COVID-19, Renin-Angiotensin System and Endothelial Dysfunction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Novel Severe Acute Respiratory Syndrome Coronavirus-19
3. ACE2 Peptidase Activity and Signal Transduction
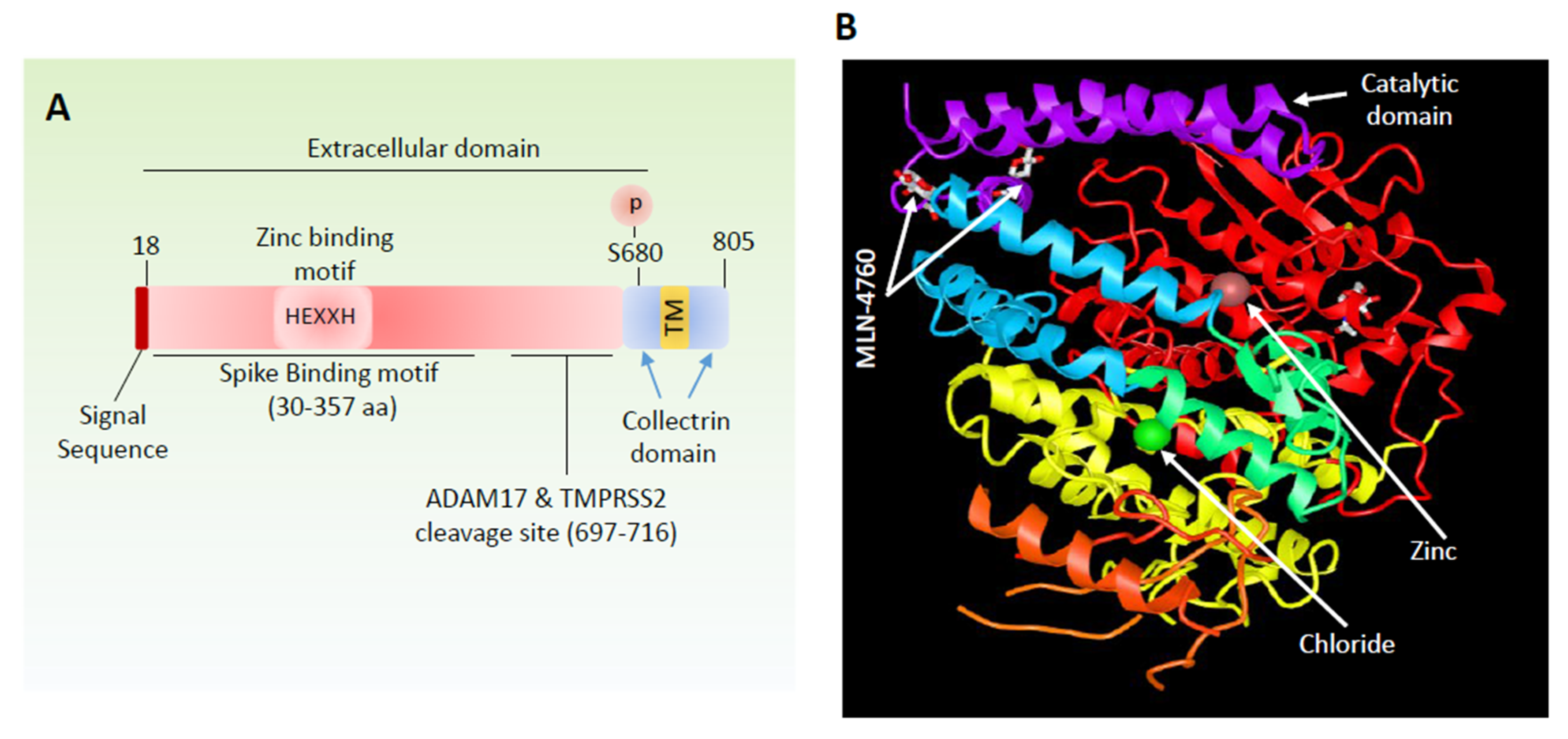
4. Angiotensin-Converting Enzyme 2 is a Viral Recognition Receptor
5. Viral–Host Interaction Activates Spike Protein through Proteolytic Activation
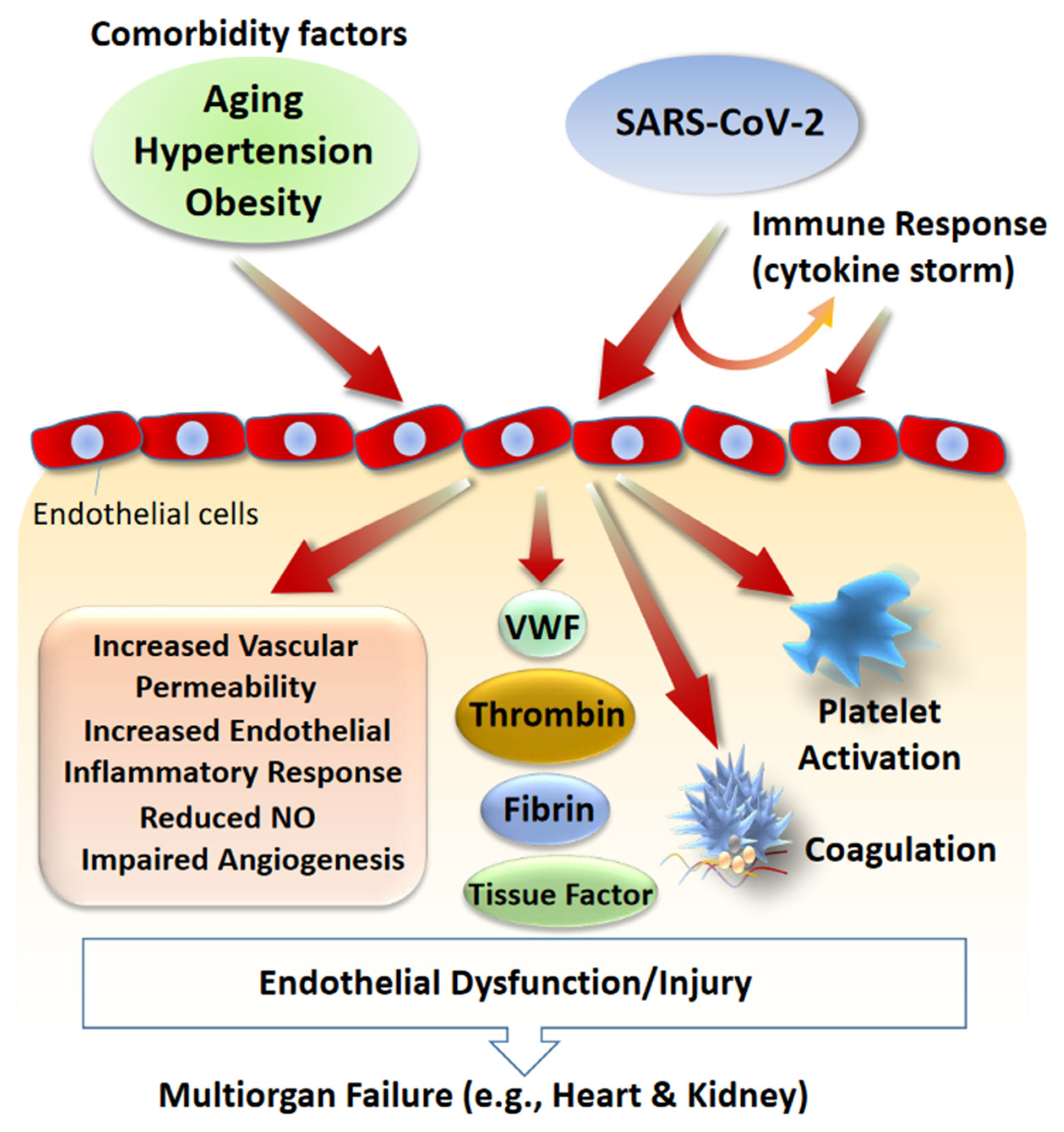
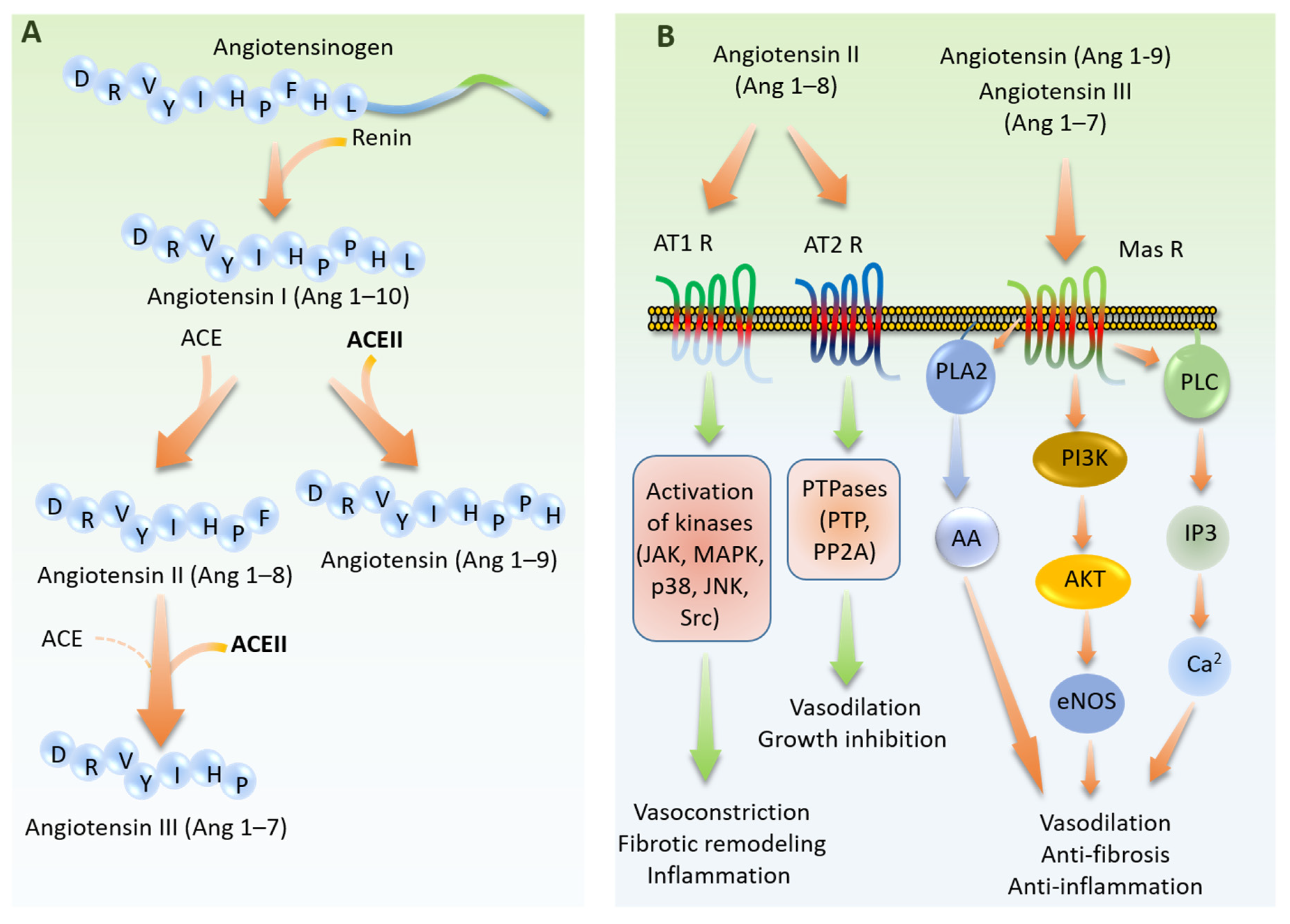
6. Role of Endothelial Dysfunction in SARS-CoV-2 Infections: Circulating RAS
7. Role of Endothelial Dysfunction in SARS-CoV-2 Infections: Local RAS
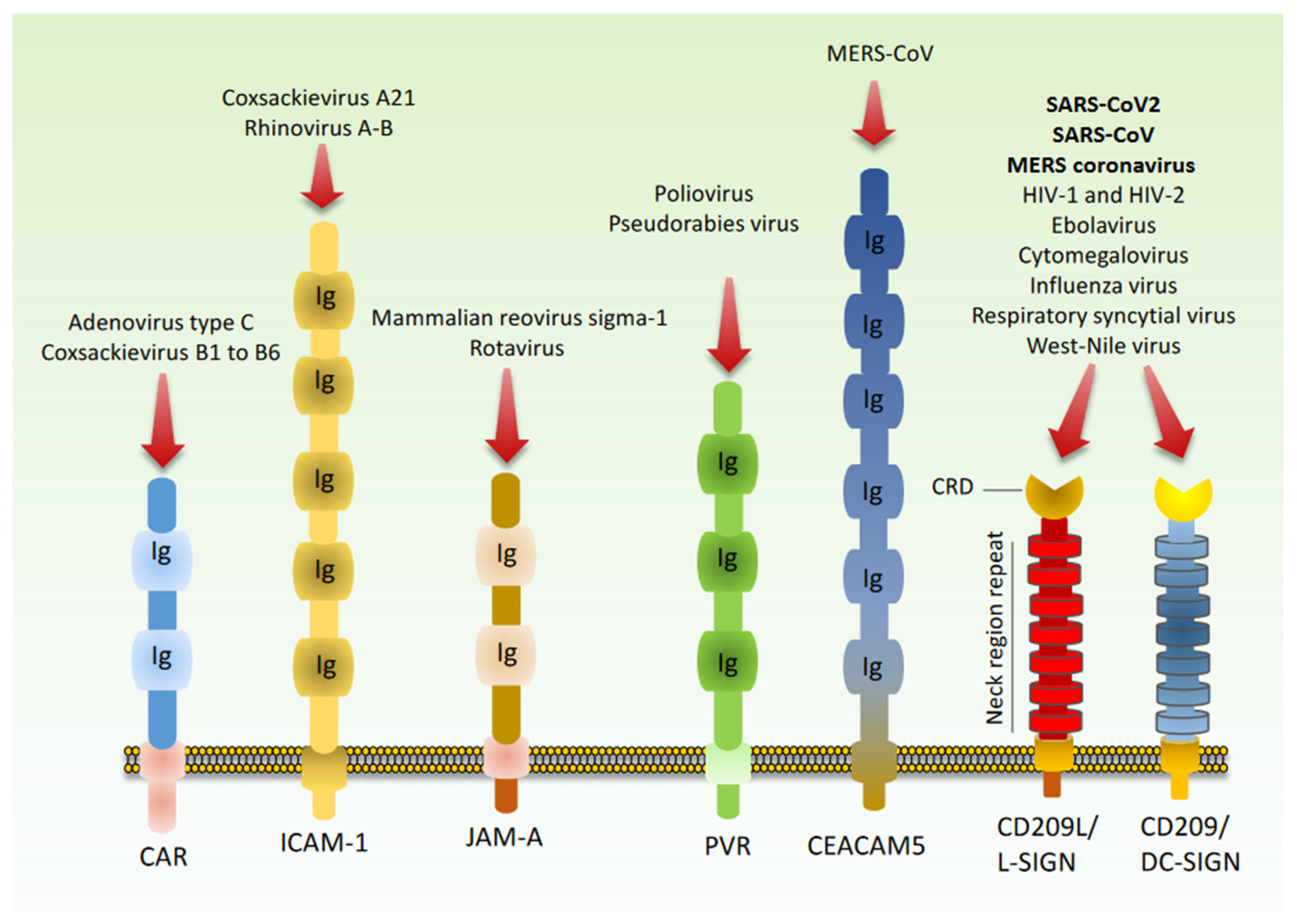
8. Cell Adhesion Molecules as Potential Receptors for SARS-CoV2 Host Recognition
9. Therapeutic Targets for Treatment of COVID-19
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Lee, N.; Hui, D.; Wu, A.; Chan, P.; Cameron, P.; Joynt, G.M.; Ahuja, A.; Yung, M.Y.; Leung, C.B.; To, K.F.; et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.T.; Li, Y.; Wong, T.W.; Tam, W.; Chan, A.T.; Lee, J.H.; Leung, D.Y.; Ho, T. Evidence of airborne transmission of the severe acute respiratory syndrome virus. N. Engl. J. Med. 2004, 350, 1731–1739. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Ren, L.L.; Wang, Y.M.; Wu, Z.Q.; Xiang, Z.C.; Guo, L.; Xu, T.; Jiang, Y.Z.; Xiong, Y.; Li, Y.J.; Li, X.W.; et al. Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chin. Med. J. (Engl.) 2020. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.; Lai, S.T.; Poon, L.L.; Guan, Y.; Yam, L.Y.; Lim, W.; Nicholls, J.; Yee, W.K.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Wenham, C.; Smith, J.; Morgan, R.; Gender and COVID-19 Working Group. COVID-19: The gendered impacts of the outbreak. Lancet (Lond. Engl.) 2020, 395, 846–848. [Google Scholar] [CrossRef]
- Taylor, F.B., Jr.; Toh, C.H.; Hoots, W.K.; Wada, H.; Levi, M.; Scientific Subcommittee on Disseminated Intravascular Coagulation (DIC) of the International Society on Thrombosis and Haemostasis (ISTH). Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb. Haemost. 2001, 86, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Spiezia, L.; Boscolo, A.; Poletto, F.; Cerruti, L.; Tiberio, I.; Campello, E.; Navalesi, P.; Simioni, P. COVID-19-Related Severe Hypercoagulability in Patients Admitted to Intensive Care Unit for Acute Respiratory Failure. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in Intensive Care Unit. A Report of Thromboelastography Findings and other Parameters of Hemostasis. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Tang, N. Response to “Lupus anticoagulant is frequent in patients with Covid-19”. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef]
- Haverkate, F.; Thompson, S.G.; Duckert, F. Haemostasis factors in angina pectoris; relation to gender, age and acute-phase reaction. Results of the ECAT Angina Pectoris Study Group. Thromb. Haemost. 1995, 73, 561–567. [Google Scholar]
- Stehouwer, C.D.; Nauta, J.J.; Zeldenrust, G.C.; Hackeng, W.H.; Donker, A.J.; den Ottolander, G.J. Urinary albumin excretion, cardiovascular disease, and endothelial dysfunction in non-insulin-dependent diabetes mellitus. Lancet 1992, 340, 319–323. [Google Scholar] [CrossRef]
