Endoglin Trafficking/Exosomal Targeting in Liver Cells Depends on N-Glycosylation



Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning and Purification of Adenoviral Expression Vectors
2.2. Culturing of Immortalized Cell Lines
2.3. Isolation and Culturing of Primary Cells from Rat and Mouse
2.4. Remarks on Ethical Issues on Animal Experimentation
2.5. Stimulation of Cells
2.6. Transient Transfection
2.7. Sucrose Gradient Analysis
2.8. Adenoviral Infection
2.9. Isolation and Characterization of Exosomes from Conditioned Media
2.10. Tunicamycin Treatment
2.11. Precipitation of Glycosylated Proteins with ConA Beads
2.12. Co-Immunoprecipitation of Endoglin and TGF-β1 with PPabE2 from Supernatants of sol-Eng and Mock-Transfected HepG2
2.13. Sodium Dodecylsulfate Polyacrylamide Gel Electrophoresis and Western Blot Analysis
3. Results
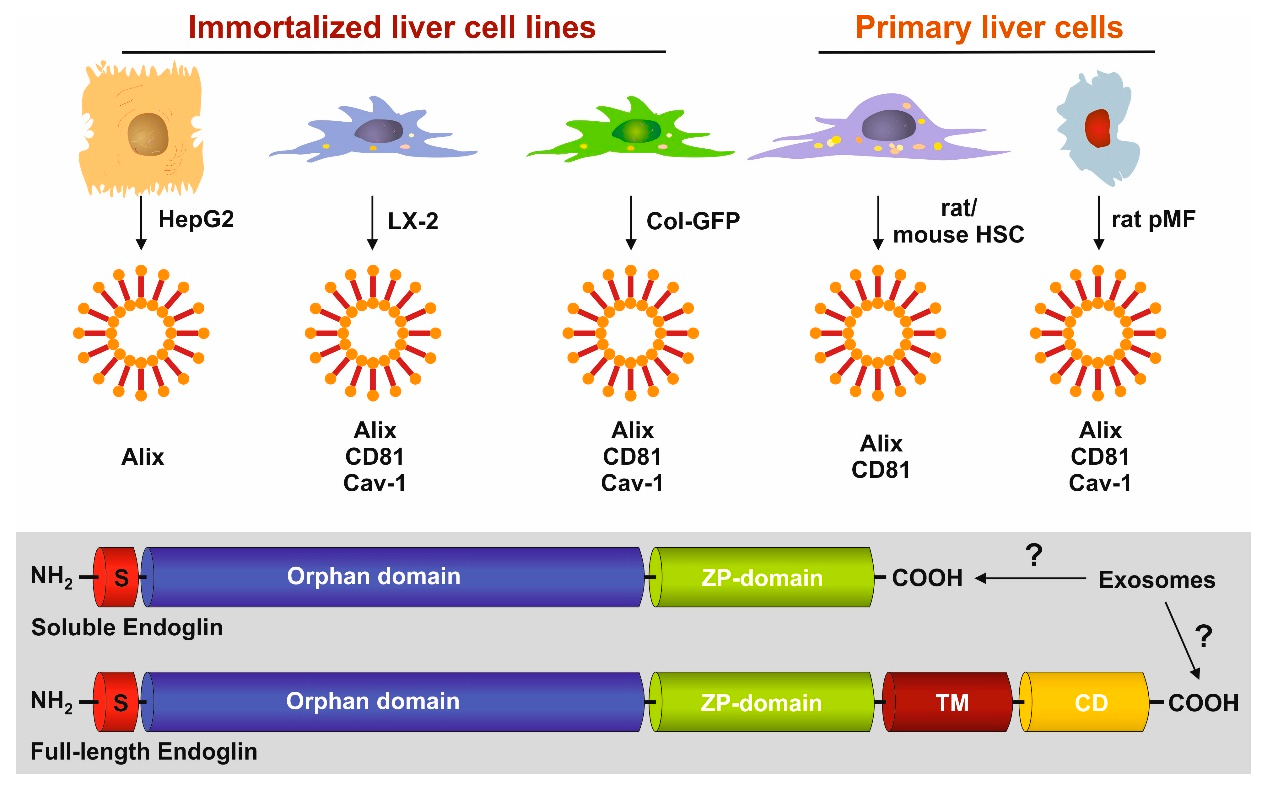
3.1. FL-Endoglin Is Co-Localized with Caveolin-1 in the Lipid Raft Fraction
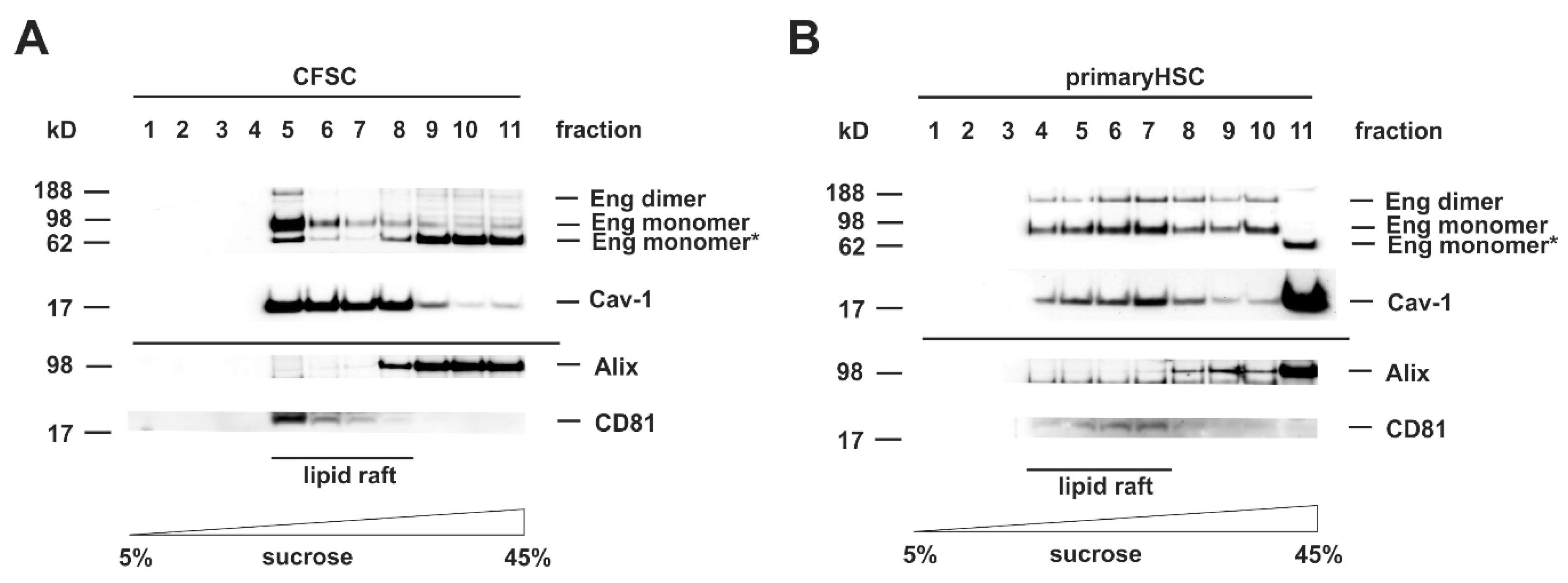
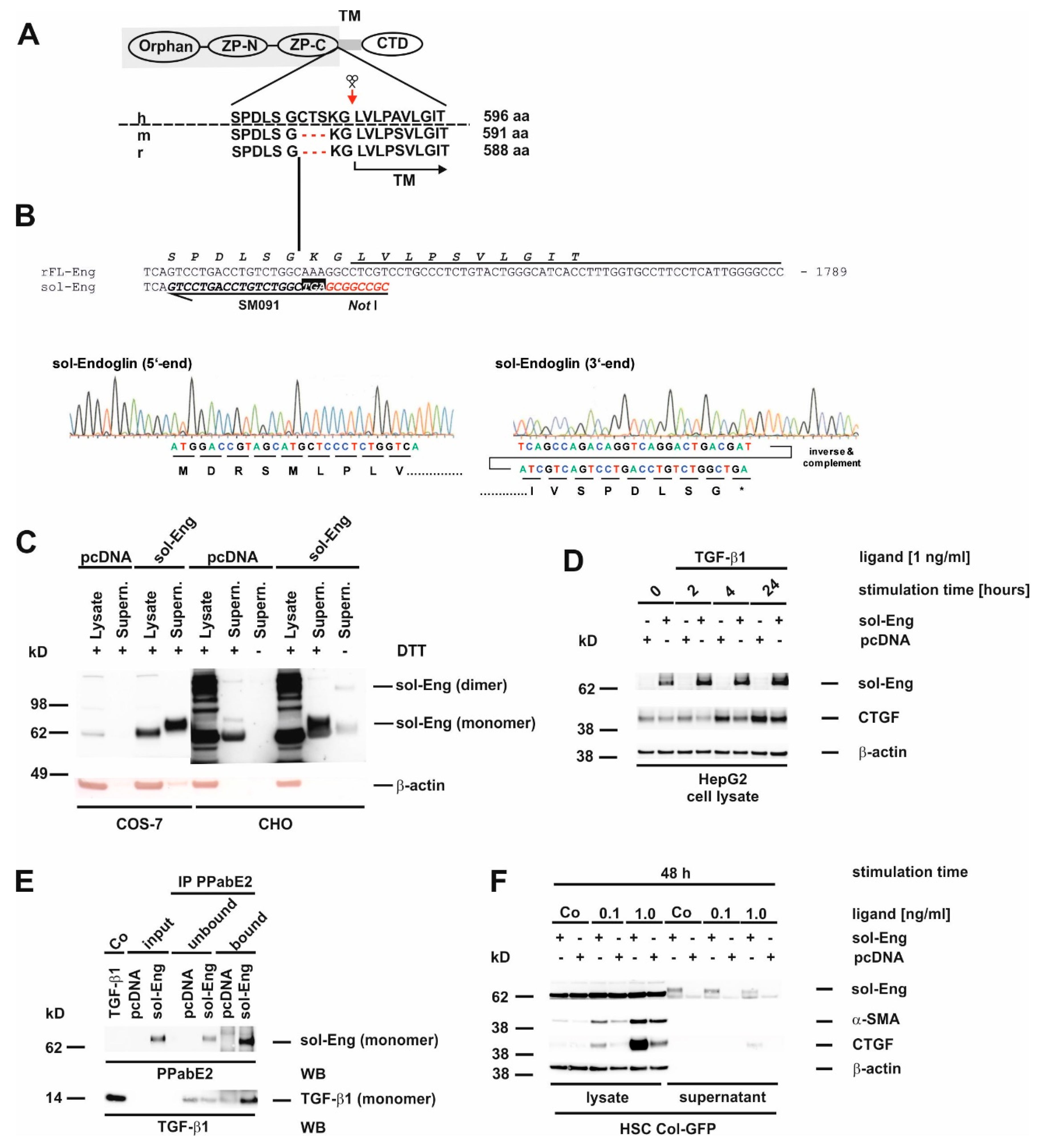
3.2. Cloning of an Adenoviral Vector for Expression of Rat Full-Length Endoglin
