2020. [Google Scholar] [CrossRef]
- Amraei, R.; Rahimi, N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef]
- Levi, M.; Scully, M. How I treat disseminated intravascular coagulation. Blood 2018, 131, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; O’Meara, M.J.; Guo, J.Z.; Swaney, D.L.; Tummino, T.A.; Huttenhain, R.; et al. A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kannemeier, C.; Shibamiya, A.; Nakazawa, F.; Trusheim, H.; Ruppert, C.; Markart, P.; Song, Y.; Tzima, E.; Kennerknecht, E.; Niepmann, M.; et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc. Natl. Acad. Sci. USA 2007, 104, 6388–6393. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wu, X.; Zheng, X.; Luo, S.; Xu, S.; Weng, J. Targeting inflammation and cytokine storm in COVID-19. Pharm. Res. 2020, 159, 105051. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; Hlh Across Speciality Collaboration, U.K. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Wright, D.J.M. Prevention of the cytokine storm in COVID-19. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef]
- Szotowski, B.; Antoniak, S.; Poller, W.; Schultheiss, H.P.; Rauch, U. Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ. Res. 2005, 96, 1233–1239. [Google Scholar] [CrossRef]
- Wan, S.; Yi, Q.; Fan, S.; Lv, J.; Zhang, X.; Guo, L.; Lang, C.; Xiao, Q.; Xiao, K.; Yi, Z.; et al. Characteristics of lymphocyte subsets and cytokines in peripheral blood of 123 hospitalized patients with 2019 novel coronavirus pneumonia (NCP). medRxiv 2020. [Google Scholar] [CrossRef]
- Stouthard, J.M.; Levi, M.; Hack, C.E.; Veenhof, C.H.; Romijn, H.A.; Sauerwein, H.P.; van der Poll, T. Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb. Haemost. 1996, 76, 738–742. [Google Scholar] [CrossRef]
- Kerr, R.; Stirling, D.; Ludlam, C.A. Interleukin 6 and haemostasis. Br. J. Haematol. 2001, 115, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Thomas, S.; Mistry, Y.; Poole, S. Inhibition of tissue factor and cytokine release. Haemostasis 1996, 26, 92–95. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, E.; Friederich, P.W.; Vlasuk, G.P.; Rote, W.E.; Vroom, M.B.; Levi, M.; van der Poll, T. Activation of coagulation by administration of recombinant factor VIIa elicits interleukin 6 (IL-6) and IL-8 release in healthy human subjects. Clin. Diagn. Lab. Immunol. 2003, 10, 495–497. [Google Scholar] [CrossRef]
- Noris, M.; Benigni, A.; Remuzzi, G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 2020, 98, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Wilk, C.M. Coronaviruses hijack the complement system. Nat. Rev. Immunol. 2020, 20, 350. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Laudes, I.J.; Chu, J.C.; Sikranth, S.; Huber-Lang, M.; Guo, R.F.; Riedemann, N.; Sarma, J.V.; Schmaier, A.H.; Ward, P.A. Anti-c5a ameliorates coagulation/fibrinolytic protein changes in a rat model of sepsis. Am. J. Pathol. 2002, 160, 1867–1875. [Google Scholar] [CrossRef]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus-host interactome and proteomic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. BioRxiv 2020, 10. [Google Scholar] [CrossRef]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv 2020, 10. [Google Scholar] [CrossRef]
- Mastaglio, S.; Ruggeri, A.; Risitano, A.M.; Angelillo, P.; Yancopoulou, D.; Mastellos, D.C.; Huber-Lang, M.; Piemontese, S.; Assanelli, A.; Garlanda, C.; et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin. Immunol. 2020, 215, 108450. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M.; Mastellos, D.C.; Huber-Lang, M.; Yancopoulou, D.; Garlanda, C.; Ciceri, F.; Lambris, J.D. Complement as a target in COVID-19? Nat. Rev. Immunol. 2020, 20, 343–344. [Google Scholar] [CrossRef] [PubMed]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.; PG, D.E.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; McCrae, K.R. Diagnosis and management of the antiphospholipid syndrome. Blood Rev. 2017, 31, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Harzallah, I.; Debliquis, A.; Drenou, B. Lupus anticoagulant is frequent in patients with Covid-19. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef]
- Uthman, I.W.; Gharavi, A.E. Viral infections and antiphospholipid antibodies. Semin. Arthritis Rheum 2002, 31, 256–263. [Google Scholar] [CrossRef]
- Shoenfeld, Y.; Blank, M.; Cervera, R.; Font, J.; Raschi, E.; Meroni, P.L. Infectious origin of the antiphospholipid syndrome. Ann. Rheum Dis. 2006, 65, 2–6. [Google Scholar] [CrossRef]