- Lof, A.; Muller, J.P.; Brehm, M.A. A biophysical view on von Willebrand factor activation. J. Cell. Physiol. 2018, 233, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Butera, D.; Passam, F.; Ju, L.; Cook, K.M.; Woon, H.; Aponte-Santamaria, C.; Gardiner, E.; Davis, A.K.; Murphy, D.A.; Bronowska, A.; et al. Autoregulation of von Willebrand factor function by a disulfide bond switch. Sci. Adv. 2018, 4, eaaq1477. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.J.; Gattinoni, L. Management of COVID-19 Respiratory Distress. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.M.; Gomez-Puertas, P.; Cavanagh, D.; Gorbalenya, A.E.; Enjuanes, L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch. Virol. 2003, 148, 2207–2235. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch. Virol. 2016, 161, 2921–2949. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Weinstein, R.A. Planning for epidemics--the lessons of SARS. N. Engl. J. Med. 2004, 350, 2332–2334. [Google Scholar] [CrossRef]
- Bolles, M.; Donaldson, E.; Baric, R. SARS-CoV and emergent coronaviruses: Viral determinants of interspecies transmission. Curr. Opin. Virol. 2011, 1, 624–634. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Hofmann, H.; Pyrc, K.; van der Hoek, L.; Geier, M.; Berkhout, B.; Pohlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.W.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Fouchier, R.A.M.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Zhang, H.; Wada, J.; Hida, K.; Tsuchiyama, Y.; Hiragushi, K.; Shikata, K.; Wang, H.; Lin, S.; Kanwar, Y.S.; Makino, H. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J. Biol. Chem. 2001, 276, 17132–17139. [Google Scholar] [CrossRef]
- Verrey, F.; Singer, D.; Ramadan, T.; Vuille-dit-Bille, R.N.; Mariotta, L.; Camargo, S.M. Kidney amino acid transport. Pflug. Arch. 2009, 458, 53–60. [Google Scholar] [CrossRef]
- Danilczyk, U.; Sarao, R.; Remy, C.; Benabbas, C.; Stange, G.; Richter, A.; Arya, S.; Pospisilik, J.A.; Singer, D.; Camargo, S.M.; et al. Essential role for collectrin in renal amino acid transport. Nature 2006, 444, 1088–1091. [Google Scholar] [CrossRef]
- Lambert, D.W.; Clarke, N.E.; Hooper, N.M.; Turner, A.J. Calmodulin interacts with angiotensin-converting enzyme-2 (ACE2) and inhibits shedding of its ectodomain. Febs. Lett. 2008, 582, 385–390. [Google Scholar] [CrossRef]
- Lai, Z.W.; Lew, R.A.; Yarski, M.A.; Mu, F.T.; Andrews, R.K.; Smith, A.I. The identification of a calmodulin-binding domain within the cytoplasmic tail of angiotensin-converting enzyme-2. Endocrinology 2009, 150, 2376–2381. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Dales, N.A.; Gould, A.E.; Brown, J.A.; Calderwood, E.F.; Guan, B.; Minor, C.A.; Gavin, J.M.; Hales, P.; Kaushik, V.K.; Stewart, M.; et al. Substrate-based design of the first class of angiotensin-converting enzyme-related carboxypeptidase (ACE2) inhibitors. J. Am. Chem. Soc. 2002, 124, 11852–11853. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed]
