Notch Signaling Function in the Angiocrine Regulation of Tumor Development



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: Angiogenesis
2. Angiocrine Signaling
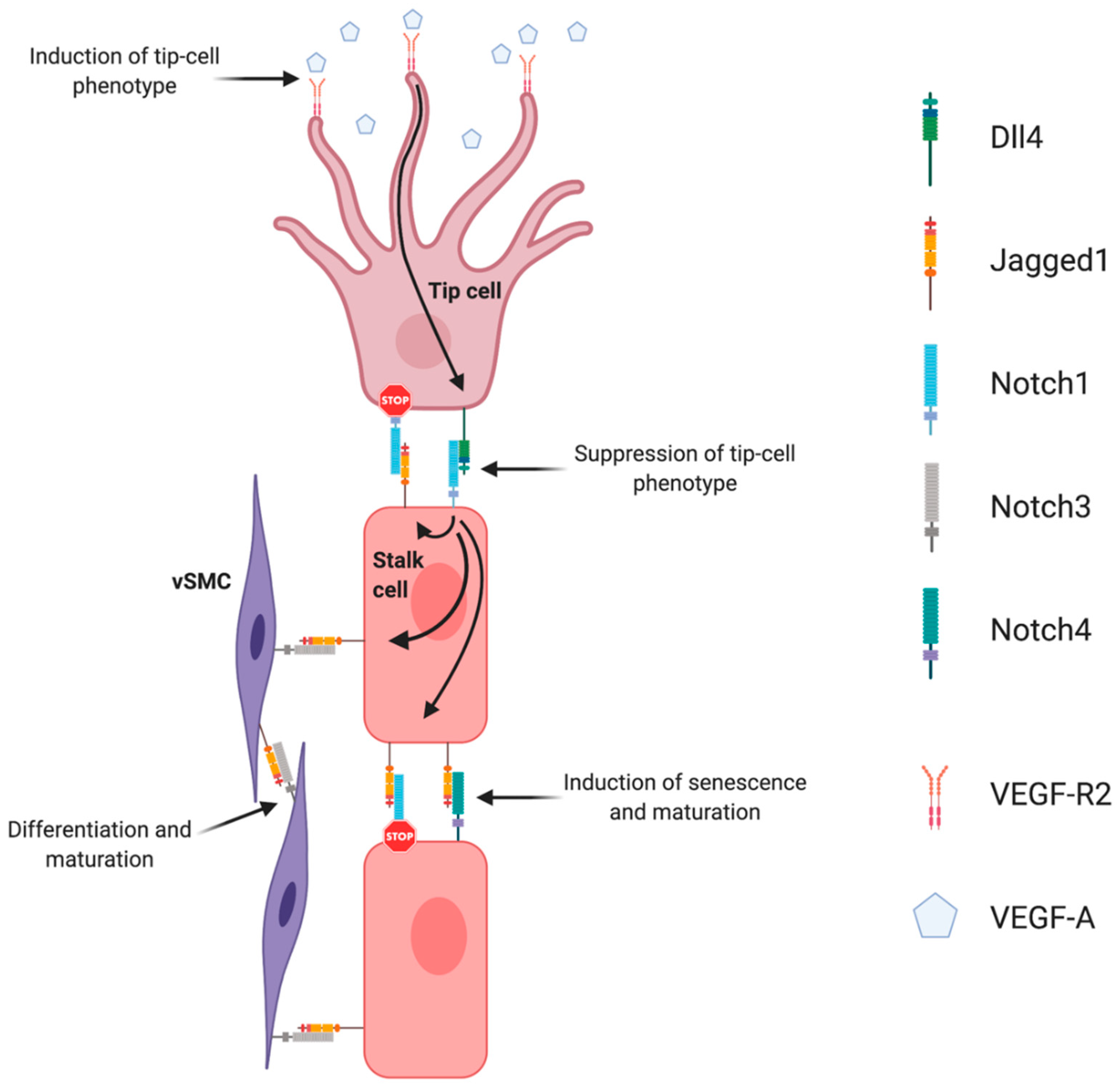
3. Notch Signaling Pathway in Angiogenesis and Tumoral Neoangiogenesis
4. Notch Signaling Function in Solid Tumors
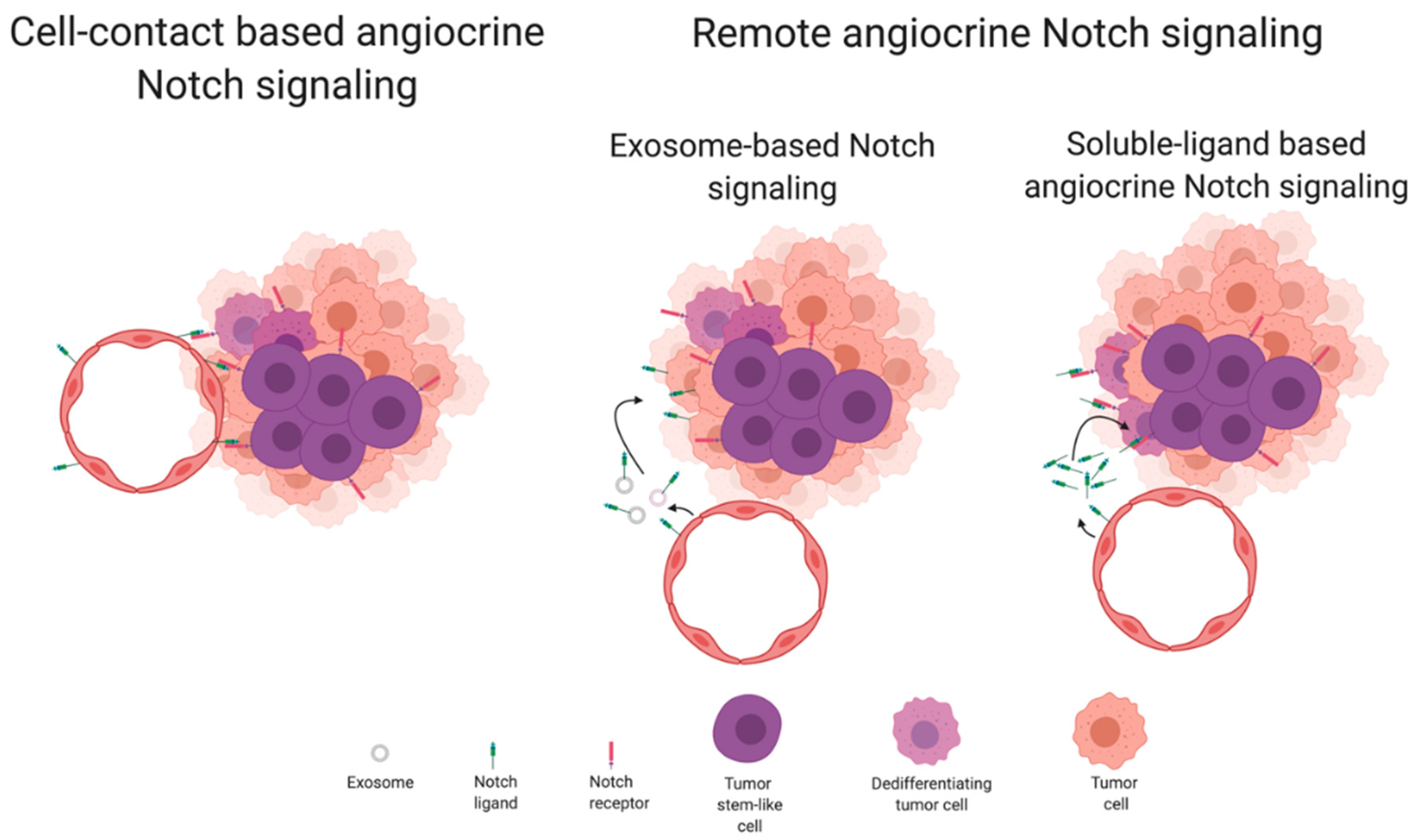
5. Angiocrine Functions of Notch Signaling
5.1. Remote Angiocrine Notch Signaling in Cancer
5.2. Cell-Contact-Based Angiocrine Notch Signaling in Cancer
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hunter, J. A Treatise on the Blood, Inflammation, and Gun-Shot Wounds. Clin. Orthop. Relat. Res. 1794. [Google Scholar] [CrossRef]
- Flamme, I.; Frölich, T.; Risau, W. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J. Cell Physiol. 1997, 173, 206–210. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Karamysheva, A.F. Mechanisms of angiogenesis. Biochemistry 2008, 73, 751. [Google Scholar] [CrossRef] [PubMed]
- Moses, M.A. The Regulation of Neovascularization by Matrix Metalloproteinases and Their Inhibitors. Stem Cells 1997, 15, 180–189. [Google Scholar] [CrossRef]
- Papetti, M.; Herman, I.M. Mechanisms of normal and tumor-derived angiogenesis. Am. J. Physiol. Cell Physiol. 2002, 282, 947–970. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Fraser, H.M.; Lunn, S.F. Angiogenesis and its control in the female reproductive system. Br. Med. Bull. 2000, 56, 787–797. [Google Scholar] [CrossRef]
- Arbiser, J.L. Angiogenesis and the skin: A primer. J. Am. Acad. Dermatol. 1996, 34, 486–497. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- ALGIRE, G.H. An adaptation of the transparent-chamber technique to the mouse. J. Natl. Cancer Inst. 1943, 4, 1–11. [Google Scholar]
- Ide, A.G. Vascularization of the Brown Pearce rabbit epithelioma transplant as seen in the transparent ear chamber. Am. J. Roentg. 1939, 42, 891–899. [Google Scholar]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Kerbel, R.S. Antiangiogenic therapy: A universal chemosensitization strategy for cancer? Science 2006, 312, 1171–1175. [Google Scholar] [CrossRef]
- Meadows, K.L.; Hurwitz, H.I. Anti-VEGF Therapies in the Clinic. Cold Spring Harb. Perspect. Med. 2012, 2, a006577. [Google Scholar] [CrossRef]
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 2010, 10, 138–146. [Google Scholar] [CrossRef]
- Kerbel, R.S.; Benezra, R.; Lyden, D.C.; Hattori, K.; Heissig, B.; Nolan, D.J.; Mittal, V.; Shaked, Y.; Dias, S.; Bertolini, F.; et al. Endothelial progenitor cells are cellular hubs essential for neoangiogenesis of certain aggressive adenocarcinomas and metastatic transition but not adenomas. Proc. Natl. Acad. Sci. USA 2008, 105, E54. [Google Scholar] [CrossRef]
- Pasquier, J.; Ghiabi, P.; Chouchane, L.; Razzouk, K.; Rafii, S.; Rafii, A. Angiocrine endothelium: From physiology to cancer. J. Transl. Med. 2020, 18, 52. [Google Scholar] [CrossRef]
- Singhal, M.; Augustin, H.G. Beyond angiogenesis: Exploiting angiocrine factors to restrict tumor progression and metastasis. Cancer Res. 2020, 80, 659–662. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Schweisguth, F. Regulation of Notch Signaling Activity. Curr. Biol. 2004, 14, R129–R138. [Google Scholar] [CrossRef] [PubMed]
