Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma


{kind=link}
Abstract
1. Context-Dependent Roles of NOTCH Signaling in Cancer
2. NOTCH Signaling in Neural Stem/Progenitor Cells and Glioma Formation
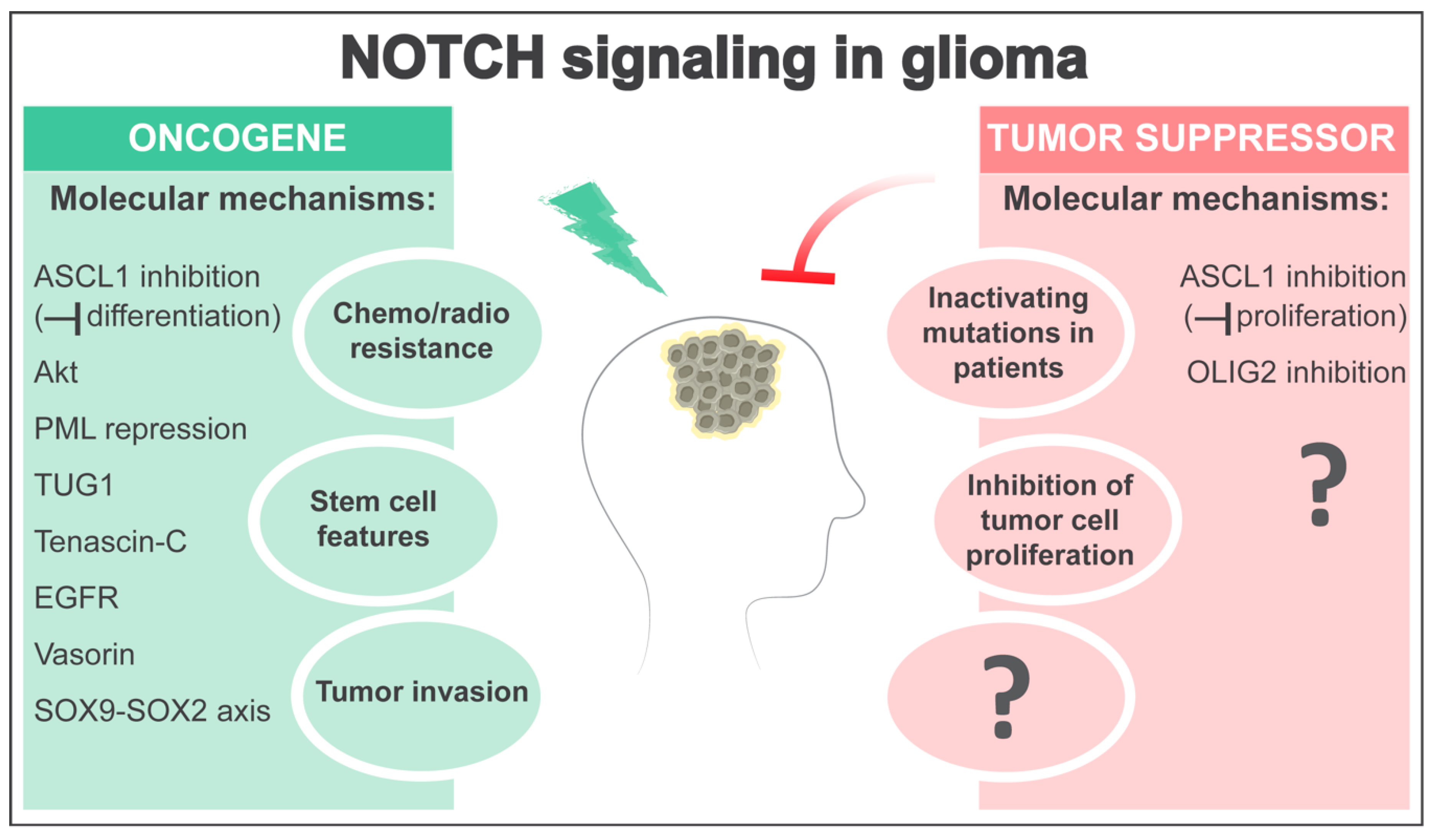
3. NOTCH as an Oncogene in Glioma
4. NOTCH as a Tumor Suppressor in Glioma
5. Open Questions and Perspectives
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Gessler, M. Delta-Notch–and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic. Acids. Res. 2007, 35, 4583–4596. [Google Scholar] [CrossRef] [PubMed]
- Pierfelice, T.; Alberi, L.; Gaiano, N. Notch in the vertebrate nervous system: An old dog with new tricks. Neuron 2011, 69, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Boareto, M.; Iber, D.; Taylor, V. Differential interactions between Notch and ID factors control neurogenesis by modulating Hes factor autoregulation. Development 2017, 144, 3465–3474. [Google Scholar] [CrossRef]
- Imayoshi, I.; Isomura, A.; Harima, Y.; Kawaguchi, K.; Kori, H.; Miyachi, H.; Fujiwara, T.; Ishidate, F.; Kageyama, R. Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 2013, 342, 1203–1208. [Google Scholar] [CrossRef]
- Bai, G.; Sheng, N.; Xie, Z.; Bian, W.; Yokota, Y.; Benezra, R.; Kageyama, R.; Guillemot, F.; Jing, N. Id sustains Hes1 expression to inhibit precocious neurogenesis by releasing negative autoregulation of Hes1. Dev. Cell 2007, 13, 283–297. [Google Scholar] [CrossRef]
- Nam, H.S.; Benezra, R. High levels of Id1 expression define B1 type adult neural stem cells. Cell Stem. Cell 2009, 5, 515–526. [Google Scholar] [CrossRef]
- Castel, D.; Mourikis, P.; Bartels, S.J.; Brinkman, A.B.; Tajbakhsh, S.; Stunnenberg, H.G. Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 2013, 27, 1059–1071. [Google Scholar] [CrossRef]
- Wang, H.; Zou, J.; Zhao, B.; Johannsen, E.; Ashworth, T.; Wong, H.; Pear, W.S.; Schug, J.; Blacklow, S.C.; Arnett, K.L.; et al. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14908–14913. [Google Scholar] [CrossRef]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navas, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.S.; et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953. [Google Scholar] [CrossRef] [PubMed]
- Nowell, C.S.; Radtke, F. Notch as a tumour suppressor. Nat. Rev. Cancer 2017, 17, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Lim, J.S.; Sage, J.; Aifantis, I. From fly wings to targeted cancer therapies: A centennial for notch signaling. Cancer Cell 2014, 25, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Egloff, A.M.; Grandis, J.R. Molecular pathways: Context-dependent approaches to Notch targeting as cancer therapy. Clin. Cancer Res. 2012, 18, 5188–5195. [Google Scholar] [CrossRef]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.T.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordonez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Bea, S.; Gonzalez-Diaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.A.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Q.; Li, D.; Ching, K.; Zhang, C.; Zheng, X.; Ozeck, M.; Shi, S.; Li, X.; Wang, H.; et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a gamma-secretase inhibitor. Clin. Cancer Res. 2015, 21, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Rampias, T.; Vgenopoulou, P.; Avgeris, M.; Polyzos, A.; Stravodimos, K.; Valavanis, C.; Scorilas, A.; Klinakis, A. A new tumor suppressor role for the Notch pathway in bladder cancer. Nat. Med. 2014, 20, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Harmanci, A.S.; Erson-Omay, E.Z.; Li, J.; Coskun, S.; Simon, M.; Krischek, B.; Ozduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef]
- Radtke, F.; Fasnacht, N.; Macdonald, H.R. Notch signaling in the immune system. Immunity 2010, 32, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Jhappan, C.; Gallahan, D.; Stahle, C.; Chu, E.; Smith, G.H.; Merlino, G.; Callahan, R. Expression of an activated Notch-related int-3 transgene interferes with cell differentiation and induces neoplastic transformation in mammary and salivary glands. Genes Dev. 1992, 6, 345–355. [Google Scholar] [CrossRef]