- Keidar, S.; Kaplan, M.; Gamliel-Lazarovich, A. ACE2 of the heart: From angiotensin I to angiotensin (1-7). Cardiovasc. Res. 2007, 73, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.; Alenina, N.; Andrade-Navarro, M.A.; Santos, R.A. MAS and its related G protein-coupled receptors, Mrgprs. Pharm. Rev. 2014, 66, 1080–1105. [Google Scholar] [CrossRef]
- Solinski, H.J.; Gudermann, T.; Breit, A. Pharmacology and signaling of MAS-related G protein-coupled receptors. Pharm. Rev. 2014, 66, 570–597. [Google Scholar] [CrossRef]
- Lin, C.S.; Pan, C.H. Regulatory mechanisms of atrial fibrotic remodeling in atrial fibrillation. Cell Mol. Life Sci. 2008, 65, 1489–1508. [Google Scholar] [CrossRef]
- Karnik, S.S.; Unal, H.; Kemp, J.R.; Tirupula, K.C.; Eguchi, S.; Vanderheyden, P.M.; Thomas, W.G. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli [corrected]. Pharm. Rev. 2015, 67, 754–819. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dong, J.; Martin, M.; He, M.; Gongol, B.; Marin, T.L.; Chen, L.; Shi, X.; Yin, Y.; Shang, F.; et al. AMP-activated Protein Kinase Phosphorylation of Angiotensin-Converting Enzyme 2 in Endothelium Mitigates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef]
- Wang, S.; Guo, F.; Liu, K.; Wang, H.; Rao, S.; Yang, P.; Jiang, C. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 2008, 136, 8–15. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef]
- Haga, S.; Nagata, N.; Okamura, T.; Yamamoto, N.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antivir. Res. 2010, 85, 551–555. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pohlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef]
- Bertram, S.; Dijkman, R.; Habjan, M.; Heurich, A.; Gierer, S.; Glowacka, I.; Welsch, K.; Winkler, M.; Schneider, H.; Hofmann-Winkler, H.; et al. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 2013, 87, 6150–6160. [Google Scholar] [CrossRef]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef]
- Gierer, S.; Bertram, S.; Kaup, F.; Wrensch, F.; Heurich, A.; Kramer-Kuhl, A.; Welsch, K.; Winkler, M.; Meyer, B.; Drosten, C.; et al. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J. Virol. 2013, 87, 5502–5511. [Google Scholar] [CrossRef]
- Shirato, K.; Kawase, M.; Matsuyama, S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J. Virol. 2013, 87, 12552–12561. [Google Scholar] [CrossRef]
- Park, J.-E.; Li, K.; Barlan, A.; Fehr, A.R.; Perlman, S.; McCray, P.B., Jr.; Gallagher, T. Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc. Natl. Acad. Sci. USA 2016, 113, 12262–12267. [Google Scholar] [CrossRef]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar] [CrossRef]
- Yamada, Y.; Liu, D.X. Proteolytic activation of the spike protein at a novel RRRR/S motif is implicated in furin-dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J. Virol. 2009, 83, 8744–8758. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Dominguez, S.R.; Holmes, K.V. Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS-CoV) in virus entry and syncytia formation. PLoS ONE 2013, 8, e76469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, D.; Song, J.W.; Zullo, J.; Lipphardt, M.; Coneh-Gould, L.; Goligorsky, M.S. Endothelial cell dysfunction and glycocalyx—A vicious circle. Matrix. Biol. 2018, 71–72, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.R.; Rachel, G.; Parthasarathy, D. HIV Proteins and Endothelial Dysfunction: Implications in Cardiovascular Disease. Front. Cardiovasc. Med. 2018, 5, 185. [Google Scholar] [CrossRef]
