Splicing Players Are Differently Expressed in Sporadic Amyotrophic Lateral Sclerosis Molecular Clusters and Brain Regions



Abstract
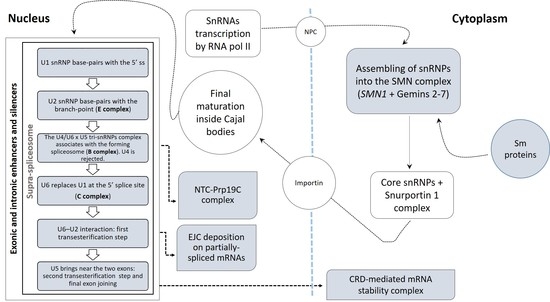
1. Introduction
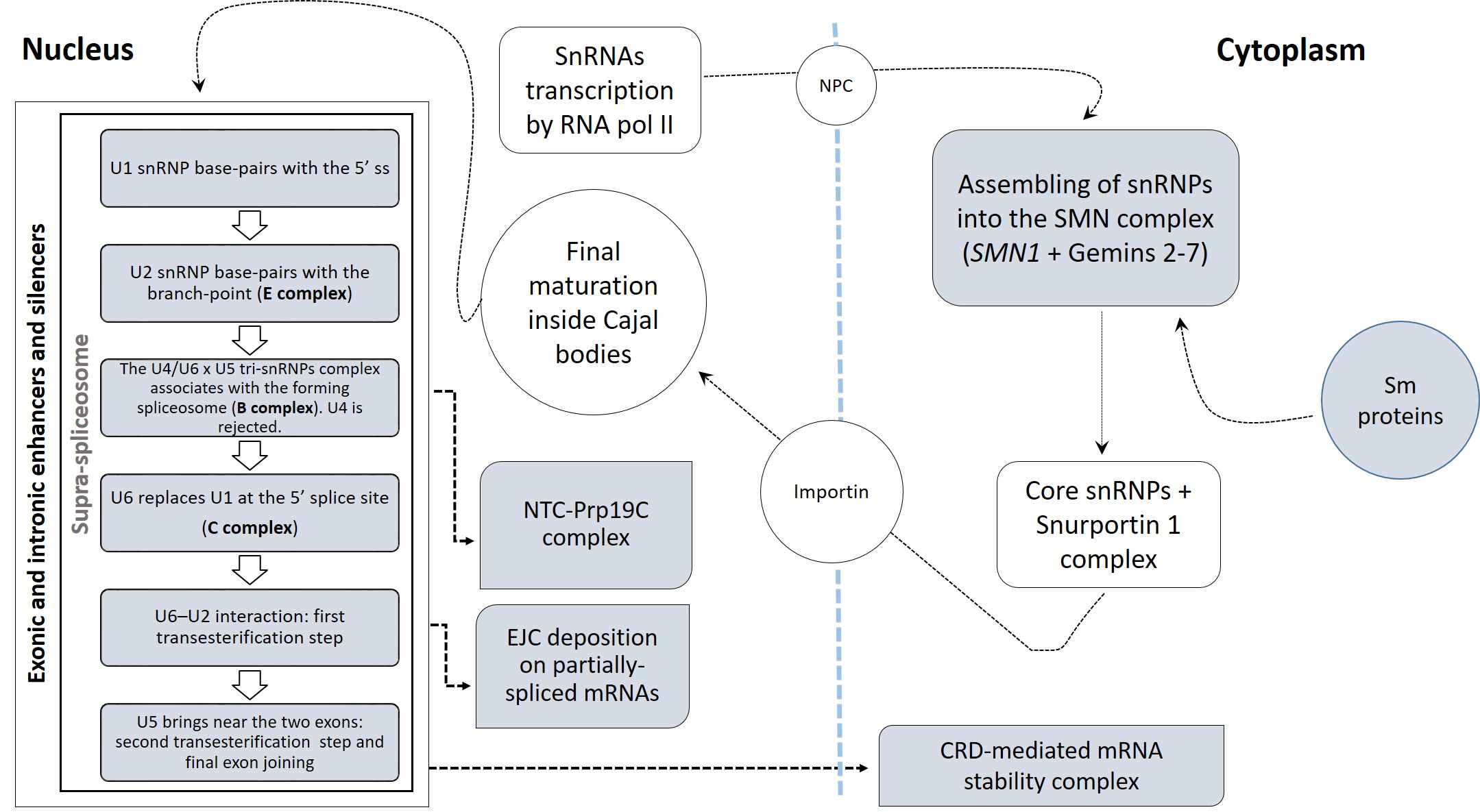
2. Materials and Methods
2.1. Data Source and Gene Selection
2.2. Gene List Filtering and Differential Expression Analysis
2.3. Functional Characterization of Differentially Expressed Splicing Factors
2.4. Protein–Protein Interaction Network Analysis
3. Results and Discussion
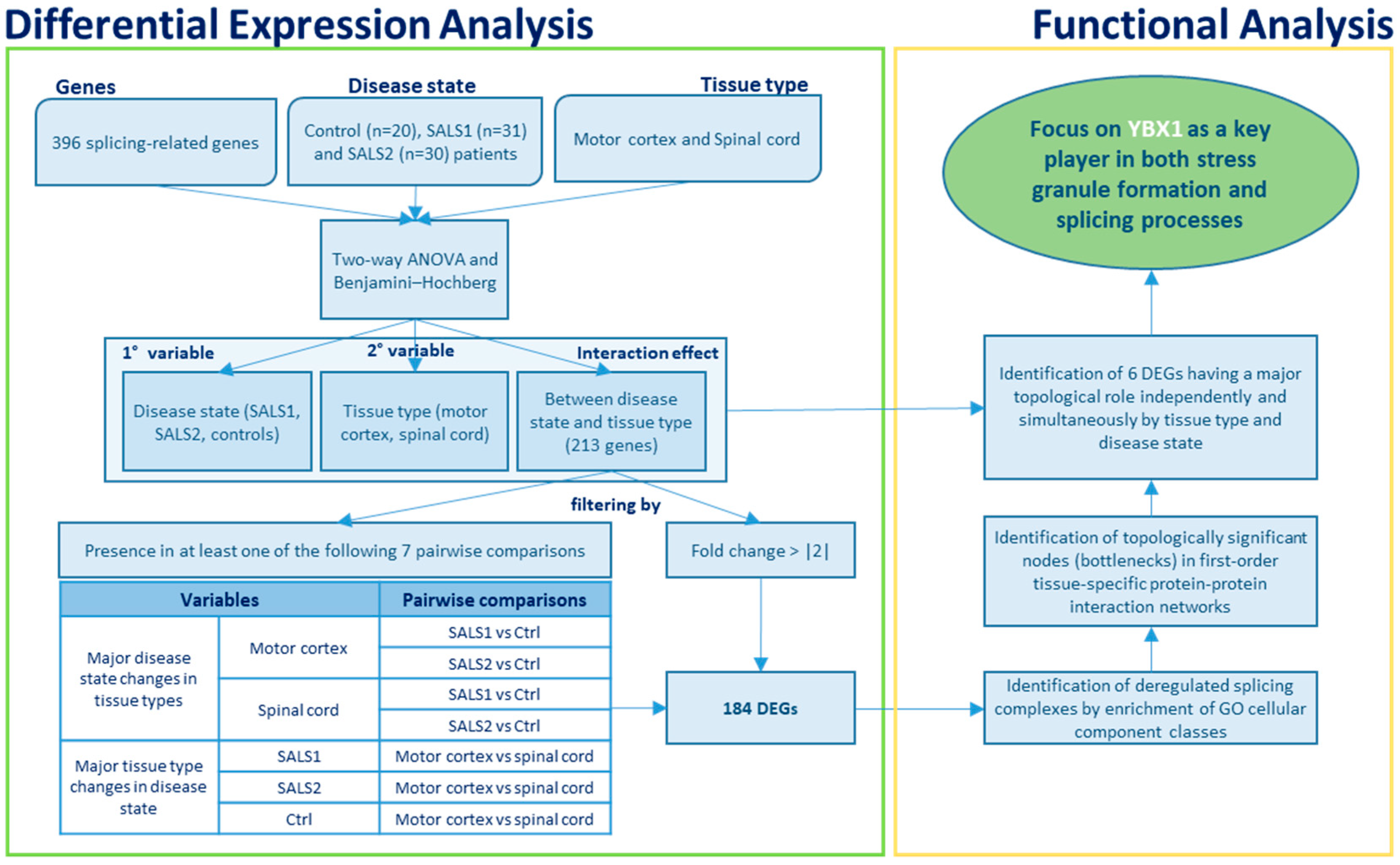
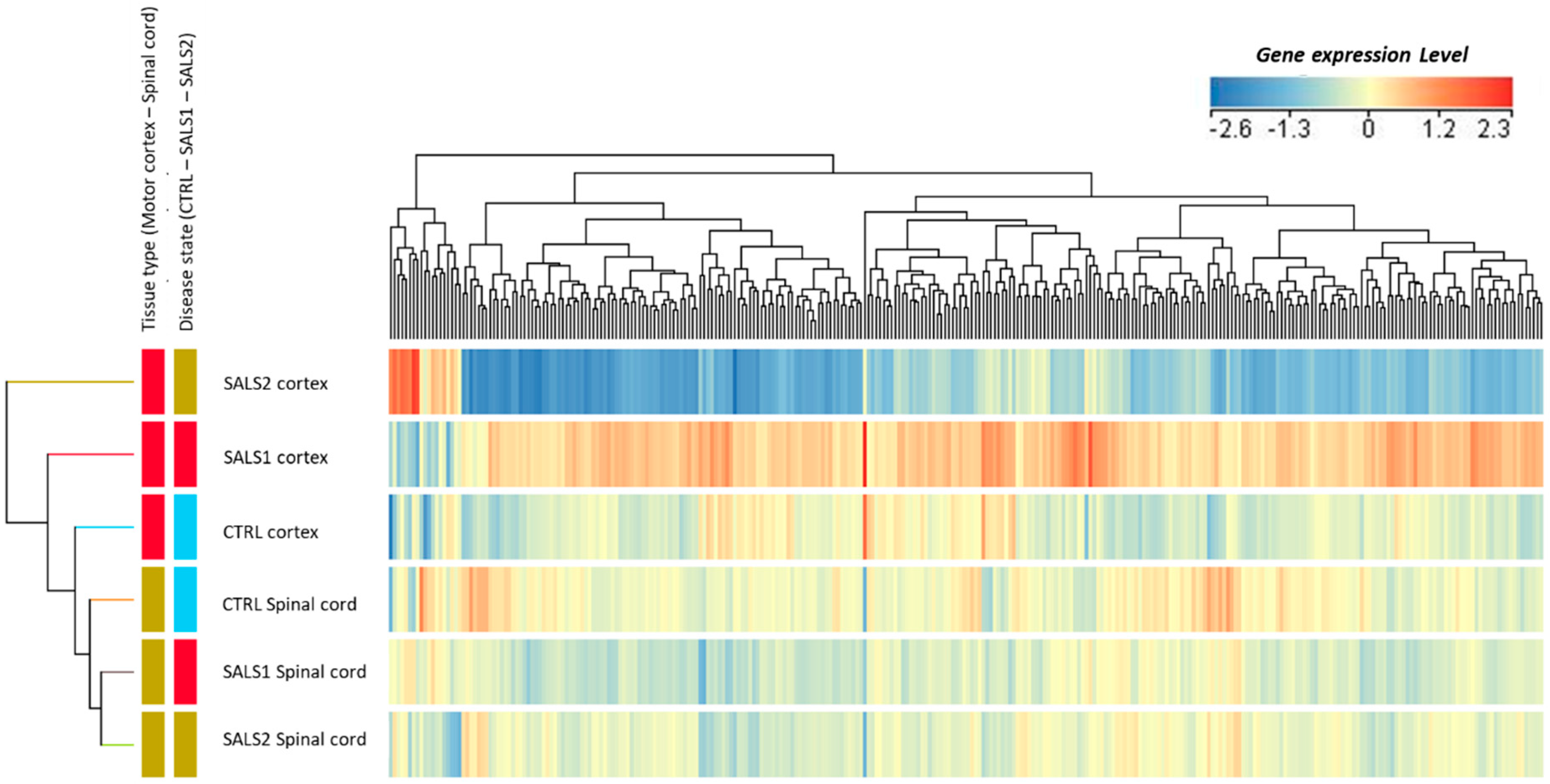
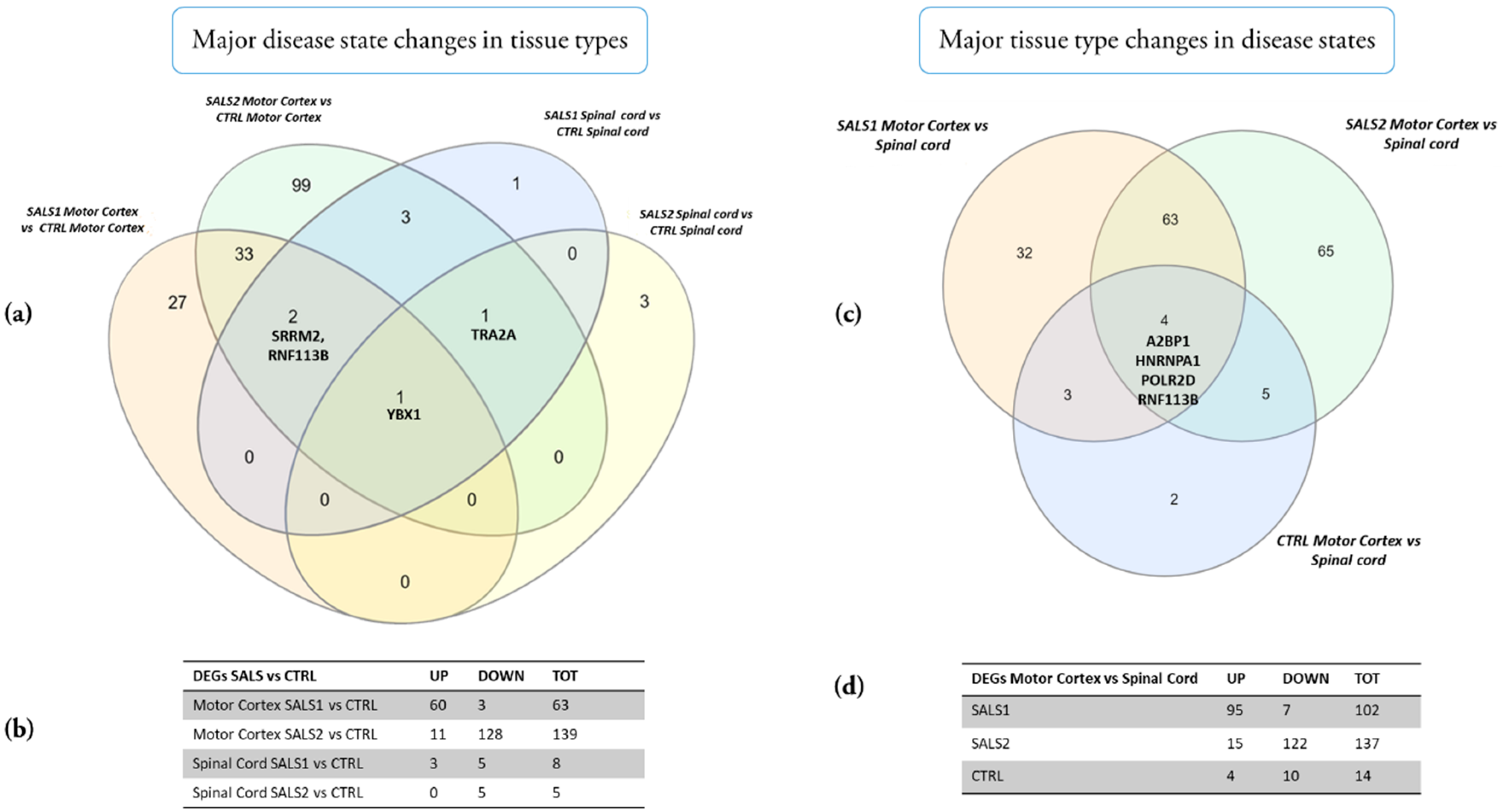
3.1. Identification of Differentially Expressed Genes in Tissue Types and Disease States
3.2. Deregulated Splicing Complexes
3.2.1. Spliceosomal Complex Core
U1 snRNP
U2 snRNP
U4, U5, U6 snRNPs and tri-snRNPs Complex
3.2.2. U12-Dependent Minor Splicing Pathway
3.2.3. Exon–Exon Junction Complex (EJC)
3.2.4. CRD-Mediated mRNA Stability Complex
3.2.5. RNA Polymerase II, Core Complex
3.2.6. Prp19 Complex
3.2.7. SMN–Sm Protein Complex
3.2.8. Supraspliceosomal Complex
3.3. Tissue-Specific Networks of Spliceosome-Related Gene Products
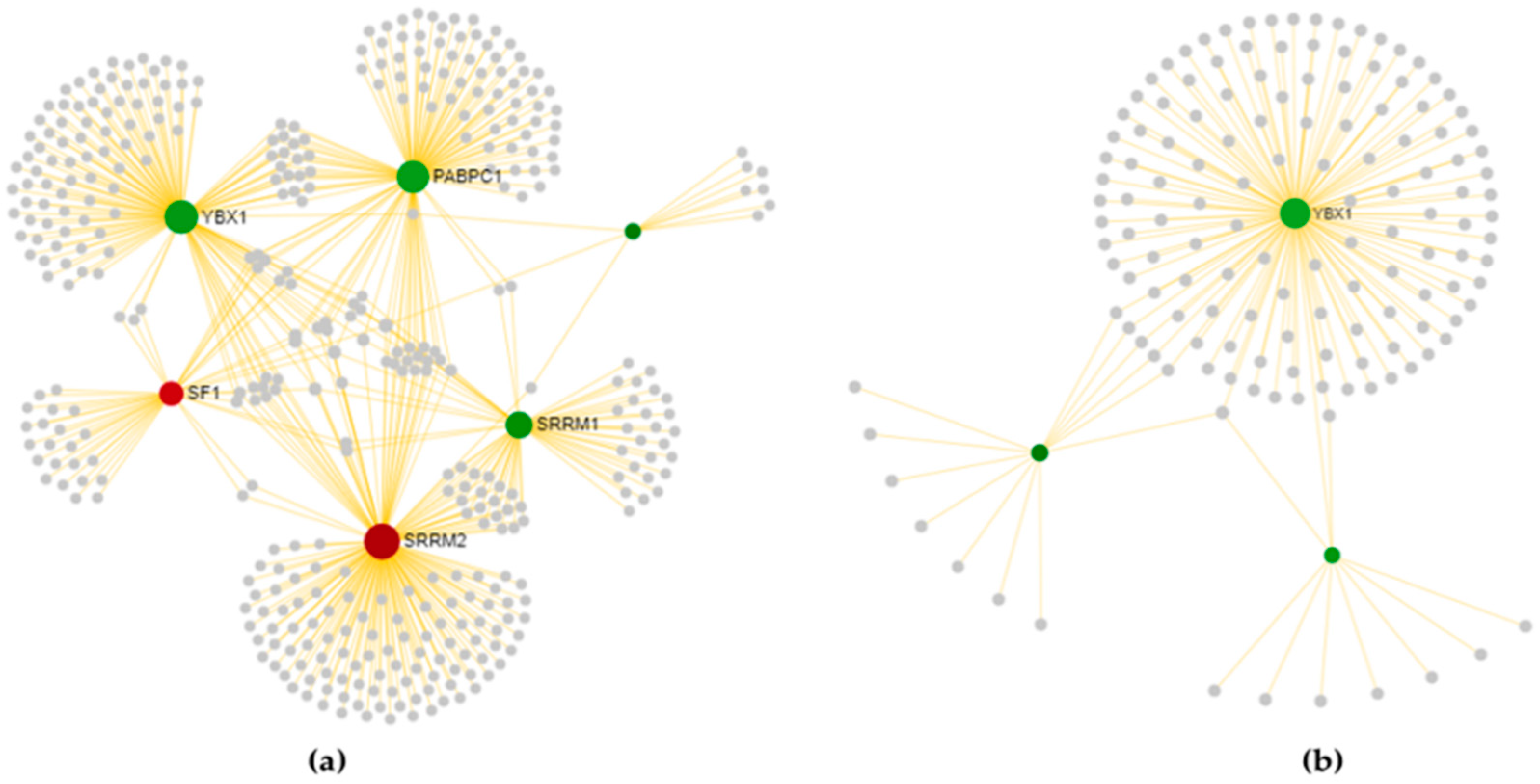
3.3.1. Cortex-Specific PPI Networks
3.3.2. Spinal Cord-Specific PPI Networks
3.3.3. The Most Relevant Splicing-Related Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Perrone, B.; La Cognata, V.; Sprovieri, T.; Ungaro, C.; Conforti, F.L.; Andò, S.; Cavallaro, S. Alternative Splicing of ALS Genes: Misregulation and Potential Therapies. Cell. Mol. Neurobiol. 2019, 40, 1–14. [Google Scholar] [CrossRef]
- Su, C.-H.; Tarn, W.-Y. Alternative Splicing in Neurogenesis and Brain Development. Front. Mol. Biosci. 2018, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Raj, B.; Blencowe, B.J. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 2015, 87, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Weyn-Vanhentenryck, S.M.; Feng, H.; Ustianenko, D.; Duffié, R.; Yan, Q.; Jacko, M.; Martinez, J.C.; Goodwin, M.; Zhang, X.; Hengst, U.; et al. Precise temporal regulation of alternative splicing during neural development. Nat. Commun. 2018, 9, 2189. [Google Scholar] [CrossRef]
- Porter, R.S.; Jaamour, F.; Iwase, S. Neuron-specific alternative splicing of transcriptional machineries: Implications for neurodevelopmental disorders. Mol. Cell. Neurosci. 2018, 87, 35–45. [Google Scholar] [CrossRef]
- Hoskins, A.A.; Moore, M.J. The spliceosome: A flexible, reversible macromolecular machine. Trends Biochem. Sci. 2012, 37, 179–188. [Google Scholar] [CrossRef]
- Dvinge, H. Regulation of alternative mRNA splicing: Old players and new perspectives. FEBS Lett. 2018, 592, 2987–3006. [Google Scholar] [CrossRef]
- Jutzi, D.; Akinyi, M.V.; Mechtersheimer, J.; Frilander, M.J.; Ruepp, M.-D. The emerging role of minor intron splicing in neurological disorders. Cell Stress 2018, 2, 40–54. [Google Scholar] [CrossRef]