- Dexter, J.S. The analysis of a case of continuous variation in drosophila by a study of its linkage relations. Am. Nat. 1914, 48, 712. [Google Scholar] [CrossRef]
- Morgan, T.H. The Theory of the Gene. Am. Nat. 1917, 51, 513. [Google Scholar] [CrossRef]
- Wharton, K.A.; Johansen, K.M.; Xu, T.; Artavanis-Tsakonas, S. Nucleotide sequence from the neurogenic locus notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell 1985, 43, 567–581. [Google Scholar] [CrossRef]
- Kidd, S.; Kelley, M.R.; Young, M.W. Sequence of the notch locus of Drosophila melanogaster: Relationship of the encoded protein to mammalian clotting and growth factors. Mol. Cell. Biol. 1986, 6, 3094–3108. [Google Scholar] [CrossRef]
- Fortini, M.E. Notch signaling: The core pathway and its posttranslational regulation. Dev. Cell 2009, 16, 633–647. [Google Scholar] [CrossRef]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat Rev Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef]
- Logeat, F.; Bessia, C.; Brou, C.; LeBail, O.; Jarriault, S.; Seidah, N.G.; Israël, A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad. Sci. USA 1998, 95, 8108–8112. [Google Scholar] [CrossRef]
- Rana, N.A.; Haltiwanger, R.S. Fringe benefits: Functional and structural impacts of O-glycosylation on the extracellular domain of Notch receptors. Curr. Opin. Struct. Biol. 2011, 21, 583–589. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Lim, J.S.; Sage, J.; Aifantis, I. From fly wings to targeted cancer therapies: A centennial for notch signaling. Cancer Cell 2014, 25, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Brou, C.; Logeat, F.; Gupta, N.; Bessia, C.; LeBail, O.; Doedens, J.R.; Cumano, A.; Roux, P.; Black, R.A.; Israël, A. A novel proteolytic cleavage involved in Notch signaling: The role of the disintegrin-metalloprotease TACE. Mol. Cell 2000, 5, 207–216. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.; Sliz, P.; Pear, W.S.; Aster, J.C.; Blacklow, S.C. Cooperative assembly of higher-order Notch complexes functions as a switch to induce transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 2103–2108. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.J.; Kovall, R.A. Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell 2006, 124, 985–996. [Google Scholar] [CrossRef]
- Kao, H.Y.; Ordentlich, P.; Koyano-Nakagawa, N.; Tang, Z.; Downes, M.; Kintner, C.R.; Evans, R.M.; Kadesch, T. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev. 1998, 12, 2269–2277. [Google Scholar] [CrossRef]
- Swiatek, P.J.; Lindsell, C.E.; Amo, F.F.D.; Weinmaster, G.; Gridley, T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994, 8, 707–719. [Google Scholar] [CrossRef]
- Weinmaster, G.; Roberts, V.J.; Lemke, G. Notch2: A second mammalian Notch gene. Cell Death Differ. 1992, 116, 931–941. [Google Scholar]
- McCright, B.; Gao, X.; Shen, L.; Lozier, J.; Lan, Y.; Maguire, M.; Herzlinger, D.; Weinmaster, G.; Jiang, R.; Gridley, T. Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development 2001, 128, 491–502. [Google Scholar]
- Varadkar, P.; Kraman, M.; Despres, D.; Ma, G.; Lozier, J.; McCright, B. Notch2 is required for the proliferation of cardiac neural crest-derived smooth muscle cells. Dev. Dyn. 2008, 237, 1144–1152. [Google Scholar] [CrossRef]