- Robbins, J.; Blondel, B.J.; Gallahan, D.; Callahan, R. Mouse mammary tumor gene int-3: A member of the notch gene family transforms mammary epithelial cells. J. Virol. 1992, 66, 2594–2599. [Google Scholar] [CrossRef]
- Klinakis, A.; Szabolcs, M.; Politi, K.; Kiaris, H.; Artavanis-Tsakonas, S.; Efstratiadis, A. Myc is a Notch1 transcriptional target and a requisite for Notch1-induced mammary tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9262–9267. [Google Scholar] [CrossRef]
- Miao, K.; Lei, J.H.; Valecha, M.V.; Zhang, A.; Xu, J.; Wang, L.; Lyu, X.; Chen, S.; Miao, Z.; Zhang, X.; et al. NOTCH1 activation compensates BRCA1 deficiency and promotes triple-negative breast cancer formation. Nat. Commun. 2020, 11, 3256. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef]
- Lefort, K.; Mandinova, A.; Ostano, P.; Kolev, V.; Calpini, V.; Kolfschoten, I.; Devgan, V.; Lieb, J.; Raffoul, W.; Hohl, D.; et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007, 21, 562–577. [Google Scholar] [CrossRef]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Sriuranpong, V.; Borges, M.W.; Ravi, R.K.; Arnold, D.R.; Nelkin, B.D.; Baylin, S.B.; Ball, D.W. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001, 61, 3200–3205. [Google Scholar] [PubMed]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Ouadah, Y.; Rojas, E.R.; Riordan, D.P.; Capostagno, S.; Kuo, C.S.; Krasnow, M.A. Rare Pulmonary Neuroendocrine Cells Are Stem Cells Regulated by Rb, p53, and Notch. Cell 2019, 179, 403–416.e23. [Google Scholar] [CrossRef]
- Kimura, F.; Florl, A.R.; Seifert, H.H.; Louhelainen, J.; Maas, S.; Knowles, M.A.; Schulz, W.A. Destabilization of chromosome 9 in transitional cell carcinoma of the urinary bladder. Br. J. Cancer 2001, 85, 1887–1893. [Google Scholar] [CrossRef]
- Loganathan, S.K.; Schleicher, K.; Malik, A.; Quevedo, R.; Langille, E.; Teng, K.; Oh, R.H.; Rathod, B.; Tsai, R.; Samavarchi-Tehrani, P.; et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 2020, 367, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Xia, R.; Li, J.; Long, Z.; Ren, H.; Chen, W.; Mao, L. Common and complex Notch1 mutations in Chinese oral squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Klinakis, A.; Lobry, C.; Abdel-Wahab, O.; Oh, P.; Haeno, H.; Buonamici, S.; van De Walle, I.; Cathelin, S.; Trimarchi, T.; Araldi, E.; et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature 2011, 473, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Lobry, C.; Ntziachristos, P.; Ndiaye-Lobry, D.; Oh, P.; Cimmino, L.; Zhu, N.; Araldi, E.; Hu, W.; Freund, J.; Abdel-Wahab, O.; et al. Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J. Exp. Med. 2013, 210, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.J.; Bhandoola, A. The earliest thymic progenitors for T cells possess myeloid lineage potential. Nature 2008, 452, 764–767. [Google Scholar] [CrossRef]
- Feyerabend, T.B.; Terszowski, G.; Tietz, A.; Blum, C.; Luche, H.; Gossler, A.; Gale, N.W.; Radtke, F.; Fehling, H.J.; Rodewald, H.R. Deletion of Notch1 converts pro-T cells to dendritic cells and promotes thymic B cells by cell-extrinsic and cell-intrinsic mechanisms. Immunity 2009, 30, 67–79. [Google Scholar] [CrossRef]
- Wada, H.; Masuda, K.; Satoh, R.; Kakugawa, K.; Ikawa, T.; Katsura, Y.; Kawamoto, H. Adult T-cell progenitors retain myeloid potential. Nature 2008, 452, 768–772. [Google Scholar] [CrossRef]
- Kim, Y.W.; Koo, B.K.; Jeong, H.W.; Yoon, M.J.; Song, R.; Shin, J.; Jeong, D.C.; Kim, S.H.; Kong, Y.Y. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood 2008, 112, 4628–4638. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, H.; Rodriguez, S.; Cao, L.; Parish, J.; Mumaw, C.; Zollman, A.; Kamoka, M.M.; Mu, J.; Chen, D.Z.; et al. Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-kappaB-dependent manner. Cell Stem. Cell 2014, 15, 51–65. [Google Scholar] [CrossRef]
- Yao, D.; Huang, Y.; Huang, X.; Wang, W.; Yan, Q.; Wei, L.; Xin, W.; Gerson, S.; Stanley, P.; Lowe, J.B.; et al. Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood 2011, 117, 5652–5662. [Google Scholar] [CrossRef]