- Lerman, A.; Burnett, J.C., Jr. Intact and altered endothelium in regulation of vasomotion. Circulation 1992, 86, III12–III19. [Google Scholar]
- Anderson, T.J. Assessment and treatment of endothelial dysfunction in humans. J. Am. Coll. Cardiol. 1999, 34, 631–638. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef]
- Odegaard, A.O.; Jacobs, D.R., Jr.; Sanchez, O.A.; Goff, D.C., Jr.; Reiner, A.P.; Gross, M.D. Oxidative stress, inflammation, endothelial dysfunction and incidence of type 2 diabetes. Cardiovasc. Diabetol. 2016, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.; Videla, L.A. Impact of the Co-Administration of N-3 Fatty Acids and Olive Oil Components in Preclinical Nonalcoholic Fatty Liver Disease Models: A Mechanistic View. Nutrients 2020, 12, 499. [Google Scholar] [CrossRef] [PubMed]
- Dupin, N.; Fisher, C.; Kellam, P.; Ariad, S.; Tulliez, M.; Franck, N.; Van Marck, E.; Salmon, D.; Gorin, I.; Escande, J.-P. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 1999, 96, 4546–4551. [Google Scholar] [CrossRef]
- Mackow, E.R.; Gavrilovskaya, I.N. Hantavirus regulation of endothelial cell functions. Thromb. Haemost. 2009, 102, 1030–1041. [Google Scholar]
- Steinberg, B.; Goldenberg, N.; Lee, W. Do viral infections mimic bacterial sepsis? The role of microvascular permeability: A review of mechanisms and methods. Antivir. Res. 2012, 93, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.N.; Gorbunova, E.E.; Mackow, N.A.; Mackow, E.R. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J. Virol. 2008, 82, 5797–5806. [Google Scholar] [CrossRef] [PubMed]
- Vervaeke, P.; Vermeire, K.; Liekens, S. Endothelial dysfunction in dengue virus pathology. Rev. Med. Virol. 2015, 25, 50–67. [Google Scholar] [CrossRef]
- Stacey, K.J.; Watterson, D.; Modhiran, N.; Young, P.R. Response to comment on “Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity” and “Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination”. Sci. Transl. Med. 2015, 7, 318lr314. [Google Scholar] [CrossRef][Green Version]
- Beatty, P.R.; Puerta-Guardo, H.; Killingbeck, S.S.; Glasner, D.R.; Hopkins, K.; Harris, E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med. 2015, 7, 304ra141. [Google Scholar] [CrossRef]
- Lafon, M.-E.; Steffan, A.-M.; Royer, C.; Jaeck, D.; Beretz, A.; Kirn, A.; Gendrault, J.-L. HIV-1 infection induces functional alterations in human liver endothelial cells in primary culture. Aids (Lond. Engl.) 1994, 8, 747–752. [Google Scholar] [CrossRef]
- Corbeil, J.; Evans, L.A.; McQueen, P.W.; Vasak, E.; Edward, P.D.; Richman, D.D.; Penny, R.; Cooper, D.A. Productive in vitro infection of human umbilical vein endothelial cells and three colon carcinoma cell lines with HIV-1. Immunol. Cell Biol. 1995, 73, 140–145. [Google Scholar] [CrossRef]