- Hamada, Y.; Kadokawa, Y.; Okabe, M.; Ikawa, M.; Coleman, J.R.; Tsujimoto, Y. Mutation in ankyrin repeats of the mouse Notch2 gene induces early embryonic lethality. Development 1999, 126, 3415–3424. [Google Scholar] [PubMed]
- Wang, Q.; Zhao, N.; Kennard, S.; Lilly, B. Notch2 and Notch3 Function Together to Regulate Vascular Smooth Muscle Development. PLoS ONE 2012, 7, e37365. [Google Scholar] [CrossRef] [PubMed]
- Domenga, V.; Fardoux, P.; Lacombe, P.; Monet, M.; Maciazek, J.; Krebs, L.T.; Klonjkowski, B.; Berrou, E.; Mericskay, M.; Li, Z.; et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004, 18, 2730–2735. [Google Scholar] [CrossRef] [PubMed]
- Uyttendaele, H.; Marazzi, G.; Wu, G.; Yan, Q.; Sassoon, D.; Kitajewski, J. Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development 1996, 122, 2251–2259. [Google Scholar] [PubMed]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar]
- Gridley, T. Notch signaling during vascular development. Proc. Natl. Acad. Sci. USA 2001, 98, 5377–5378. [Google Scholar] [CrossRef]
- Limbourg, A.; Ploom, M.; Elligsen, D.; Sörensen, I.; Ziegelhoeffer, T.; Gossler, A.; Drexler, H.; Limbourg, F.P. Notch ligand Delta-like 1 is essential for postnatal arteriogenesis. Circ. Res. 2007, 100, 363–371. [Google Scholar] [CrossRef]
- Sörensen, I.; Adams, R.H.; Gossler, A. DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 2009, 113, 5680–5688. [Google Scholar] [CrossRef]
- Doi, H.; Iso, T.; Sato, H.; Yamazaki, M.; Matsui, H.; Tanaka, T.; Manabe, I.; Arai, M.; Nagai, R.; Kurabayashi, M. Jagged1-selective Notch Signaling Induces Smooth Muscle Differentiation via a RBP-Jκ-dependent Pathway. J. Biol. Chem. 2006, 281, 28555–28564. [Google Scholar] [CrossRef]
- Xue, Y.; Gao, X.; Lindsell, C.E.; Norton, C.R.; Chang, B.; Hicks, C.; Gendron-Maguire, M.; Rand, E.B.; Weinmaster, G.; Gridley, T. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 1999, 8, 723–730. [Google Scholar] [CrossRef]
- Shutter, J.R.; Scully, S.; Fan, W.; Richards, W.G.; Kitajewski, J.; Deblandre, G.A.; Kintner, C.R.; Stark, K.L. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev. 2000, 14, 1313–1318. [Google Scholar] [PubMed]
- Duarte, A.; Hirashima, M.; Benedito, R.; Trindade, A.; Diniz, P.; Bekman, E.; Costa, L.; Henrique, D.; Rossant, J. Dosage-sensitive requirement for mouse Dll4 in artery development. Gene Dev. 2004, 18, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Shutter, J.R.; Tanigaki, K.; Honjo, T.; Stark, K.L.; Gridley, T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Gene Dev. 2004, 18, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Gale, N.W.; Dominguez, M.G.; Noguera, I.; Pan, L.; Hughes, V.; Valenzuela, D.M.; Murphy, A.J.; Adams, N.C.; Lin, H.C.; Holash, J.; et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc. Natl. Acad. Sci. USA 2004, 101, 15949–15954. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Shirakawa, T.; Li, Y.; Soma, A.; Oka, M.; Dotto, G.P.; Fairman, R.M.; Velazquez, O.C.; Herlyn, M. Regulation of Notch1 and Dll4 by vascular endothelial growth Factor in arterial endothelial Cells: Implications for modulating arteriogenesis and angiogenesis. Mol. Cell Biol. 2003, 23, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.K.; Li, J.-L.; Murga, M.; Harris, A.L.; Tosato, G. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 2006, 107, 931–939. [Google Scholar] [CrossRef]