- Yoda, M.; Kimura, T.; Tohmonda, T.; Uchikawa, S.; Koba, T.; Takito, J.; Morioka, H.; Matsumoto, M.; Link, D.C.; Chiba, K.; et al. Dual functions of cell-autonomous and non-cell-autonomous ADAM10 activity in granulopoiesis. Blood 2011, 118, 6939–6942. [Google Scholar] [CrossRef] [PubMed]
- Licciulli, S.; Avila, J.L.; Hanlon, L.; Troutman, S.; Cesaroni, M.; Kota, S.; Keith, B.; Simon, M.C.; Puré, E.; Radtke, F.; et al. Notch1 is required for Kras-induced lung adenocarcinoma and controls tumor cell survival via p53. Cancer Res. 2013, 73, 5974–5984. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, B.; Colaluca, I.N.; D’Ario, G.; Donzelli, M.; Tosoni, D.; Volorio, S.; Pelosi, G.; Spaggiari, L.; Mazzarol, G.; Viale, G.; et al. Alterations of the Notch pathway in lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 22293–22298. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; de la Cruz, C.C.; Sayles, L.C.; Alleyne-Chin, C.; Vaka, D.; Knaak, T.D.; Bigos, M.; Xu, Y.; Hoang, C.D.; Shrager, J.B.; et al. A rare population of CD24(+)ITGB4(+)Notch(hi) cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell 2013, 24, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C.; et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro. Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Reifenberger, G.; Wirsching, H.G.; Knobbe-Thomsen, C.B.; Weller, M. Advances in the molecular genetics of gliomas—Implications for classification and therapy. Nat. Rev. Clin. Oncol. 2017, 14, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.; Sun, D.; Pedraza, A.M.; Vera, E.; Wang, Z.; Burns, D.K.; Parada, L.F. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat. Neurosci. 2019, 22, 545–555. [Google Scholar] [CrossRef]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef]
- Bond, A.M.; Ming, G.L.; Song, H. Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem. Cell 2015, 17, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Giachino, C.; Taylor, V. Notching up neural stem cell homogeneity in homeostasis and disease. Front. Neurosci. 2014, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H.; Temple, S. Neural stem cells: Generating and regenerating the brain. Neuron 2013, 80, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Gotz, M.; Nakafuku, M.; Petrik, D. Neurogenesis in the Developing and Adult Brain-Similarities and Key Differences. Cold Spring Harb. Perspect Biol. 2016, 8. [Google Scholar] [CrossRef]
- Navarro Negredo, P.; Yeo, R.W.; Brunet, A. Aging and Rejuvenation of Neural Stem Cells and Their Niches. Cell Stem. Cell 2020, 27, 202–223. [Google Scholar] [CrossRef]
- Andreu-Agullo, C.; Morante-Redolat, J.M.; Delgado, A.C.; Farinas, I. Vascular niche factor PEDF modulates Notch-dependent stemness in the adult subependymal zone. Nat. Neurosci. 2009, 12, 1514–1523. [Google Scholar] [CrossRef]
- Ehm, O.; Goritz, C.; Covic, M.; Schaffner, I.; Schwarz, T.J.; Karaca, E.; Kempkes, B.; Kremmer, E.; Pfrieger, F.W.; Espinosa, L.; et al. RBPJkappa-dependent signaling is essential for long-term maintenance of neural stem cells in the adult hippocampus. J. Neurosci. 2010, 30, 13794–13807. [Google Scholar] [CrossRef]
- Imayoshi, I.; Sakamoto, M.; Yamaguchi, M.; Mori, K.; Kageyama, R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J. Neurosci. 2010, 30, 3489–3498. [Google Scholar] [CrossRef]
- Mizutani, K.; Yoon, K.; Dang, L.; Tokunaga, A.; Gaiano, N. Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature 2007, 449, 351–355. [Google Scholar] [CrossRef]
- Lugert, S.; Basak, O.; Knuckles, P.; Haussler, U.; Fabel, K.; Gotz, M.; Haas, C.A.; Kempermann, G.; Taylor, V.; Giachino, C. Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem. Cell 2010, 6, 445–456. [Google Scholar] [CrossRef]
- Lugert, S.; Vogt, M.; Tchorz, J.S.; Muller, M.; Giachino, C.; Taylor, V. Homeostatic neurogenesis in the adult hippocampus does not involve amplification of Ascl1(high) intermediate progenitors. Nat. Commun. 2012, 3, 670. [Google Scholar] [CrossRef] [PubMed]