- Verma, B.; Akinyi, M.V.; Norppa, A.J.; Frilander, M.J. Minor spliceosome and disease. Semin. Cell Dev. Boil. 2018, 79, 103–112. [Google Scholar] [CrossRef]
- La Cognata, V.; Iemmolo, R.; D’Agata, V.; Scuderi, S.; Drago, F.; Zappia, M.; Cavallaro, S. Increasing the Coding Potential of Genomes Through Alternative Splicing: The Case of PARK2 Gene. Curr. Genom. 2014, 15, 203–216. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Turunen, J.J.; Niemelä, E.H.; Verma, B.; Frilander, M.J. The significant other: Splicing by the minor spliceosome. Wiley Interdiscip. Rev. RNA 2013, 4, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.B.; Bellini, M. The assembly of a spliceosomal small nuclear ribonucleoprotein particle. Nucleic Acids Res. 2008, 36, 6482–6493. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; D’Agata, V.; Cavalcanti, F.; Cavallaro, S. Splicing: Is there an alternative contribution to Parkinson’s disease? Neurogenetics 2015, 16, 245–263. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Zhou, Z.; Fu, X.-D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef]
- Aronica, E.; Baas, F.; Iyer, A.; Asbroek, A.L.T.; Morello, G.; Cavallaro, S. Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol. Dis. 2015, 74, 359–376. [Google Scholar] [CrossRef]
- Giulietti, M.; Piva, F.; D’Antonio, M.; D’Onorio De Meo, P.; Paoletti, D.; Castrignano, T.; D’Erchia, A.M.; Picardi, E.; Zambelli, F.; Principato, G.; et al. SpliceAid-F: A database of human splicing factors and their RNA-binding sites. Nucleic. Acids Res. 2013, 41, D125–D131. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; Da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed]
- Basha, O.; Shpringer, R.; Argov, C.M.; Yeger-Lotem, E. The DifferentialNet database of differential protein-protein interactions in human tissues. Nucleic Acids Res. 2018, 46, D522–D526. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, H.; Rathi, N.; Roy, D. Construction and Analysis of the Protein-Protein Interaction Networks Based on Gene Expression Profiles of Parkinson’s Disease. PLoS ONE 2014, 9, e103047. [Google Scholar] [CrossRef] [PubMed]
- Morello, G.; Spampinato, A.G.; Cavallaro, S. Molecular Taxonomy of Sporadic Amyotrophic Lateral Sclerosis Using Disease-Associated Genes. Front. Neurol. 2017, 8, 3. [Google Scholar] [CrossRef]
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; Fagegaltier, D.; Ostrow, L.; Phatnani, H.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177.e5. [Google Scholar] [CrossRef] [PubMed]
- Morello, G.; Guarnaccia, M.; Spampinato, A.G.; Salomone, S.; D’Agata, V.; Conforti, F.L.; Aronica, E.; Cavallaro, S. Integrative multi-omic analysis identifies new drivers and pathways in molecularly distinct subtypes of ALS. Sci. Rep. 2019, 9, 9968. [Google Scholar] [CrossRef]
- Yamazaki, T.; Chen, S.; Yu, Y.; Yan, B.; Haertlein, T.C.; Carrasco, M.A.; Tapia, J.C.; Zhai, B.; Das, R.; Lalancette-Hebert, M.; et al. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2012, 2, 799–806. [Google Scholar] [CrossRef]