- Siekmann, A.F.; Covassin, L.; Lawson, N.D. Modulation of VEGF signalling output by the Notch pathway. Bioessays 2008, 30, 303–313. [Google Scholar] [CrossRef]
- Suchting, S.; Freitas, C.; le Noble, F.; Benedito, R.; Bréant, C.; Duarte, A.; Eichmann, A. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc. Natl. Acad. Sci. USA 2007, 104, 3225–3230. [Google Scholar] [CrossRef]
- Hellström, M.; Phng, L.K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef]
- Roca, C.; Adams, R.H. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007, 21, 2511–2524. [Google Scholar] [CrossRef]
- Sainson, R.C.A.; Aoto, J.; Nakatsu, M.N.; Holderfield, M.; Conn, E.; Koller, E.; Hughes, C.C. Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J. 2005, 19, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 2009, 137, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, A.-R.; Trindade, A.; Fernandes, A.C.; Carvalho, C.; Gigante, J.; Tavares, A.T.; Diéguez-Hurtado, R.; Yagita, H.; Adams, R.H.; Duarte, A. Endothelial Jagged1 antagonizes Dll4 regulation of endothelial branching and promotes vascular maturation downstream of Dll4/Notch1. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef]
- Thurston, G.; Noguera-Troise, I.; Yancopoulos, G.D. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat. Rev. Cancer 2007, 7, 327–331. [Google Scholar] [CrossRef]
- Scehnet, J.S.; Jiang, W.; Ram Kumar, S.; Krasnoperov, V.; Trindade, A.; Benedito, R.; Djokovic, D.; Borges, C.; Ley, E.J.; Duarte, A.; et al. Inhibition of Dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood 2007, 109, 4753–4760. [Google Scholar] [CrossRef]
- Trindade, A.; Djokovic, D.; Gigante, J.; Badenes, M.; Pedrosa, A.R.; Fernandes, A.C.; Lopes-da-Costa, L.; Krasnoperov, V.; Liu, R.; Gill, P.S.; et al. Low-dosage inhibition of Dll4 signaling promotes wound healing by inducing functional neo-angiogenesis. PLoS ONE 2012, 7, e29863. [Google Scholar] [CrossRef]
- Urs, S.; Roudabush, A.; O’Neill, C.F.; Pinz, I.; Prudovsky, I.; Kacer, D.; Tang, Y.; Liaw, L.; Small, D. Soluble Forms of the Notch Ligands Delta1 and Jagged1 Promote in Vivo Tumorigenicity in NIH3T3 Fibroblasts with Distinct Phenotypes. Am. J. Pathol. 2008, 173, 865–878. [Google Scholar] [CrossRef]
- Pedrosa, A.R.; Trindade, A.; Carvalho, C.; Graça, J.; Carvalho, S.; Peleteiro, M.C.; Adams, R.H.; Duarte, A. Endothelial Jagged1 promotes solid tumor growth through both pro-angiogenic and angiocrine functions. Oncotarget 2015, 6, 24404–24423. [Google Scholar] [CrossRef]
- Zeng, Q.; Li, S.; Chepeha, D.B.; Giordano, T.J.; Li, J.; Zhang, H.; Polverini, P.J.; Nor, J.; Kitajewski, J.; Wang, C.Y. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell 2005, 8, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, Y.; Hernandez, S.L.; Das, I.; Ahn, A.; Huang, J.; Vorontchikhina, M.; Sharma, A.; Kanamaru, E.; Borisenko, V.; DeSilva, D.M.; et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008, 68, 4727–4735. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia Requires Notch Signaling to Maintain the Undifferentiated Cell State. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Sainson, R.C.A.; Harris, A.L. Hypoxia-regulated differentiation: Let’s step it up a Notch. Trends Mol. Med. 2006, 12, 141–143. [Google Scholar] [CrossRef]