- Giachino, C.; Basak, O.; Lugert, S.; Knuckles, P.; Obernier, K.; Fiorelli, R.; Frank, S.; Raineteau, O.; Alvarez-Buylla, A.; Taylor, V. Molecular diversity subdivides the adult forebrain neural stem cell population. Stem. Cells 2014, 32, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.; Rolando, C.; Giachino, C.; Saotome, I.; Erni, A.; Brien, C.; Zhang, R.; Zimber-Strobl, U.; Radtke, F.; Artavanis-Tsakonas, S.; et al. Notch2 Signaling Maintains NSC Quiescence in the Murine Ventricular-Subventricular Zone. Cell Rep. 2018, 22, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Ables, J.L.; Decarolis, N.A.; Johnson, M.A.; Rivera, P.D.; Gao, Z.; Cooper, D.C.; Radtke, F.; Hsieh, J.; Eisch, A.J. Notch1 is required for maintenance of the reservoir of adult hippocampal stem cells. J. Neurosci. 2010, 30, 10484–10492. [Google Scholar] [CrossRef]
- Basak, O.; Giachino, C.; Fiorini, E.; Macdonald, H.R.; Taylor, V. Neurogenic subventricular zone stem/progenitor cells are Notch1-dependent in their active but not quiescent state. J. Neurosci. 2012, 32, 5654–5666. [Google Scholar] [CrossRef]
- Kawai, H.; Kawaguchi, D.; Kuebrich, B.D.; Kitamoto, T.; Yamaguchi, M.; Gotoh, Y.; Furutachi, S. Area-Specific Regulation of Quiescent Neural Stem Cells by Notch3 in the Adult Mouse Subependymal Zone. J. Neurosci. 2017, 37, 11867–11880. [Google Scholar] [CrossRef]
- Zhang, R.; Boareto, M.; Engler, A.; Louvi, A.; Giachino, C.; Iber, D.; Taylor, V. Id4 Downstream of Notch2 Maintains Neural Stem Cell Quiescence in the Adult Hippocampus. Cell Rep. 2019, 28, 1485–1498.e6. [Google Scholar] [CrossRef]
- Sueda, R.; Imayoshi, I.; Harima, Y.; Kageyama, R. High Hes1 expression and resultant Ascl1 suppression regulate quiescent vs. active neural stem cells in the adult mouse brain. Genes Dev. 2019, 33, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; Urban, N.; Achimastou, A.; Ito, A.; Simic, M.; Ullom, K.; Martynoga, B.; Lebel, M.; Goritz, C.; Frisen, J.; et al. A transcriptional mechanism integrating inputs from extracellular signals to activate hippocampal stem cells. Neuron 2014, 83, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Urban, N.; van den Berg, D.L.; Forget, A.; Andersen, J.; Demmers, J.A.; Hunt, C.; Ayrault, O.; Guillemot, F. Return to quiescence of mouse neural stem cells by degradation of a proactivation protein. Science 2016, 353, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Blomfield, I.M.; Rocamonde, B.; Masdeu, M.D.M.; Mulugeta, E.; Vaga, S.; van den Berg, D.L.; Huillard, E.; Guillemot, F.; Urban, N. Id4 promotes the elimination of the pro-activation factor Ascl1 to maintain quiescence of adult hippocampal stem cells. eLife 2019, 8. [Google Scholar] [CrossRef]
- Ge, W.; Martinowich, K.; Wu, X.; He, F.; Miyamoto, A.; Fan, G.; Weinmaster, G.; Sun, Y.E. Notch signaling promotes astrogliogenesis via direct CSL-mediated glial gene activation. J. Neurosci. Res. 2002, 69, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Anthony, T.E.; Mason, H.A.; Gridley, T.; Fishell, G.; Heintz, N. Brain lipid-binding protein is a direct target of Notch signaling in radial glial cells. Genes Dev. 2005, 19, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Deneen, B.; Ho, R.; Lukaszewicz, A.; Hochstim, C.J.; Gronostajski, R.M.; Anderson, D.J. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 2006, 52, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Benner, E.J.; Luciano, D.; Jo, R.; Abdi, K.; Paez-Gonzalez, P.; Sheng, H.; Warner, D.S.; Liu, C.; Eroglu, C.; Kuo, C.T. Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature 2013, 497, 369–373. [Google Scholar] [CrossRef]
- Magnusson, J.P.; Goritz, C.; Tatarishvili, J.; Dias, D.O.; Smith, E.M.; Lindvall, O.; Kokaia, Z.; Frisen, J. A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science 2014, 346, 237–241. [Google Scholar] [CrossRef]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef]
- John, G.R.; Shankar, S.L.; Shafit-Zagardo, B.; Massimi, A.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Multiple sclerosis: Re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat. Med. 2002, 8, 1115–1121. [Google Scholar] [CrossRef]