3.3. Cloning of an Adenoviral Vector for Expression of Rat Soluble Endoglin
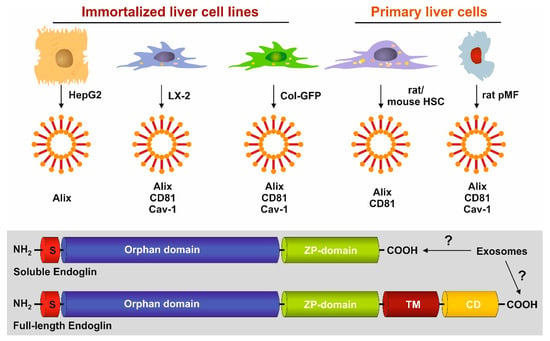
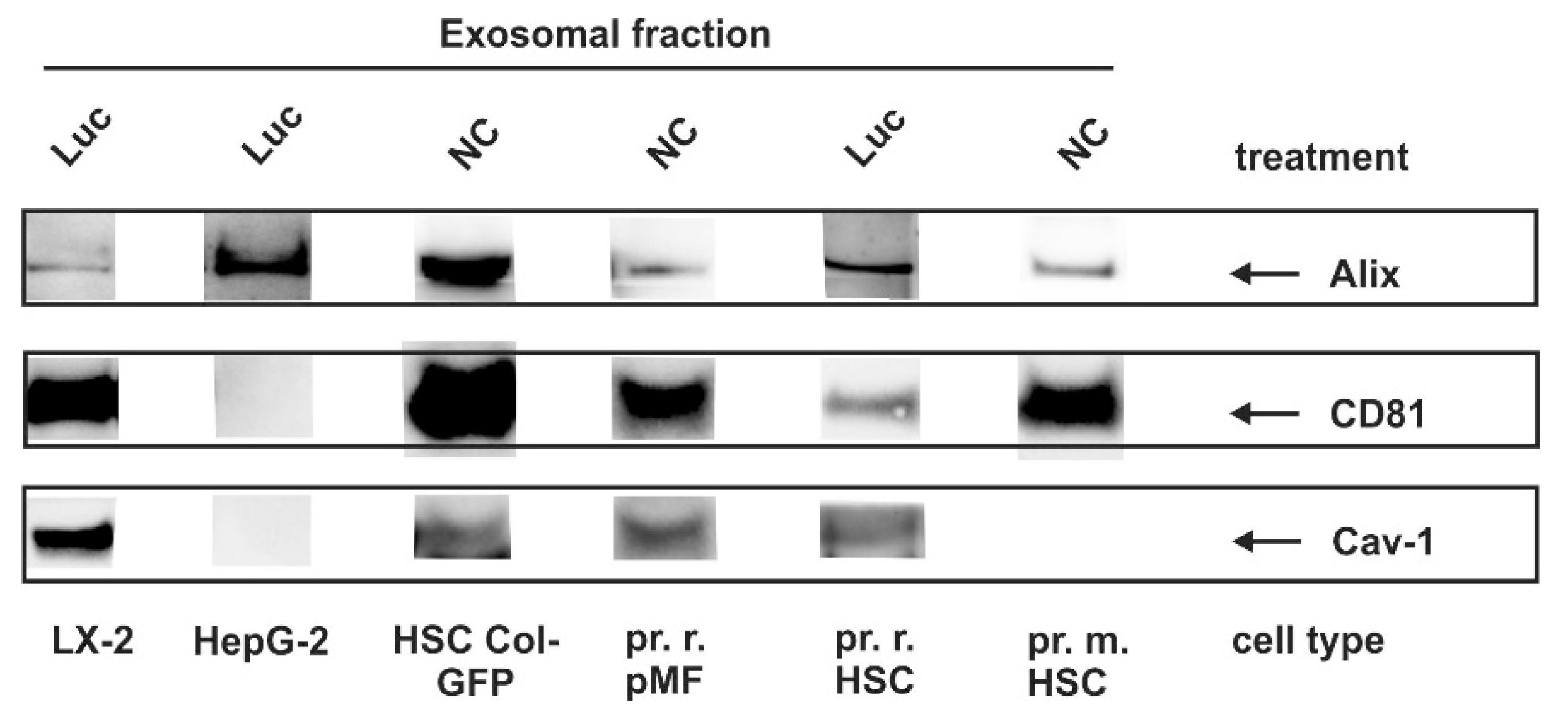
3.4. Identification of Exosomal Marker Proteins
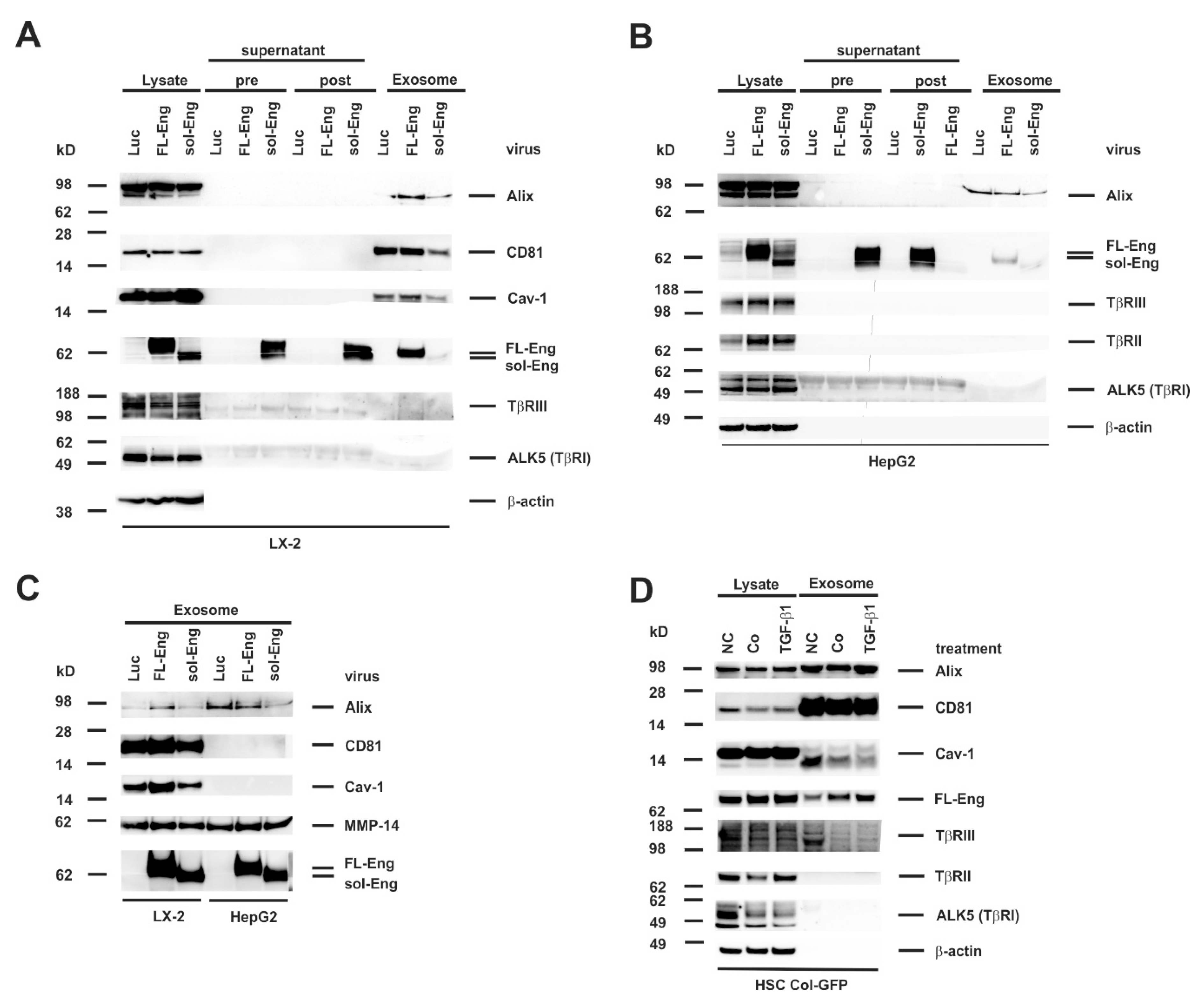
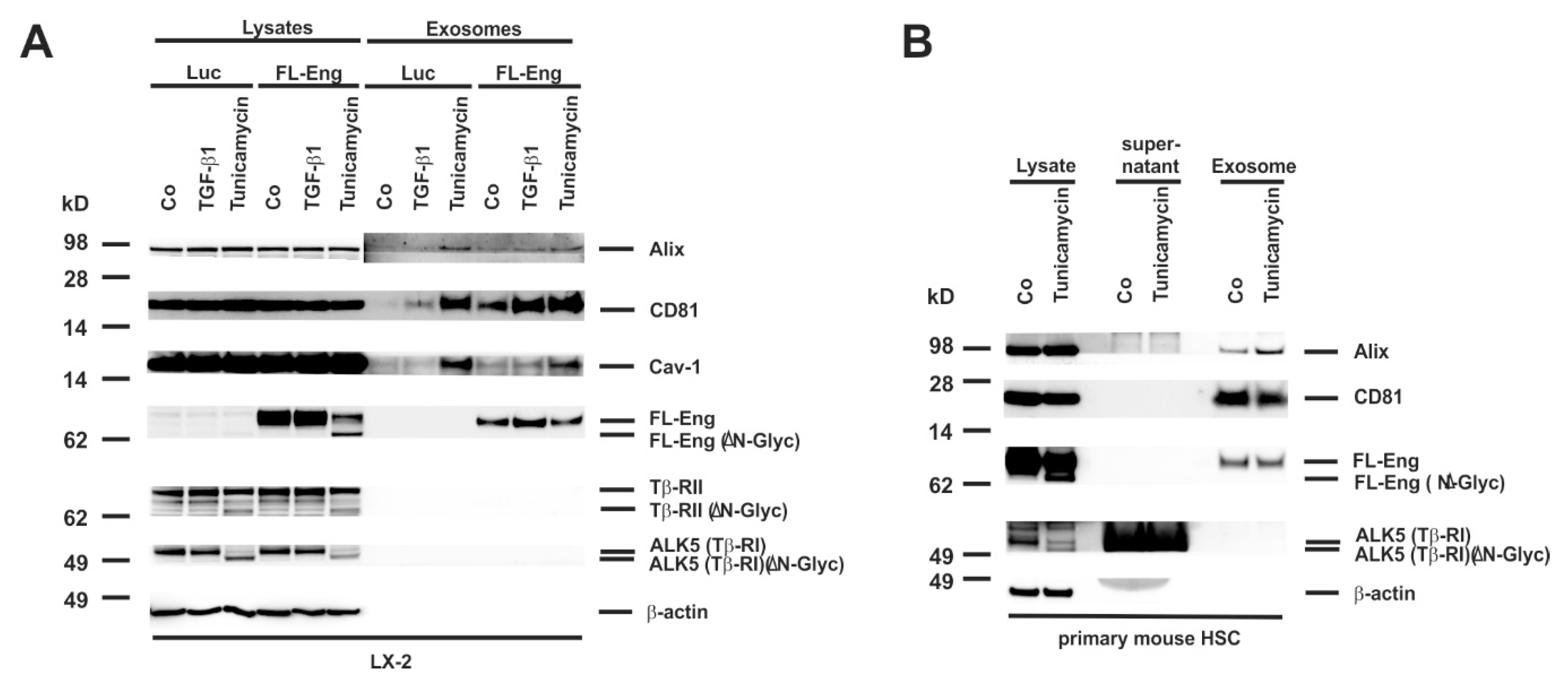
3.5. Full-Length and Soluble Endoglin Are Targeted to the Exosome Compartment of Permanent Cell Lines
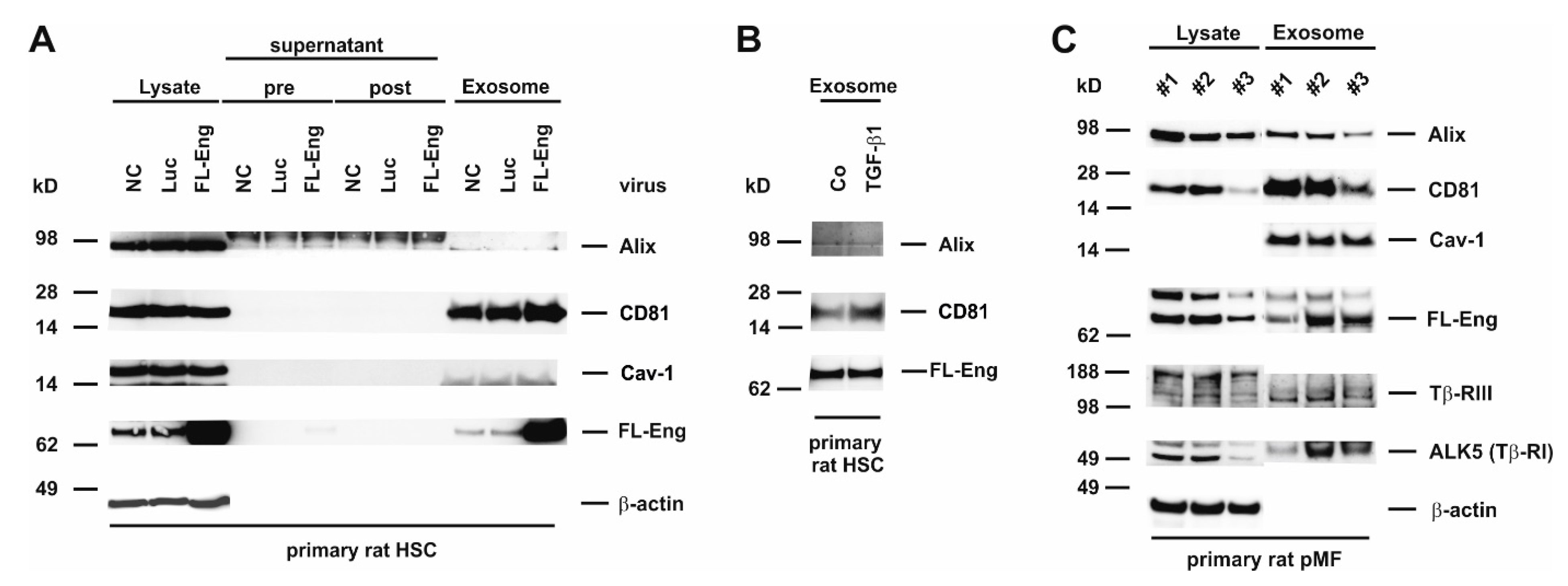
3.6. Full-Length Endoglin Is Targeted to the Exosome Compartment of Primary Mesenchymal Liver Cells
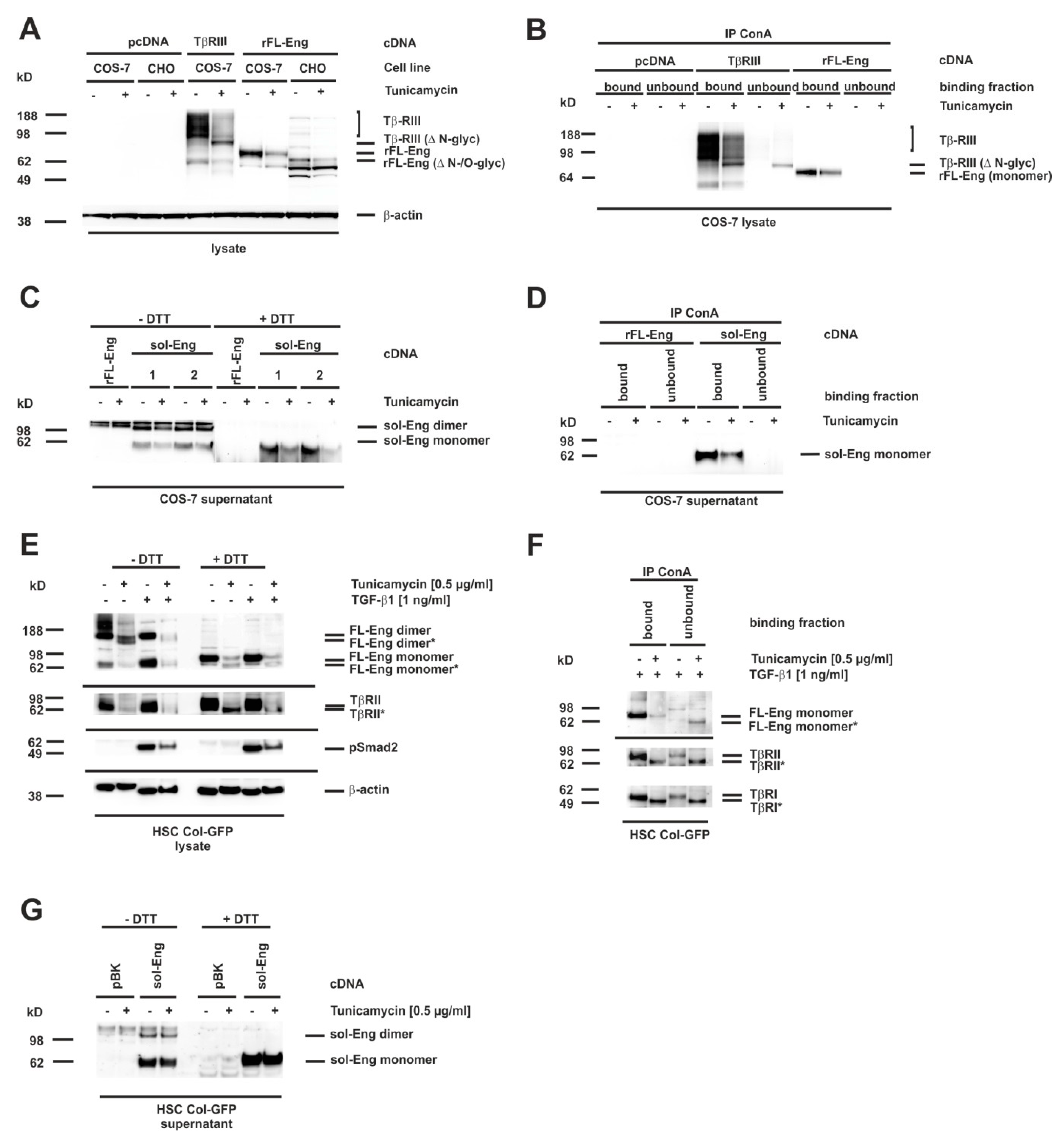