- Poland, S.; Rice, G.; Dekaban, G. HIV-1 infection of human brain-derived microvascular endothelial cells in vitro. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. Off. Publ. Int. Retrovirol. Assoc. 1995, 8, 437–445. [Google Scholar] [CrossRef]
- de Gaetano Donati, K.; Rabagliati, R.; Iacoviello, L.; Cauda, R. HIV infection, HAART, and endothelial adhesion molecules: Current perspectives. Lancet Infect. Dis. 2004, 4, 213–222. [Google Scholar] [CrossRef]
- Stefano, G.B.; Salzet, M.; Bilfinger, T.V. Long-term exposure of human blood vessels to HIV gp120, morphine, and anandamide increases endothelial adhesion of monocytes: Uncoupling of nitric oxide release. J. Cardiovasc. Pharmacol. 1998, 31, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Keshav, S.; † OLLIN, M.; Montaner, L.; Gordon, S. HIV-1 induces tumour necrosis factor and IL-1 gene expression in primary human macrophages independent of productive infection. Clin. Exp. Immunol. 1994, 95, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Galea, P.; Vermot-Desroches, C.; Le Contel, C.; Wijdenes, J.; Chermann, J.-C. Circulating cell adhesion molecules in HIV1-infected patients as indicator markers for AIDS progression. Res. Immunol. 1997, 148, 109–117. [Google Scholar] [CrossRef]
- Zietz, C.; Hotz, B.; Stürzl, M.; Rauch, E.; Penning, R.; Löhrs, U. Aortic endothelium in HIV-1 infection: Chronic injury, activation, and increased leukocyte adherence. Am. J. Pathol. 1996, 149, 1887. [Google Scholar]
- Graham, S.M.; Rajwans, N.; Jaoko, W.; ESTAMBALE, B.B.; Mcclelland, R.S.; Overbaugh, J.; LILES, W.C. Endothelial activation biomarkers increase after HIV-1 acquisition: Plasma VCAM-1 predicts disease progression. Aids (Lond. Engl.) 2013, 27, 1803–1813. [Google Scholar] [CrossRef]
- Tikellis, C.; Thomas, M. Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef]
- Atlas, S.A. The renin-angiotensin aldosterone system: Pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 2007, 13, 9–20. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Gurley, S.B.; Allred, A.; Le, T.H.; Griffiths, R.; Mao, L.; Philip, N.; Haystead, T.A.; Donoghue, M.; Breitbart, R.E.; Acton, S.L. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J. Clin. Investig. 2006, 116, 2218–2225. [Google Scholar] [CrossRef]
- Rabelo, L.A.; Todiras, M.; Nunes-Souza, V.; Qadri, F.; Szijarto, I.A.; Gollasch, M.; Penninger, J.M.; Bader, M.; Santos, R.A.; Alenina, N. Genetic deletion of ACE2 induces vascular dysfunction in C57BL/6 mice: Role of nitric oxide imbalance and oxidative stress. PLoS ONE 2016, 11, e0150255. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.E.; Kim, E.N.; Kim, M.Y.; Lim, J.H.; Jang, I.; Ban, T.H.; Shin, S.J.; Park, C.W.; Chang, Y.S.; Choi, B.S. Age-associated changes in the vascular renin-angiotensin system in mice. Oxidative Med. Cell. Longev. 2016, 2016, 6731093. [Google Scholar] [CrossRef] [PubMed]
- Novel, C.P.E.R.E. The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China. Zhonghua Liu Xing Bing Xue Za Zhi = Zhonghua Liuxingbingxue Zazhi 2020, 41, 145. [Google Scholar]