- Wang, S.; Sdrulla, A.D.; diSibio, G.; Bush, G.; Nofziger, D.; Hicks, C.; Weinmaster, G.; Barres, B.A. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 1998, 21, 63–75. [Google Scholar] [CrossRef]
- Hu, Q.D.; Ang, B.T.; Karsak, M.; Hu, W.P.; Cui, X.Y.; Duka, T.; Takeda, Y.; Chia, W.; Sankar, N.; Ng, Y.K.; et al. F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell 2003, 115, 163–175. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.R.; Wang, Z.; Sun, D.; Chen, J.; Xu, J.; Kim, E.; Hatanpaa, K.J.; Raisanen, J.M.; Burns, D.K.; Johnson, J.E.; et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 2015, 28, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Bachoo, R.M.; Maher, E.A.; Ligon, K.L.; Sharpless, N.E.; Chan, S.S.; You, M.J.; Tang, Y.; DeFrances, J.; Stover, E.; Weissleder, R.; et al. Epidermal growth factor receptor and Ink4a/Arf: Convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 2002, 1, 269–277. [Google Scholar] [CrossRef]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef]
- Cuevas, I.C.; Slocum, A.L.; Jun, P.; Costello, J.F.; Bollen, A.W.; Riggins, G.J.; McDermott, M.W.; Lal, A. Meningioma transcript profiles reveal deregulated Notch signaling pathway. Cancer Res. 2005, 65, 5070–5075. [Google Scholar] [CrossRef]
- Fan, X.; Mikolaenko, I.; Elhassan, I.; Ni, X.; Wang, Y.; Ball, D.; Brat, D.J.; Perry, A.; Eberhart, C.G. Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004, 64, 7787–7793. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Purow, B.W.; Haque, R.M.; Noel, M.W.; Su, Q.; Burdick, M.J.; Lee, J.; Sundaresan, T.; Pastorino, S.; Park, J.K.; Mikolaenko, I.; et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005, 65, 2353–2363. [Google Scholar] [CrossRef]
- Shih, A.H.; Holland, E.C. Notch signaling enhances nestin expression in gliomas. Neoplasia 2006, 8, 1072–1082. [Google Scholar] [CrossRef]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem. Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Giachino, C.; Boulay, J.L.; Ivanek, R.; Alvarado, A.; Tostado, C.; Lugert, S.; Tchorz, J.; Coban, M.; Mariani, L.; Bettler, B.; et al. A Tumor Suppressor Function for Notch Signaling in Forebrain Tumor Subtypes. Cancer Cell 2015, 28, 730–742. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem. Cells 2010, 28, 5–16. [Google Scholar] [CrossRef]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem. Cells 2010, 28, 17–28. [Google Scholar] [CrossRef]
- Xie, Q.; Wu, Q.; Kim, L.; Miller, T.E.; Liau, B.B.; Mack, S.C.; Yang, K.; Factor, D.C.; Fang, X.; Huang, Z.; et al. RBPJ maintains brain tumor-initiating cells through CDK9-mediated transcriptional elongation. J. Clin. Investig. 2016, 126, 2757–2772. [Google Scholar] [CrossRef]
- Natarajan, S.; Li, Y.; Miller, E.E.; Shih, D.J.; Taylor, M.D.; Stearns, T.M.; Bronson, R.T.; Ackerman, S.L.; Yoon, J.K.; Yun, K. Notch1-induced brain tumor models the sonic hedgehog subgroup of human medulloblastoma. Cancer Res. 2013, 73, 5381–5390. [Google Scholar] [CrossRef]
- Pierfelice, T.J.; Schreck, K.C.; Dang, L.; Asnaghi, L.; Gaiano, N.; Eberhart, C.G. Notch3 activation promotes invasive glioma formation in a tissue site-specific manner. Cancer Res. 2011, 71, 1115–1125. [Google Scholar] [CrossRef]
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; Van Der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J.; et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem. Cells 2010, 28, 1019–1029. [Google Scholar] [CrossRef]
- Wu, H.C.; Lin, Y.C.; Liu, C.H.; Chung, H.C.; Wang, Y.T.; Lin, Y.W.; Ma, H.I.; Tu, P.H.; Lawler, S.E.; Chen, R.H. USP11 regulates PML stability to control Notch-induced malignancy in brain tumours. Nat. Commun. 2014, 5, 3214. [Google Scholar] [CrossRef]