- Groen, E.J.; Fumoto, K.; Blokhuis, A.M.; Engelen-Lee, J.; Zhou, Y.; Heuvel, D.M.V.D.; Koppers, M.; Van Diggelen, F.; Van Heest, J.; Demmers, J.A.; et al. ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum. Mol. Genet. 2013, 22, 3690–3704. [Google Scholar] [CrossRef]
- Therrien, M.; Rouleau, G.A.; Dion, P.A.; Parker, J.A. FET proteins regulate lifespan and neuronal integrity. Sci. Rep. 2016, 6, 25159. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.; O’Connell, J.D.; Yamazaki, T.; Gangopadhyay, J.; Gygi, S.P.; Reed, R. Interactome analyses revealed that the U1 snRNP machinery overlaps extensively with the RNAP II machinery and contains multiple ALS/SMA-causative proteins. Sci. Rep. 2018, 8, 8755. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Ling, S.-C.; Qiu, J.; Albuquerque, C.P.; Zhou, Y.; Tokunaga, S.; Li, H.; Qiu, H.; Bui, A.; Yeo, G.W.; et al. ALS-causative mutations in FUS/TLS confer gain- and loss-of-function by altered association with SMN and U1-snRNP. Nat. Commun. 2015, 6, 6171. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Chi, B.; Xia, W.; Gangopadhyay, J.; Yamazaki, T.; Winkelbauer-Hurt, M.E.; Yin, S.; Eliasse, Y.; Adams, E.; Shaw, C.E.; et al. U1 snRNP is mislocalized in ALS patient fibroblasts bearing NLS mutations in FUS and is required for motor neuron outgrowth in zebrafish. Nucleic Acids Res. 2015, 43, 3208–3218. [Google Scholar] [CrossRef]
- Yin, S.; Lopez-Gonzalez, R.; Kunz, R.C.; Gangopadhyay, J.; Borufka, C.; Gygi, S.P.; Gao, F.-B.; Reed, R. Evidence that C9ORF72 Dipeptide Repeat Proteins Associate with U2 snRNP to Cause Mis-splicing in ALS/FTD Patients. Cell Rep. 2017, 19, 2244–2256. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Chitta, R.K.; High, A.A.; Taylor, J.P. Global Analysis of TDP-43 Interacting Proteins Reveals Strong Association with RNA Splicing and Translation Machinery. J. Proteome Res. 2010, 9, 1104–1120. [Google Scholar] [CrossRef]
- Pons, M.; Miguel, L.; Miel, C.; Avequin, T.; Juge, F.; Frebourg, T.; Campion, D.; Lecourtois, M. Splicing factors act as genetic modulators of TDP-43 production in a new autoregulatory TDP-43 Drosophila model. Hum. Mol. Genet. 2017, 26, 3396–3408. [Google Scholar] [CrossRef]
- Rode, S.; Ohm, H.; Zipfel, J.; Rumpf, S. The spliceosome-associated protein Mfap1 binds to VCP in Drosophila. PLoS ONE 2017, 12, e0183733. [Google Scholar] [CrossRef]
- Kim, H.J.; Raphael, A.R.; LaDow, E.S.; McGurk, L.; Weber, R.A.; Trojanowski, J.Q.; Lee, V.M.; Finkbeiner, S.; Gitler, A.D.; Bonini, N.M. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 2014, 46, 152–160. [Google Scholar] [CrossRef]
- Xu, W.; Cao, M.; Zheng, H.; Tan, X.; Li, L.; Cui, G.; Xu, J.; Cao, J.; Ke, K.; Wu, Q. Upregulation of SYF2 is associated with neuronal apoptosis caused by reactive astrogliosis to neuroinflammation. J. Neurosci. Res. 2014, 92, 318–328. [Google Scholar] [CrossRef]