- Diez, H.; Fischer, A.; Winkler, A.; Hu, C.J.; Hatzopoulos, A.K.; Breier, G.; Gessler, M. Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp. Cell Res. 2007, 313, 1–9. [Google Scholar] [CrossRef]
- Kangsamaksin, T.; Murtomaki, A.; Kofler, N.M.; Cuervo, H.; Chaudhri, R.A.; Tattersall, I.W.; Rosenstiel, P.E.; Shawber, C.J.; Kitajewski, J. Notch Decoys that Selectively Block Dll/Notch or Jagged/Notch Disrupt Angiogenesis by Unique Mechanisms to Inhibit Tumor Growth. Cancer Discov. 2014, 5, 182–197. [Google Scholar] [CrossRef]
- Previs, R.A.; Coleman, R.L.; Harris, A.L.; Sood, A.K. Molecular Pathways: Translational and Therapeutic Implications of the Notch Signaling Pathway in Cancer. Clin. Cancer Res. 2014, 21, 955–961. [Google Scholar] [CrossRef]
- Yin, L.; Velazquez, O. Notch signaling: Emerging molecular targets for cancer therapy. Biochem. Pharmacol. 2010, 80, 690–701. [Google Scholar] [CrossRef]
- Gallahan, D.; Callahan, R. The mouse mammary tumor associated gene INT3 is a unique member of the NOTCH gene family (NOTCH4). Oncogene 1997, 14, 1883–1890. [Google Scholar] [CrossRef]
- Hu, C.; Diévart, A.; Lupien, M.; Calvo, E.; Tremblay, G.; Jolicoeur, P. Overexpression of activated murine Notch1 and Notch3 in transgenic mice blocks mammary gland development and induces mammary tumors. Am. J. Pathol. 2006, 168, 973–990. [Google Scholar] [CrossRef]
- Reedijk, M.; Odorcic, S.; Chang, L.; Zhang, H.; Miller, N.; McCready, D.R.; Lockwood, G.; Egan, S.E. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005, 65, 8530–8537. [Google Scholar] [CrossRef] [PubMed]
- South, A.P.; Cho, R.J.; Aster, J.C. The double-edged sword of Notch signaling in cancer. Semin. Cell Dev. Biol. 2012, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Li, M.; Nakayama, K.; Mao, T.L.; Davidson, B.; Zhang, Z.; Kurman, R.J.; Eberhart, C.G.; Shih, I.M.; Wang, T.L. Notch3 Gene Amplification in Ovarian Cancer. Cancer Res. 2006, 66, 6312–6318. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.A.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef]
- Whelan, J.T.; Kellogg, A.; Shewchuk, B.M.; Hewan-Lowe, K.; Bertrand, F.E. Notch-1 signaling is lost in prostate adenocarcinoma and promotes PTEN gene expression. J. Cell Biochem. 2009, 107, 992–1001. [Google Scholar] [CrossRef]
- Hafeez, B.B.; Adhami, V.M.; Asim, M.; Siddiqui, I.A.; Bhat, K.M.; Zhong, W.; Saleem, M.; Din, M.; Setaluri, V.; Mukhtar, H. Targeted knockdown of Notch1 inhibits invasion of human prostate cancer cells concomitant with inhibition of matrix metalloproteinase-9 and urokinase plasminogen activator. Clin. Cancer Res. 2009, 15, 452–459. [Google Scholar] [CrossRef]
- Christofori, G. New signals from the invasive front. Nature 2006, 441, 444–450. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Christiansen, J.J.; Rajasekaran, A.K. Reassessing Epithelial to Mesenchymal Transition as a Prerequisite for Carcinoma Invasion and Metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef]