- Katsushima, K.; Natsume, A.; Ohka, F.; Shinjo, K.; Hatanaka, A.; Ichimura, N.; Sato, S.; Takahashi, S.; Kimura, H.; Totoki, Y.; et al. Targeting the Notch-regulated non-coding RNA TUG1 for glioma treatment. Nat. Commun. 2016, 7, 13616. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Mirzaei, R.; Zemp, F.J.; Wei, W.; Senger, D.L.; Robbins, S.M.; Yong, V.W. Activation of NOTCH Signaling by Tenascin-C Promotes Growth of Human Brain Tumor-Initiating Cells. Cancer Res. 2017, 77, 3231–3243. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, E.; Taylor, V.; Giachino, C. Tenascin-C induces expression of the Notch ligand Jagged1 to promote glioma growth. Transl. Cancer Res. 2017, 6, S1057–S1060. [Google Scholar] [CrossRef]
- Sivasankaran, B.; Degen, M.; Ghaffari, A.; Hegi, M.E.; Hamou, M.F.; Ionescu, M.C.; Zweifel, C.; Tolnay, M.; Wasner, M.; Mergenthaler, S.; et al. Tenascin-C is a novel RBPJkappa-induced target gene for Notch signaling in gliomas. Cancer Res. 2009, 69, 458–465. [Google Scholar] [CrossRef]
- Lim, K.J.; Brandt, W.D.; Heth, J.A.; Muraszko, K.M.; Fan, X.; Bar, E.E.; Eberhart, C.G. Lateral inhibition of Notch signaling in neoplastic cells. Oncotarget 2015, 6, 1666–1677. [Google Scholar] [CrossRef]
- Doetsch, F.; Caille, I.; Lim, D.A.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef]
- Doetsch, F.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Regeneration of a germinal layer in the adult mammalian brain. Proc. Natl. Acad. Sci. USA 1999, 96, 11619–11624. [Google Scholar] [CrossRef]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem. Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef]
- Eyler, C.E.; Matsunaga, H.; Hovestadt, V.; Vantine, S.J.; van Galen, P.; Bernstein, B.E. Single-cell lineage analysis reveals genetic and epigenetic interplay in glioblastoma drug resistance. Genome Biol. 2020, 21, 174. [Google Scholar] [CrossRef]
- Park, N.I.; Guilhamon, P.; Desai, K.; McAdam, R.F.; Langille, E.; O’Connor, M.; Lan, X.; Whetstone, H.; Coutinho, F.J.; Vanner, R.J.; et al. ASCL1 Reorganizes Chromatin to Direct Neuronal Fate and Suppress Tumorigenicity of Glioblastoma Stem Cells. Cell Stem. Cell 2017, 21, 411. [Google Scholar] [CrossRef]
- Rajakulendran, N.; Rowland, K.J.; Selvadurai, H.J.; Ahmadi, M.; Park, N.I.; Naumenko, S.; Dolma, S.; Ward, R.J.; So, M.; Lee, L.; et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 2019, 33, 498–510. [Google Scholar] [CrossRef]
- Shen, Q.; Wang, Y.; Kokovay, E.; Lin, G.; Chuang, S.M.; Goderie, S.K.; Roysam, B.; Temple, S. Adult SVZ stem cells lie in a vascular niche: A quantitative analysis of niche cell-cell interactions. Cell Stem. Cell 2008, 3, 289–300. [Google Scholar] [CrossRef]
- Tavazoie, M.; Van der Veken, L.; Silva-Vargas, V.; Louissaint, M.; Colonna, L.; Zaidi, B.; Garcia-Verdugo, J.M.; Doetsch, F. A specialized vascular niche for adult neural stem cells. Cell Stem. Cell 2008, 3, 279–288. [Google Scholar] [CrossRef]
- Ottone, C.; Krusche, B.; Whitby, A.; Clements, M.; Quadrato, G.; Pitulescu, M.E.; Adams, R.H.; Parrinello, S. Direct cell-cell contact with the vascular niche maintains quiescent neural stem cells. Nat. Cell Biol. 2014, 16, 1045–1056. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Man, J.; Yu, X.; Huang, H.; Zhou, W.; Xiang, C.; Miele, L.; Liu, Z.; Bebek, G.; Bao, S.; Yu, J.S. Hypoxic Induction of Vasorin Regulates Notch1 Turnover to Maintain Glioma Stem-like Cells. Cell Stem. Cell 2018, 22, 104–118.e6. [Google Scholar] [CrossRef]
- Rusu, P.; Shao, C.; Neuerburg, A.; Acikgoz, A.A.; Wu, Y.; Zou, P.; Phapale, P.; Shankar, T.S.; Doring, K.; Dettling, S.; et al. GPDfile. Cell Stem. Cell 2019, 25, 241–257.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, S.L.; Duan, J.J.; Yi, L.; Guo, Y.F.; Shi, Y.; Li, L.; Yang, Z.Y.; Liao, X.M.; Cai, J.; et al. Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1-SOX2 positive-feedback loop. Nat. Neurosci. 2019, 22, 91–105. [Google Scholar] [CrossRef]
- Bettegowda, C.; Agrawal, N.; Jiao, Y.; Sausen, M.; Wood, L.D.; Hruban, R.H.; Rodriguez, F.J.; Cahill, D.P.; McLendon, R.; Riggins, G.; et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011, 333, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- Halani, S.H.; Yousefi, S.; Velazquez Vega, J.; Rossi, M.R.; Zhao, Z.; Amrollahi, F.; Holder, C.A.; Baxter-Stoltzfus, A.; Eschbacher, J.; Griffith, B.; et al. Multi-faceted computational assessment of risk and progression in oligodendroglioma implicates NOTCH and PI3K pathways. Npj Precis Oncol. 2018, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Nakamura, H.; Suzuki, H.; Matsuo, K.; Kataoka, K.; Shimamura, T.; Motomura, K.; Ohka, F.; Shiina, S.; Yamamoto, T.; et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 2018, 20, 66–77. [Google Scholar] [CrossRef]