- Shehadeh, L.A.; Yu, K.; Wang, L.; Guevara, A.; Singer, C.; Vance, J.; Papapetropoulos, S. SRRM2, a Potential Blood Biomarker Revealing High Alternative Splicing in Parkinson’s Disease. PLoS ONE 2010, 5, e9104. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.D.; Galej, W.P.; Bai, X.-C.; Savva, C.G.; Newman, A.J.; Scheres, S.H.W.; Nagai, K. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 2015, 523, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Yahara, M.; Kitamura, A.; Kinjo, M. U6 snRNA expression prevents toxicity in TDP-43-knockdown cells. PLoS ONE 2017, 12, e0187813. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Boil. 2011, 9, e1000614. [Google Scholar] [CrossRef] [PubMed]
- Raman, R.; Allen, S.P.; Goodall, E.F.; Kramer, S.; Ponger, L.-L.; Heath, P.R.; Milo, M.; Hollinger, H.C.; Walsh, T.; Highley, J.R.; et al. Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol. Appl. Neurobiol. 2015, 41, 201–226. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Han, L.-H.; Fan, X.-Y.; Guo, H.; Wei, W.; Chen, M.-M.; Yan, S.-F. Network strategy to investigate differential pathways in sporadic amyotrophic lateral sclerosis. J. Cancer Res. Ther. 2018, 14, 1057. [Google Scholar] [CrossRef]
- Iida, A.; Takahashi, A.; Kubo, M.; Saito, S.; Hosono, N.; Ohnishi, Y.; Kiyotani, K.; Mushiroda, T.; Nakajima, M.; Ozaki, K.; et al. A functional variant in ZNF512B is associated with susceptibility to amyotrophic lateral sclerosis in Japanese. Hum. Mol. Genet. 2011, 20, 3684–3692. [Google Scholar] [CrossRef]
- Reber, S.; Stettler, J.; Filosa, G.; Colombo, M.; Jutzi, D.; Lenzken, S.C.; Schweingruber, C.; Bruggmann, R.; Bachi, A.; Barabino, S.M.; et al. Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J. 2016, 35, 1504–1521. [Google Scholar] [CrossRef]
- Ishihara, T.; Ariizumi, Y.; Shiga, A.; Kato, T.; Tan, C.-F.; Sato, T.; Miki, Y.; Yokoo, M.; Fujino, T.; Koyama, A.; et al. Decreased number of Gemini of coiled bodies and U12 snRNA level in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 4136–4147. [Google Scholar] [CrossRef]
- Shibata, T.; Tokunaga, E.; Hattori, S.; Watari, K.; Murakami, Y.; Yamashita, N.; Oki, E.; Itou, J.; Toi, M.; Maehara, Y.; et al. Y-box binding protein YBX1 and its correlated genes as biomarkers for poor outcomes in patients with breast cancer. Oncotarget 2018, 9, 37216–37228. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, M.; Shibata, T.; Watari, K.; Ono, M. Oncogenic Y-box binding protein-1 as an effective therapeutic target in drug-resistant cancer. Cancer Sci. 2019, 110, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Nijssen, J.; Benitez, J.A.; Hoogstraaten, R.; Kee, N.; Hedlund, E. Axon-seq decodes the motor axon transcriptome and its modulation in response to ALS. Stem Cell Rep. 2018, 11, 321596. [Google Scholar] [CrossRef] [PubMed]