- Klymkowsky, M.W.; Savagner, P. Epithelial-Mesenchymal Transition A Cancer Researcher’s Conceptual Friend and Foe. Am. J. Pathol. 2009, 174, 1588–1593. [Google Scholar] [CrossRef]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-κB and epithelial to mesenchymal transition of cancer. J. Cell Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Banerjee, S.; Sarkar, F.H. Emerging role of Notch in stem cells and cancer. Cancer Lett. 2009, 279, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The role of notch signaling pathway in epithelial-mesenchymal transition (emt) during development and tumor aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3117. [Google Scholar] [CrossRef]
- Cannito, S.; Novo, E.; Compagnone, A.; Valfrè di Bonzo, L.; Busletta, C.; Zamara, E.; Paternostro, C.; Povero, D.; Bandino, A.; Bozzo, F.; et al. Redox mechanisms switch on hypoxia-dependent epithelial–mesenchymal transition in cancer cells. Carcinogenesis 2008, 29, 2267–2278. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Gene Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef]
- Yang, M.-H.; Wu, K.-J. TWIST activation by hypoxia inducible factor-1 (HIF-1): Implications in metastasis and development. Cell Cycle 2008, 7, 2090–2096. [Google Scholar] [CrossRef]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.; Pochampally, R.; Xing, F.; Watabe, K.; Miele, L. Notch signaling: Targeting cancer stem cells and epithelial-to-mesenchymal transition. Oncotargets Ther. 2013, 6, 1249–1259. [Google Scholar]
- Huang, Q.B.; Ma, X.; Li, H.Z.; Ai, Q.; Liu, S.W.; Zhang, Y.; Gao, Y.; Fan, Y.; Ni, D.; Wang, B.J.; et al. Endothelial Delta-like 4 (DLL4) promotes renal cell carcinoma hematogenous metastasis. Oncotarget 2014, 5, 3066–3075. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kuramoto, T.; Goto, H.; Mitsuhashi, A.; Tabata, S.; Ogawa, H.; Uehara, H.; Saijo, A.; Kakiuchi, S.; Maekawa, Y.; Yasutomo, K.; et al. Dll4-Fc, an Inhibitor of Dll4-Notch Signaling, Suppresses Liver Metastasis of Small Cell Lung Cancer Cells through the Downregulation of the NF-κB Activity. Mol. Cancer Ther. 2012, 11, 2578–2587. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Z.; Jia, X.; Wang, L.; Chen, Z.; Wang, S.; Wang, M.; Zhang, J.; Wu, M. MMGZ01, an anti-DLL4 monoclonal antibody, promotes nonfunctional vessels and inhibits breast tumor growth. Cancer Lett. 2016, 372, 118–127. [Google Scholar] [CrossRef]
- Koch, U.; Lehal, R.; Radtke, F. Stem cells living with a Notch. Development 2013, 140, 689–704. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing notch ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef]
- Jiang, L.-Y.; Zhang, X.-L.; Du, P.; Zheng, J.-H. γ-Secretase Inhibitor, DAPT Inhibits Self-renewal and Stemness Maintenance of Ovarian Cancer Stem-like Cells In Vitro. Chin. J. Cancer Res. 2011, 23, 140–146. [Google Scholar] [CrossRef]
- McAuliffe, S.M.; Morgan, S.L.; Wyant, G.A.; Tran, L.T.; Muto, K.W.; Chen, Y.S.; Chin, K.T.; Partridge, J.C.; Poole, B.B.; Cheng, K.H.; et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E2939–E2948. [Google Scholar] [CrossRef]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, R.C.; Ouzounova, M.; Davis, A.; Choi, D.; Tchuenkam, S.M.; Kim, G.; Luther, T.; Quraishi, A.A.; Senbabaoglu, Y.; Conley, S.J.; et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol. Cancer Ther. 2015, 14, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Wohlfeil, S.A.; Häfele, V.; Dietsch, B.; Schledzewski, K.; Winkler, M.; Zierow, J.; Leibing, T.; Mohammadi, M.M.; Heineke, J.; Sticht, C.; et al. Hepatic endothelial Notch activation protects against liver metastasis by regulating endothelial-tumor cell adhesion independent of angiocrine signaling. Cancer Res. 2018, 79, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Sonoshita, M.; Aoki, M.; Fuwa, H.; Aoki, K.; Hosogi, H.; Sakai, Y.; Hashida, H.; Takabayashi, A.; Sasaki, M.; Robine, S.; et al. Suppression of colon cancer metastasis by aes through inhibition of notch signaling. Cancer Cell 2011, 19, 125–137. [Google Scholar] [CrossRef]