- Boulay, J.L.; Miserez, A.R.; Zweifel, C.; Sivasankaran, B.; Kana, V.; Ghaffari, A.; Luyken, C.; Sabel, M.; Zerrouqi, A.; Wasner, M.; et al. Loss of NOTCH2 positively predicts survival in subgroups of human glial brain tumors. PLoS ONE 2007, 2, e576. [Google Scholar] [CrossRef]
- Kondo, T. Molecular mechanisms involved in gliomagenesis. Brain Tumor. Pathol. 2017, 34, 1–7. [Google Scholar] [CrossRef]
- Ge, X.; Xiao, G.; Huang, H.; Du, J.; Tao, Y.; Yang, A.; Wu, H.; Zhang, Z.; Qiu, M. Stage-dependent regulation of oligodendrocyte development and enhancement of myelin repair by dominant negative Master-mind 1 protein. Glia 2019, 67, 1654–1666. [Google Scholar] [CrossRef]
- Galvao, R.P.; Kasina, A.; McNeill, R.S.; Harbin, J.E.; Foreman, O.; Verhaak, R.G.; Nishiyama, A.; Miller, C.R.; Zong, H. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc. Natl. Acad. Sci. USA 2014, 111, E4214–E4223. [Google Scholar] [CrossRef]
- Ichimura, K.; Vogazianou, A.P.; Liu, L.; Pearson, D.M.; Backlund, L.M.; Plant, K.; Baird, K.; Langford, C.F.; Gregory, S.G.; Collins, V.P. 1p36 is a preferential target of chromosome 1 deletions in astrocytic tumours and homozygously deleted in a subset of glioblastomas. Oncogene 2008, 27, 2097–2108. [Google Scholar] [CrossRef]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L.; et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Urban, N.; Blomfield, I.M.; Guillemot, F. Quiescence of Adult Mammalian Neural Stem Cells: A Highly Regulated Rest. Neuron 2019, 104, 834–848. [Google Scholar] [CrossRef]
- Somasundaram, K.; Reddy, S.P.; Vinnakota, K.; Britto, R.; Subbarayan, M.; Nambiar, S.; Hebbar, A.; Samuel, C.; Shetty, M.; Sreepathi, H.K.; et al. Upregulation of ASCL1 and inhibition of Notch signaling pathway characterize progressive astrocytoma. Oncogene 2005, 24, 7073–7083. [Google Scholar] [CrossRef]
- Narayanan, A.; Gagliardi, F.; Gallotti, A.L.; Mazzoleni, S.; Cominelli, M.; Fagnocchi, L.; Pala, M.; Piras, I.S.; Zordan, P.; Moretta, N.; et al. The proneural gene ASCL1 governs the transcriptional subgroup affiliation in glioblastoma stem cells by directly repressing the mesenchymal gene NDRG1. Cell Death Differ. 2019, 26, 1813–1831. [Google Scholar] [CrossRef]
- Rheinbay, E.; Suva, M.L.; Gillespie, S.M.; Wakimoto, H.; Patel, A.P.; Shahid, M.; Oksuz, O.; Rabkin, S.D.; Martuza, R.L.; Rivera, M.N.; et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013, 3, 1567–1579. [Google Scholar] [CrossRef]
- Vue, T.Y.; Kollipara, R.K.; Borromeo, M.D.; Smith, T.; Mashimo, T.; Burns, D.K.; Bachoo, R.M.; Johnson, J.E. ASCL1 regulates neurodevelopmental transcription factors and cell cycle genes in brain tumors of glioma mouse models. Glia 2020. [Google Scholar] [CrossRef]
- Barone, C.; Buccarelli, M.; Alessandrini, F.; Pagin, M.; Rigoldi, L.; Sambruni, I.; Favaro, R.; Ottolenghi, S.; Pallini, R.; Ricci-Vitiani, L.; et al. Sox2-dependent maintenance of mouse oligodendroglioma involves the Sox2-mediated downregulation of Cdkn2b, Ebf1, Zfp423, and Hey2. Glia 2020. [Google Scholar] [CrossRef] [PubMed]
- Havrda, M.C.; Paolella, B.R.; Ran, C.; Jering, K.S.; Wray, C.M.; Sullivan, J.M.; Nailor, A.; Hitoshi, Y.; Israel, M.A. Id2 mediates oligodendrocyte precursor cell maturation arrest and is tumorigenic in a PDGF-rich microenvironment. Cancer Res. 2014, 74, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Huillard, E.; Kesari, S.; Maire, C.L.; Golebiowski, D.; Harrington, E.P.; Alberta, J.A.; Kane, M.F.; Theisen, M.; Ligon, K.L.; et al. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell 2011, 19, 359–371. [Google Scholar] [CrossRef]