- Nasrin, F.; Rahman, M.A.; Masuda, A.; Ohe, K.; Takeda, J.-I.; Ohno, K. HnRNP C, YB-1 and hnRNP L coordinately enhance skipping of human MUSK exon 10 to generate a Wnt-insensitive MuSK isoform. Sci. Rep. 2014, 4, 6841. [Google Scholar] [CrossRef]
- Le Hir, H.; Saulière, J.; Wang, Z. The exon junction complex as a node of post-transcriptional networks. Nat. Rev. Mol. Cell Biol. 2016, 17, 41–54. [Google Scholar] [CrossRef]
- Jackson, K.L.; Dayton, R.D.; Orchard, E.A.; Ju, S.; Ringe, D.; Petsko, G.A.; Maquat, L.E.; Klein, R.L. Preservation of forelimb function by UPF1 gene therapy in a rat model of TDP-43-induced motor paralysis. Gene. Ther. 2015, 22, 20–28. [Google Scholar] [CrossRef]
- Barmada, S.J.; Ju, S.; Arjun, A.; Batarse, A.; Archbold, H.C.; Peisach, D.; Li, X.; Zhang, Y.; Tank, E.M.H.; Qiu, H.; et al. Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc. Natl. Acad. Sci. USA 2015, 112, 7821–7826. [Google Scholar] [CrossRef]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Jin, H.; Kasarskis, E.J.; Zhu, H. ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. USA 2018, 115, E11904–E11913. [Google Scholar] [CrossRef]
- Xu, W.; Bao, P.; Jiang, X.; Wang, H.; Qin, M.; Wang, R.; Wang, T.; Yang, Y.; Lorenzini, I.; Liao, L.; et al. Reactivation of nonsense-mediated mRNA decay protects against C9orf72 dipeptide-repeat neurotoxicity. Brain 2019, 142, 1349–1364. [Google Scholar] [CrossRef]
- Sun, Y.; Eshov, A.; Guo, J.U. C9orf72 Dipeptide Repeats Inhibit UPF1-Mediated RNA Decay Independent of Stress Granule Formation. BioRxiv 2019, 623769. [Google Scholar] [CrossRef]
- Hautbergue, G.M.; Castelli, L.M.; Ferraiuolo, L.; Sanchez-Martinez, A.; Cooper-Knock, J.; Higginbottom, A.; Lin, Y.-H.; Bauer, C.S.; Dodd, J.E.; Myszczynska, M.A.; et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat. Commun. 2017, 8, 16063. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Mihevc, S.P.; Baralle, M.; Buratti, E.; Rogelj, B. TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci. Rep. 2016, 6, 33996. [Google Scholar] [CrossRef] [PubMed]
- Bakkar, N.; Kovalik, T.; Lorenzini, I.; Spangler, S.; Lacoste, A.; Sponaugle, K.; Ferrante, P.; Argentinis, E.; Sattler, R.; Bowser, R. Artificial intelligence in neurodegenerative disease research: Use of IBM Watson to identify additional RNA-binding proteins altered in amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 227–247. [Google Scholar] [CrossRef]
- Suzuki, H.; Shibagaki, Y.; Hattori, S.; Matsuoka, M. Nuclear TDP-43 causes neuronal toxicity by escaping from the inhibitory regulation by hnRNPs. Hum. Mol. Genet. 2015, 24, 1513–1527. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.; Nizzardo, M.; Vashisht, S.; Molteni, E.; Melzi, V.; Taiana, M.; Salani, S.; Santonicola, P.; Di Schiavi, E.; Bucchia, M.; et al. Key role of SMN/SYNCRIP and RNA-Motif 7 in spinal muscular atrophy: RNA-Seq and motif analysis of human motor neurons. Brain 2019, 142, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Dufu, K.; Romney, B.; Feldt, M.; Elenko, M.; Reed, R. Functional coupling of RNAP II transcription to spliceosome assembly. Genome Res. 2006, 20, 1100–1109. [Google Scholar] [CrossRef]