- Xing, F.; Kobayashi, A.; Okuda, H.; Watabe, M.; Pai, S.K.; Pandey, P.R.; Hirota, S.; Wilber, A.; Mo, Y.Y.; Moore, B.E.; et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol. Med. 2013, 5, 384–396. [Google Scholar] [CrossRef]
- Yang, Y.; Ahn, Y.H.; Gibbons, D.L.; Zang, Y.; Lin, W.; Thilaganathan, N.; Alvarez, C.A.; Moreira, D.C.; Creighton, C.J.; Gregory, P.A.; et al. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J. Clin. Investig. 2011, 121, 1373–1385. [Google Scholar] [CrossRef]
- Capaccione, K.M.; Pine, S.R. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 2013, 34, 1420–1430. [Google Scholar] [CrossRef]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.A.; Rodriguez-Barrueco, R.; Bonal, D.M.; Charytonowicz, E.; Gladoun, N.; de la Iglesia-Vicente, J.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar] [CrossRef]
- Martz, C.A.; Ottina, K.A.; Singleton, K.R.; Jasper, J.S.; Wardell, S.E.; Peraza-Penton, A.; Anderson, G.R.; Winter, P.S.; Wang, T.; Alley, H.M.; et al. Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Sci. Signal. 2014, 7. [Google Scholar] [CrossRef]
- Xie, M.; He, C.-S.; Wei, S.-H.; Zhang, L. Notch-1 contributes to epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance in non-small cell lung cancer in vitro and in vivo. Eur. J. Cancer 2013, 49, 3559–3572. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.W.; Rizzo, P.; Pannuti, A.; Golde, T.; Osborne, B.; Miele, L. Notch signals in the endothelium and cancer “stem-like” cells: Opportunities for cancer therapy. Vasc. Cell 2012, 4, 7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Henrique, D.; Schweisguth, F. Mechanisms of Notch signaling: A simple logic deployed in time and space. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Musse, A.A.; Meloty-Kapella, L.; Weinmaster, G. Notch ligand endocytosis: Mechanistic basis of signaling activity. Semin. Cell Dev. Biol. 2012, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.; Ladi, E.; Lindsell, C.; Hsieh, J.J.D.; Hayward, S.D.; Collazo, A.; Weinmaster, G. A secreted Delta1-Fc fusion protein functions both as an activator and inhibitor of Notch1 signaling. J. Neurosci. Res. 2002, 68, 655–667. [Google Scholar] [CrossRef]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial Cells Promote the Colorectal Cancer Stem Cell Phenotype through a Soluble Form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef]
- Sharghi-Namini, S.; Tan, E.; Ong, L.-L.S.; Ge, R.; Asada, H.H. Dll4-containing exosomes induce capillary sprout retraction in a 3D microenvironment. Sci Rep. 2014, 4, 1–8. [Google Scholar] [CrossRef]