- Blank, M.; Asherson, R.A.; Cervera, R.; Shoenfeld, Y. Antiphospholipid syndrome infectious origin. J. Clin. Immunol. 2004, 24, 12–23. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, X.; Jiao, Y.; Li, Z.; Liu, Q.; Ye, J.; Yang, M. Mechanisms involved in the development of thrombocytopenia in patients with COVID-19. Thromb. Res. 2020, 193, 110–115. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Q.; Wang, Y.; Wu, Y.; Xu, J.; Yu, Y.; Shang, Y. Thrombocytopenia and its association with mortality in patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Handtke, S.; Thiele, T. Large and small platelets-(When) do they differ? J. Thromb. Haemost. 2020, 18, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Handtke, S.; Steil, L.; Palankar, R.; Conrad, J.; Cauhan, S.; Kraus, L.; Ferrara, M.; Dhople, V.; Wesche, J.; Volker, U.; et al. Role of Platelet Size Revisited-Function and Protein Composition of Large and Small Platelets. Thromb. Haemost. 2019, 119, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Ranucci, M.; Ballotta, A.; Di Dedda, U.; Bayshnikova, E.; Dei Poli, M.; Resta, M.; Falco, M.; Albano, G.; Menicanti, L. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost. 2020, 18, 1747–1751. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Biondi-Zoccai, G.; Brown, T.S.; Der Nigoghossian, C.; Zidar, D.A.; Haythe, J.; et al. Cardiovascular Considerations for Patients, Health Care Workers, and Health Systems During the COVID-19 Pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef] [PubMed]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-up. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef]
- Popoola, V.O.; Tavakoli, F.; Lau, B.D.; Lankiewicz, M.; Ross, P.; Kraus, P.; Shaffer, D.; Hobson, D.B.; Aboagye, J.K.; Farrow, N.A.; et al. Exploring the impact of route of administration on medication acceptance in hospitalized patients: Implications for venous thromboembolism prevention. Thromb. Res. 2017, 160, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Dentali, F.; Mumoli, N.; Prisco, D.; Fontanella, A.; Di Minno, M.N. Efficacy and safety of extended thromboprophylaxis for medically ill patients. A meta-analysis of randomised controlled trials. Thromb. Haemost. 2017, 117, 606–617. [Google Scholar] [CrossRef]
- Schindewolf, M.; Weitz, J.I. Broadening the Categories of Patients Eligible for Extended Venous Thromboembolism Treatment. Thromb. Haemost. 2020, 120, 14–26. [Google Scholar] [CrossRef]
- Paranjpe, I.; Fuster, V.; Lala, A.; Russak, A.J.; Glicksberg, B.S.; Levin, M.A.; Charney, A.W.; Narula, J.; Fayad, Z.A.; Bagiella, E.; et al. Association of Treatment Dose Anticoagulation With In-Hospital Survival Among Hospitalized Patients With COVID-19. J. Am. Coll. Cardiol. 2020, 76, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef]
- Yamakawa, K.; Levy, J.H.; Iba, T. Recombinant human soluble thrombomodulin in patients with sepsis-associated coagulopathy (SCARLET): An updated meta-analysis. Crit. Care 2019, 23, 302. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Saitoh, D. Efficacy of antithrombin in preclinical and clinical applications for sepsis-associated disseminated intravascular coagulation. J. Intensive Care 2014, 2, 66. [Google Scholar] [CrossRef]
- Kienast, J.; Juers, M.; Wiedermann, C.J.; Hoffmann, J.N.; Ostermann, H.; Strauss, R.; Keinecke, H.O.; Warren, B.L.; Opal, S.M.; The KyberSept Investigators. Treatment effects of high-dose antithrombin without concomitant heparin in patients with severe sepsis with or without disseminated intravascular coagulation. J. Thromb. Haemost. 2006, 4, 90–97. [Google Scholar] [CrossRef]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020. [Google Scholar] [CrossRef] [PubMed]
- Fletcher-Sandersjoo, A.; Bellander, B.M. Is COVID-19 associated thrombosis caused by overactivation of the complement cascade? A literature review. Thromb. Res. 2020, 194, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H.; Levi, M.; Connors, J.M.; Thachil, J. Coagulopathy of Coronavirus Disease 2019. Crit. Care Med. 2020, 48, 1358–1364. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamel, M.H.; Yin, W.; Zavaro, C.; Francis, J.M.; Chitalia, V.C. Hyperthrombotic Milieu in COVID-19 Patients. Cells 2020, 9, 2392. https://doi.org/10.3390/cells9112392
Kamel MH, Yin W, Zavaro C, Francis JM, Chitalia VC. Hyperthrombotic Milieu in COVID-19 Patients. Cells. 2020; 9(11):2392. https://doi.org/10.3390/cells9112392
Chicago/Turabian StyleKamel, Mohamed Hassan, Wenqing Yin, Chris Zavaro, Jean M. Francis, and Vipul C. Chitalia. 2020. "Hyperthrombotic Milieu in COVID-19 Patients" Cells 9, no. 11: 2392. https://doi.org/10.3390/cells9112392
APA StyleKamel, M. H., Yin, W., Zavaro, C., Francis, J. M., & Chitalia, V. C. (2020). Hyperthrombotic Milieu in COVID-19 Patients. Cells, 9(11), 2392. https://doi.org/10.3390/cells9112392