- Yu, Y.; Reed, R. FUS functions in coupling transcription to splicing by mediating an interaction between RNAP II and U1 snRNP. Proc. Natl. Acad. Sci. USA 2015, 112, 8608–8613. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Ebmeier, C.C.; Podell, E.R.; Heimiller, J.; Taatjes, D.J.; Cech, T.R. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012, 26, 2690–2695. [Google Scholar] [CrossRef]
- Zhao, R.Y.; Ni, Z.; Pu, S.; Zhong, G.; Schmitges, F.W.; Braunschweig, U.; Blencowe, B.J.; Greenblatt, J.F.; Zhao, Y. Regulation of transcription termination by FUS and TDP-43. BioRXiv 2019, 788778. [Google Scholar] [CrossRef]
- Boehringer, A.; Garcia-Mansfield, K.; Singh, G.; Bakkar, N.; Pirrotte, P.; Bowser, R. ALS Associated Mutations in Matrin 3 Alter Protein-Protein Interactions and Impede mRNA Nuclear Export. Sci. Rep. 2017, 7, 14529. [Google Scholar] [CrossRef] [PubMed]
- Chanarat, S.; Sträßer, K. Splicing and beyond: The many faces of the Prp19 complex. Biochim. Biophys. Acta (BBA) 2013, 1833, 2126–2134. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Greensmith, L. Cellular Chaperones As Therapeutic Targets in ALS to Restore Protein Homeostasis and Improve Cellular Function. Front. Mol. Neuro Sci. 2017, 10, 251. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Lorenzini, I.; Chou, C.-C.; Torvund, M.; Rogers, R.S.; Starr, A.; Zaepfel, B.L.; Levy, J.; Johannesmeyer, J.; Schwartz, J.C.; et al. Post-transcriptional Inhibition of Hsc70-4/HSPA8 Expression Leads to Synaptic Vesicle Cycling Defects in Multiple Models of ALS. Cell Rep. 2017, 21, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Battle, D.; Kasim, M.; Yong, J.; Lotti, F.; Lau, C.-K.; Mouaikel, J.; Zhang, Z.; Han, K.; Wan, L.; Dreyfuss, G. The SMN Complex: An Assembly Machine for RNPs. Cold Spring Harb. Symp. Quant. Boil. 2006, 71, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Gubitz, A. The SMN complex. Exp. Cell Res. 2004, 296, 51–56. [Google Scholar] [CrossRef]
- Morello, G.; Guarnaccia, M.; Spampinato, A.G.; La Cognata, V.; D’Agata, V.; Cavallaro, S. Copy Number Variations in Amyotrophic Lateral Sclerosis: Piecing the Mosaic Tiles Together through a Systems Biology Approach. Mol. Neurobiol. 2018, 55, 1299–1322. [Google Scholar] [CrossRef]
- Zou, T.; Ilangovan, R.; Yu, F.; Xu, Z.; Zhou, J. SMN Protects cells against mutant SOD1 toxicity by increasing chaperone activity. Biochem. Biophys. Res. Commun. 2007, 364, 850–855. [Google Scholar] [CrossRef]
- Rodríguez-Muela, N.; Litterman, N.K.; Norabuena, E.M.; Mull, J.L.; Galazo, M.J.; Sun, C.; Ng, S.-Y.; Makhortova, N.R.; White, A.; Lynes, M.M.; et al. Single-Cell Analysis of SMN Reveals Its Broader Role in Neuromuscular Disease. Cell Rep. 2017, 18, 1484–1498. [Google Scholar] [CrossRef]