- Sheldon, H.; Heikamp, E.; Turley, H.; Dragovic, R.; Thomas, P.; Oon, C.E.; Leek, R.; Edelmann, M.; Kessler, B.; Sainson, R.C.; et al. New mechanism for Notch signaling to endothelium at a distance by Delta-like 4 incorporation into exosomes. Blood 2010, 116, 2385–2394. [Google Scholar] [CrossRef]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Halabi, N.; Guerrouahen, B.S.; Rafii, S.; Rafii, A. Breast cancer cells promote a notch-dependent mesenchymal phenotype in endothelial cells participating to a pro-tumoral niche. J. Transl. Med. 2015, 13, 27. [Google Scholar] [CrossRef]
- Cao, Z.; Ding, B.S.; Guo, P.; Lee, S.B.; Butler, J.M.; Casey, S.C.; Simons, M.; Tam, W.; Felsher, D.W.; Shido, K.; et al. Angiocrine Factors Deployed by Tumor Vascular Niche Induce B Cell Lymphoma Invasiveness and Chemoresistance. Cancer Cell 2014, 25, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhou, C.; Zhang, Z.; Wang, Q.; Wei, H.; Shi, W.; Li, J.; Wang, Z.; Ou, Y.; Wang, W.; et al. Jagged1-Notch1-deployed tumor perivascular niche promotes breast cancer stem cell phenotype through Zeb1. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jeon, H.M.; Jeon, H.Y.; Oh, S.Y.; Kim, E.J.; Jin, X.; Kim, S.H.; Kim, S.H.; Jin, X.; Kim, H. Conversion of glioma cells to glioma stem-like cells by angiocrine factors. Biochem. Biophys. Res. Commun. 2018, 496, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Hoarau-Véchot, J.; Touboul, C.; Halabi, N.; Blot-Dupin, M.; Lis, R.; Abi Khalil, C.; Rafii, S.; Rafii, A.; Pasquier, J. Akt-activated endothelium promotes ovarian cancer proliferation through notch activation. J. Transl. Med. 2019, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Chen, G.; Coetzee, S.; Thambi, N.; Hickey, C.; Shan, J.; Kovalenko, P.; Noguera-Troise, I.; Smith, E.; Fairhurst, J.; et al. Dll4 Blockade in Stromal Cells Mediates Antitumor Effects in Preclinical Models of Ovarian Cancer. Cancer Res. 2015, 75, 4086–4096. [Google Scholar] [CrossRef]
- Indraccolo, S.; Minuzzo, S.; Masiero, M.; Pusceddu, I.; Persano, L.; Moserle, L.; Reboldi, A.; Favaro, E.; Mecarozzi, M.; Di Mario, G.; et al. Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy. Cancer Res. 2009, 69, 1314–1323. [Google Scholar] [CrossRef]
- Mendonça, L.; Trindade, A.; Carvalho, C.; Correia, J.; Badenes, M.; Gigante, J.; Duarte, A. Metastasis is impaired by endothelial-specific Dll4 loss-of-function through inhibition of epithelial-to-mesenchymal transition and reduction of cancer stem cells and circulating tumor cells. Clin. Exp. Metastas 2019, 36, 365–380. [Google Scholar] [CrossRef]
- Wieland, E.; Rodriguez-Vita, J.; Liebler, S.S.; Mogler, C.; Moll, I.; Herberich, S.E.; Espinet, E.; Herpel, E.; Menuchin, A.; Chang-Claude, J.; et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017, 31, 355–367. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trindade, A.; Duarte, A. Notch Signaling Function in the Angiocrine Regulation of Tumor Development. Cells 2020, 9, 2467. https://doi.org/10.3390/cells9112467
Trindade A, Duarte A. Notch Signaling Function in the Angiocrine Regulation of Tumor Development. Cells. 2020; 9(11):2467. https://doi.org/10.3390/cells9112467
Chicago/Turabian StyleTrindade, Alexandre, and António Duarte. 2020. "Notch Signaling Function in the Angiocrine Regulation of Tumor Development" Cells 9, no. 11: 2467. https://doi.org/10.3390/cells9112467
APA StyleTrindade, A., & Duarte, A. (2020). Notch Signaling Function in the Angiocrine Regulation of Tumor Development. Cells, 9(11), 2467. https://doi.org/10.3390/cells9112467