- Guzzetta, G.; Poletti, P.; Ajelli, M.; Trentini, F.; Marziano, V.; Cereda, D.; Tirani, M.; Diurno, G.; Bodina, A.; Barone, A. Potential short-term outcome of an uncontrolled COVID-19 epidemic in Lombardy, Italy, February to March 2020. Eurosurveillance 2020, 25, 2000293. [Google Scholar] [CrossRef] [PubMed]
- Soneji, S.; Beltrán-Sánchez, H.; Yang, J.; Mann, C. Population-Level Mortality Rates from Novel Coronavirus (COVID-19) in South Korea. medRxiv 2020. [Google Scholar] [CrossRef]
- Guan, W.-Y.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S. Clinical characteristics of coronavirus disease 2019 in China. New Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; Yu, T. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Dong, X.; Cao, Y.-Y.; Yuan, Y.-D.; Yang, Y.-B.; Yan, Y.-Q.; Akdis, C.A.; Gao, Y.-D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.P. The pulmonary renin-angiotensin system. Curr. Pharm. Des. 2003, 9, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hafez, A.; Mohamed, T.; Omar, H.; Shemis, M.; Uhal, B.D. The renin angiotensin system in liver and lung: Impact and therapeutic potential in organ fibrosis. J. Lung Pulm. Respir. Res. 2018, 5, 00160. [Google Scholar] [PubMed]
- Hale, T.M. Persistent phenotypic shift in cardiac fibroblasts: Impact of transient renin angiotensin system inhibition. J. Mol. Cell. Cardiol. 2016, 93, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.; Kassiri, Z.; Jiang, C.; Liu, P.; Poutanen, S.; Penninger, J.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Amraie, R.; Napoleon, M.A.; Yin, W.; Berrigan, J.; Suder, E.; Zhao, G.; Olejnik, J.; Gummuluru, S.; Muhlberger, E.; Chitalia, V.; et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2 and are differentially expressed in lung and kidney epithelial and endothelial cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; Kallio, K.; Kaya, T.; Anastasina, M.; Smura, T.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and provides a possible pathway into the central nervous system. bioRxiv 2020. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Antón-Plágaro, C.; Kavanagh Williamson, M.; Shoemark, D.K.; Simón-Gracia, L.; Klein, K.; Bauer, M.; Hollandi, R.; Greber, U.F.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Maginnis, M.S. Virus-Receptor Interactions: The Key to Cellular Invasion. J. Mol. Biol. 2018, 430, 2590–2611. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.R. Cell adhesion molecules in the pathogenesis of and host defence against microbial infection. Mol. Pathol. 1999, 52, 220–230. [Google Scholar] [CrossRef][Green Version]
- Bhella, D. The role of cellular adhesion molecules in virus attachment and entry. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140035. [Google Scholar] [CrossRef]
- Carson, S.D. Receptor for the group B coxsackieviruses and adenoviruses: CAR. Rev. Med. Virol. 2001, 11, 219–226. [Google Scholar] [CrossRef]
- Staunton, D.E.; Merluzzi, V.J.; Rothlein, R.; Barton, R.; Marlin, S.D.; Springer, T.A. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell 1989, 56, 849–853. [Google Scholar] [CrossRef]