- Shefer, K.; Sperling, J.; Sperling, R. The Supraspliceosome—A Multi-Task Machine for Regulated Pre-mRNA Processing in the Cell Nucleus. Comput. Struct. Biotechnol. J. 2014, 11, 113–122. [Google Scholar] [CrossRef]
- Azubel, M.; Habib, N.; Sperling, R.; Sperling, J. Native Spliceosomes Assemble with Pre-mRNA to Form Supraspliceosomes. J. Mol. Boil. 2006, 356, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J. Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kim, P.M.; Sprecher, E.; Trifonov, V.; Gerstein, M. The Importance of Bottlenecks in Protein Networks: Correlation with Gene Essentiality and Expression Dynamics. PLoS Comput. Boil. 2007, 3, e59. [Google Scholar] [CrossRef] [PubMed]
- Van Rheenen, W.; Diekstra, F.P.; Harschnitz, O.; Westeneng, H.-J.; Van Eijk, K.R.; Saris, C.G.J.; Groen, E.J.N.; Van Es, M.A.; Blauw, H.M.; Van Vught, P.W.J.; et al. Whole blood transcriptome analysis in amyotrophic lateral sclerosis: A biomarker study. PLoS ONE 2018, 13, e0198874. [Google Scholar] [CrossRef] [PubMed]
- Guarino, A.M.; Di Mauro, G.; Ruggiero, G.; Geyer, N.; Delicato, A.; Foulkes, N.S.; Vallone, D.; Calabrò, V. YB-1 recruitment to stress granules in zebrafish cells reveals a differential adaptive response to stress. Sci. Rep. 2019, 9, 9059. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, D.; Battich, N.; Pelkmans, L. A Systems-Level Study Reveals Regulators of Membrane-less Organelles in Human Cells. Mol. Cell 2018, 72, 1035–1049.e5. [Google Scholar] [CrossRef] [PubMed]
- Abrakhi, S.; Kretov, D.A.; Desforges, B.; Dobra, I.; Bouhss, A.; Pastré, D.; Hamon, L. Nanoscale Analysis Reveals the Maturation of Neurodegeneration-Associated Protein Aggregates: Grown in mRNA Granules then Released by Stress Granule Proteins. ACS Nano 2017, 11, 7189–7200. [Google Scholar] [CrossRef]
- Morello, G.; Spampinato, A.G.; Conforti, F.L.; D’Agata, V.; Cavallaro, S. Selection and Prioritization of Candidate Drug Targets for Amyotrophic Lateral Sclerosis through a Meta-Analysis Approach. J. Mol. Neurosci. 2017, 61, 563–580. [Google Scholar] [CrossRef]
- Morello, G.; Cavallaro, S. Transcriptional analysis reveals distinct subtypes in amyotrophic lateral sclerosis: Implications for personalized therapy. Future Med. Chem. 2015, 7, 1335–1359. [Google Scholar] [CrossRef]
- Morello, G.; Conforti, F.L.; Parenti, R.; D’Agata, V.; Cavallaro, S. Selection of Potential Pharmacological Targets in ALS Based on Whole-Genome Expression Profiling. Curr. Med. Chem. 2015, 22, 2004–2021. [Google Scholar] [CrossRef]
- Yin, W.; Rogge, M. TargetingRNA: A Transformative Therapeutic Strategy. Clin. Transl. Sci. 2019, 12, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 2019, 102, 294–320. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Clusters | Motor Cortex | Spinal Cord |
|---|---|---|
| SALS1 | 18 | 17 |
| SALS2 | 13 | 13 |
| CTRL | 10 | 10 |
| TOT | 41 | 40 |
| Enriched GO Cellular Component | Major Disease State Changes in Tissue Types | Major Tissue Type Changes in Disease States | |||||