- Chow, R.D.; Guzman, C.D.; Wang, G.; Schmidt, F.; Youngblood, M.W.; Ye, L.; Errami, Y.; Dong, M.B.; Martinez, M.A.; Zhang, S.; et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 2017, 20, 1329–1341. [Google Scholar] [CrossRef]
- Andersson, E.R.; Lendahl, U. Therapeutic modulation of Notch signalling--are we there yet? Nat. Rev. Drug Discov. 2014, 13, 357–378. [Google Scholar] [CrossRef]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J.; et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef]
- Guner, G.; Lichtenthaler, S.F. The substrate repertoire of gamma-secretase/presenilin. Semin Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Lehal, R.; Zaric, J.; Vigolo, M.; Urech, C.; Frismantas, V.; Zangger, N.; Cao, L.; Berger, A.; Chicote, I.; Loubery, S.; et al. Pharmacological disruption of the Notch transcription factor complex. Proc. Natl. Acad. Sci. USA 2020, 117, 16292–16301. [Google Scholar] [CrossRef]
- Ran, Y.; Hossain, F.; Pannuti, A.; Lessard, C.B.; Ladd, G.Z.; Jung, J.I.; Minter, L.M.; Osborne, B.A.; Miele, L.; Golde, T.E. gamma-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. Embo Mol. Med. 2017, 9, 950–966. [Google Scholar] [CrossRef]
- Saito, N.; Hirai, N.; Aoki, K.; Suzuki, R.; Fujita, S.; Nakayama, H.; Hayashi, M.; Ito, K.; Sakurai, T.; Iwabuchi, S. The Oncogene Addiction Switch from NOTCH to PI3K Requires Simultaneous Targeting of NOTCH and PI3K Pathway Inhibition in Glioblastoma. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Zhang, C.; Martinez-Ledesma, E.; Gao, F.; Zhang, W.; Ding, J.; Wu, S.; Li, X.; Wu, J.; Yuan, Y.; Koul, D.; et al. Wild-type TP53 defined gamma-secretase inhibitor sensitivity and synergistic activity with doxorubicin in GSCs. Am. J. Cancer Res. 2019, 9, 1734–1745. [Google Scholar]
- Fassl, A.; Tagscherer, K.E.; Richter, J.; Berriel Diaz, M.; Alcantara Llaguno, S.R.; Campos, B.; Kopitz, J.; Herold-Mende, C.; Herzig, S.; Schmidt, M.H.; et al. Notch1 signaling promotes survival of glioblastoma cells via EGFR-mediated induction of anti-apoptotic Mcl-1. Oncogene 2012, 31, 4698–4708. [Google Scholar] [CrossRef]
- Yin, J.; Park, G.; Kim, T.H.; Hong, J.H.; Kim, Y.J.; Jin, X.; Kang, S.; Jung, J.E.; Kim, J.Y.; Yun, H.; et al. Pigment Epithelium-Derived Factor (PEDF) Expression Induced by EGFRvIII Promotes Self-renewal and Tumor Progression of Glioma Stem Cells. PLoS Biol. 2015, 13, e1002152. [Google Scholar] [CrossRef]
- Purow, B.W.; Sundaresan, T.K.; Burdick, M.J.; Kefas, B.A.; Comeau, L.D.; Hawkinson, M.P.; Su, Q.; Kotliarov, Y.; Lee, J.; Zhang, W.; et al. Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis 2008, 29, 918–925. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Lu, F.; Chen, Y.; Zhao, C.; Wang, H.; He, D.; Xu, L.; Wang, J.; He, X.; Deng, Y.; Lu, E.E.; et al. Olig2-Dependent Reciprocal Shift in PDGF and EGF Receptor Signaling Regulates Tumor Phenotype and Mitotic Growth in Malignant Glioma. Cancer Cell 2016, 29, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, M.P.; Greulich, P.; Wabik, A.; Frede, J.; Simons, B.D.; Jones, P.H. Differentiation imbalance in single oesophageal progenitor cells causes clonal immortalization and field change. Nat. Cell Biol. 2014, 16, 615–622. [Google Scholar] [CrossRef]
- Bowling, S.; Lawlor, K.; Rodriguez, T.A. Cell competition: The winners and losers of fitness selection. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Friebel, E.; Kapolou, K.; Unger, S.; Nunez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parmigiani, E.; Taylor, V.; Giachino, C. Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma. Cells 2020, 9, 2304. https://doi.org/10.3390/cells9102304
Parmigiani E, Taylor V, Giachino C. Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma. Cells. 2020; 9(10):2304. https://doi.org/10.3390/cells9102304
Chicago/Turabian StyleParmigiani, Elena, Verdon Taylor, and Claudio Giachino. 2020. "Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma" Cells 9, no. 10: 2304. https://doi.org/10.3390/cells9102304
APA StyleParmigiani, E., Taylor, V., & Giachino, C. (2020). Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma. Cells, 9(10), 2304. https://doi.org/10.3390/cells9102304