- Barton, E.S.; Forrest, J.C.; Connolly, J.L.; Chappell, J.D.; Liu, Y.; Schnell, F.J.; Nusrat, A.; Parkos, C.A.; Dermody, T.S. Junction adhesion molecule is a receptor for reovirus. Cell 2001, 104, 441–451. [Google Scholar] [CrossRef]
- Makino, A.; Shimojima, M.; Miyazawa, T.; Kato, K.; Tohya, Y.; Akashi, H. Junctional adhesion molecule 1 is a functional receptor for feline calicivirus. J. Virol. 2006, 80, 4482–4490. [Google Scholar] [CrossRef]
- Mendelsohn, C.L.; Wimmer, E.; Racaniello, V.R. Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 1989, 56, 855–865. [Google Scholar] [CrossRef]
- Chan, C.M.; Chu, H.; Wang, Y.; Wong, B.H.; Zhao, X.; Zhou, J.; Yang, D.; Leung, S.P.; Chan, J.F.; Yeung, M.L.; et al. Carcinoembryonic Antigen-Related Cell Adhesion Molecule 5 Is an Important Surface Attachment Factor That Facilitates Entry of Middle East Respiratory Syndrome Coronavirus. J. Virol. 2016, 90, 9114–9127. [Google Scholar] [CrossRef]
- Alvarez, C.P.; Lasala, F.; Carrillo, J.; Muniz, O.; Corbi, A.L.; Delgado, R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002, 76, 6841–6844. [Google Scholar] [CrossRef] [PubMed]
- Cormier, E.G.; Durso, R.J.; Tsamis, F.; Boussemart, L.; Manix, C.; Olson, W.C.; Gardner, J.P.; Dragic, T. L-SIGN (CD209L) and DC-SIGN (CD209) mediate transinfection of liver cells by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2004, 101, 14067–14072. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Hemmila, E.M.; Holmes, K.V. Human coronavirus 229E can use CD209L (L-SIGN) to enter cells. Adv. Exp. Med. Biol. 2006, 581, 265–269. [Google Scholar] [CrossRef]
- Marzi, A.; Gramberg, T.; Simmons, G.; Moller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar] [CrossRef] [PubMed]
- Londrigan, S.L.; Turville, S.G.; Tate, M.D.; Deng, Y.M.; Brooks, A.G.; Reading, P.C. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J. Virol. 2011, 85, 2990–3000. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, M.; Takenouchi, A.; Shimoda, H.; Kimura, N.; Maeda, K. Distinct usage of three C-type lectins by Japanese encephalitis virus: DC-SIGN, DC-SIGNR, and LSECtin. Arch. Virol. 2014, 159, 2023–2031. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Huang, Y.; Ganesh, L.; Leung, K.; Kong, W.P.; Schwartz, O.; Subbarao, K.; Nabel, G.J. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 2004, 78, 5642–5650. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D., Jr.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753. [Google Scholar] [CrossRef]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Zhang, Y.; Zhang, W.; Yuan, Y.; Bao, J.; et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Hulswit, R.J.; de Haan, C.A.; Bosch, B.J. Coronavirus Spike Protein and Tropism Changes. Adv. Virus Res. 2016, 96, 29–57. [Google Scholar] [CrossRef] [PubMed]
- Gaur, P.; Ranjan, P.; Sharma, S.; Patel, J.R.; Bowzard, J.B.; Rahman, S.K.; Kumari, R.; Gangappa, S.; Katz, J.M.; Cox, N.J.; et al. Influenza A virus neuraminidase protein enhances cell survival through interaction with carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) protein. J. Biol. Chem. 2012, 287, 15109–15117. [Google Scholar] [CrossRef]