|---|---|---|---|---|---|---|---|
| SALS1 Motor Cortex vs Control | SALS2 Motor Cortex vs Control | SALS1 Spinal Cord vs Control | SALS2 Spinal Cord vs Control | Ctrl | SALS1 | SALS2 | |
| U1 snRNP |  1.55 × 109 1.55 × 109 |  3.70 × 107 3.70 × 107 | 5.18 × 108 | ||||
| A2AF | 1.24 × 102 |  2.87× 102 2.87× 102 | 2.82× 102 | ||||
| U2 snRNP | 1.59 × 109 | 8.44 × 109 | 1.38 × 109 | ||||
| U2-type prespliceosome | 8.35 × 1010 | 8.33 × 106 | 7.23 × 1010 | ||||
| U2-type precatalytic spliceosome | 2.84 × 106 | 8.57 × 1028 | 2.80 × 1015 | 2.89 × 1026 | |||
| U2-type catalytic step 1 spliceosome | 4.21× 102 | 1.46 × 104 | 2.65 × 103 | 4.01 × 106 | |||
| U2-type catalytic step 2 spliceosome | 2.29 × 107 | 6.89 × 1013 | 6.47 × 1011 | 2.09 × 1011 | |||
| U4 snRNP | 3.36 × 103 | 3.22 × 103 | |||||
| U5 snRNP | 6.80 × 107 | 1.54 × 108 | 5.47 × 106 | 5.55 × 107 | |||
| U6 snRNP | 1.38 × 106 | 1.26 × 106 | |||||
| U4/U6.U5 tri-snRNP complex | 3.39 × 104 | 1.61 × 1017 | 8.01 × 1011 | 5.64 × 1016 | |||
| U12-type spliceosomal complex | 3.75× 106 | 6.35 × 109 | 4.30 × 102 | 1.09 × 106 | 1.81 × 107 | ||
| Exon-exon junction complex | 9.41 × 105 | 2.07 × 109 | 4.60 × 107 | 5.09 × 1011 | |||
| CRD-mediated mRNA stability complex | 1.90 × 104 | 1.02 × 103 | 2.70 × 102 | 2.35 × 105 | |||
| RNA polymerase II, core complex | 2.84 × 1010 | 1.65 × 104 | 2.45 × 1010 | ||||
| Prp19 complex | 2.44 × 104 | 2.38 × 104 | |||||
| SMN-Sm protein complex | 1.90 × 105 | 5.93 × 103 | 1.64 × 105 | ||||
| SMN complex | 3.31 × 103 | 3.18 × 103 | |||||
| pICln-Sm protein complex | 2.68 × 105 | 2.39 × 105 | |||||
| Supraspliceosomal complex | 2.84 × 104 | 1.64 × 104 | 1.43 × 102 | ||||
| Spliceosomal complex | 1.11 × 104 | ||||||
| Catalytic step 2 spliceosome | 6.15 × 103 | 9.80 × 103 | |||||
| Post-mRNA release spliceosomal complex | 6.29 × 103 | 3.74 × 103 | 2.34 × 104 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Cognata, V.; Gentile, G.; Aronica, E.; Cavallaro, S. Splicing Players Are Differently Expressed in Sporadic Amyotrophic Lateral Sclerosis Molecular Clusters and Brain Regions. Cells 2020, 9, 159. https://doi.org/10.3390/cells9010159
La Cognata V, Gentile G, Aronica E, Cavallaro S. Splicing Players Are Differently Expressed in Sporadic Amyotrophic Lateral Sclerosis Molecular Clusters and Brain Regions. Cells. 2020; 9(1):159. https://doi.org/10.3390/cells9010159
Chicago/Turabian StyleLa Cognata, Valentina, Giulia Gentile, Eleonora Aronica, and Sebastiano Cavallaro. 2020. "Splicing Players Are Differently Expressed in Sporadic Amyotrophic Lateral Sclerosis Molecular Clusters and Brain Regions" Cells 9, no. 1: 159. https://doi.org/10.3390/cells9010159
APA StyleLa Cognata, V., Gentile, G., Aronica, E., & Cavallaro, S. (2020). Splicing Players Are Differently Expressed in Sporadic Amyotrophic Lateral Sclerosis Molecular Clusters and Brain Regions. Cells, 9(1), 159. https://doi.org/10.3390/cells9010159