- Gao, W.; He, W.; Zhao, K.; Lu, H.; Ren, W.; Du, C.; Chen, K.; Lan, Y.; Song, D.; Gao, F. Identification of NCAM that interacts with the PHE-CoV spike protein. Virol. J. 2010, 7, 254. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Simmons, G.; Pohlmann, S.; Baribaud, F.; Ni, H.; Leslie, G.J.; Haggarty, B.S.; Bates, P.; Weissman, D.; Hoxie, J.A.; et al. Differential N-linked glycosylation of human immunodeficiency virus and Ebola virus envelope glycoproteins modulates interactions with DC-SIGN and DC-SIGNR. J. Virol. 2003, 77, 1337–1346. [Google Scholar] [CrossRef]
- Chan, V.S.; Chan, K.Y.; Chen, Y.; Poon, L.L.; Cheung, A.N.; Zheng, B.; Chan, K.H.; Mak, W.; Ngan, H.Y.; Xu, X.; et al. Homozygous L-SIGN (CLEC4M) plays a protective role in SARS coronavirus infection. Nat. Genet. 2006, 38, 38–46. [Google Scholar] [CrossRef]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Su, J.B. Vascular endothelial dysfunction and pharmacological treatment. World J. Cardiol. 2015, 7, 719–741. [Google Scholar] [CrossRef]
- South, A.M.; Tomlinson, L.; Edmonston, D.; Hiremath, S.; Sparks, M.A. Controversies of renin-angiotensin system inhibition during the COVID-19 pandemic. Nat. Rev. Nephrol. 2020. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.J.; Ye, M.; Wysocki, J.; William, J.; Lloveras, J.; Batlle, D. Localization of ACE2 in the renal vasculature: Amplification by angiotensin II type 1 receptor blockade using telmisartan. Am. J. Physiol. Ren. Physiol. 2009, 296, F398–F405. [Google Scholar] [CrossRef] [PubMed]
- Burrell, L.M.; Risvanis, J.; Kubota, E.; Dean, R.G.; MacDonald, P.S.; Lu, S.; Tikellis, C.; Grant, S.L.; Lew, R.A.; Smith, A.I.; et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur. Heart J. 2005, 26, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Ramchand, J.; Patel, S.K.; Srivastava, P.M.; Farouque, O.; Burrell, L.M. Elevated plasma angiotensin converting enzyme 2 activity is an independent predictor of major adverse cardiac events in patients with obstructive coronary artery disease. PLoS ONE 2018, 13, e0198144. [Google Scholar] [CrossRef]
- Khera, R.; Clark, C.; Lu, Y.; Guo, Y.; Ren, S.; Truax, B.; Spatz, E.S.; Murugiah, K.; Lin, Z.; Omer, S.B.; et al. Association of Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers with the Risk of Hospitalization and Death in Hypertensive Patients with Coronavirus Disease-19. medRxiv 2020. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amraei, R.; Rahimi, N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. https://doi.org/10.3390/cells9071652
Amraei R, Rahimi N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells. 2020; 9(7):1652. https://doi.org/10.3390/cells9071652
Chicago/Turabian StyleAmraei, Razie, and Nader Rahimi. 2020. "COVID-19, Renin-Angiotensin System and Endothelial Dysfunction" Cells 9, no. 7: 1652. https://doi.org/10.3390/cells9071652
APA StyleAmraei, R., & Rahimi, N. (2020). COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells, 9(7), 1652. https://doi.org/10.3390/cells9071652