3.7. Glycosylation of Endoglin
3.8. Proper Glycosylation of Endoglin Is Necessary for Exosome Targeting
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| Alix | Apoptosis-linked gene 2-interacting protein |
| BMP | bone morphogenetic proteins |
| Cav-1 | Caveolin-1 |
| ccp | clathrin coated pits |
| CD | cytosolic domain |
| CTGF | connective tissue growth factor |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DTT | dithiothreitol |
| Eng | Endoglin |
| FL-Eng | full-length endoglin |
| GFP | green fluorescent protein |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| HHT-1 | hereditary hemorrhagic telangiectasia type-1 |
| HSC | hepatic stellate cells |
| KC | Kupffer cell(s) |
| LSEC | Liver sinusoidal endothelial cell(s) |
| Luc | luciferase |
| MMP-14 | metalloproteinase-14 |
| pMF | portal myofibroblast |
| sol-Eng | soluble endoglin |
| TβRI-III | TGF-β receptors contains type I-III |
| TBST | Tris-buffered saline with Tween 20 |
| TGF-β | transforming growth factor-β |
| TM | transmembrane domain |
References
- Nickel, J.; Ten Dijke, P.; Mueller, T.D. TGF-β family co-receptor function and signaling. Acta Biochim. Biophys. Sin. 2018, 50, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Tacke, F. Liver fibrosis: From pathogenesis to novel therapies. Dig. Dis. 2016, 34, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Meurer, S.K.; Gressner, O.A.; Herrmann, J.; Borkham-Kamphorst, E.; Gressner, A.M. BMP-7 as antagonist of organ fibrosis. Front. Biosci. 2009, 14, 4992–5012. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, R.; Velazquez, V.M.; Brigstock, D.R. Fibrogenic signaling is suppressed in hepatic stellate cells through targeting of connective tissue growth factor (CCN2) by Cellular or Exosomal MicroRNA-199a-5p. Am. J. Pathol. 2016, 186, 2921–2933. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.M.; Weiskirchen, R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J. Cell. Mol. Med. 2006, 10, 76–99. [Google Scholar] [CrossRef]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming growth factor-β receptors and Smads: Regulatory complexity and functional versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Andres, J.L.; Stanley, K.; Cheifetz, S.; Massagué, J. Membrane-anchored and soluble forms of betaglycan, a polymorphic proteoglycan that binds transforming growth factor-β. J. Cell Biol. 1989, 109, 3137–3145. [Google Scholar] [CrossRef]
- Gougos, A.; Letarte, M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J. Biol. Chem. 1990, 265, 8361–8364. [Google Scholar] [PubMed]
- Lux, A.; Gallione, C.J.; Marchuk, D.A. Expression analysis of endoglin missense and truncation mutations: Insights into protein structure and disease mechanisms. Hum. Mol. Genet. 2000, 9, 745–755. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, Y.W.; Park, J.; Lee, H.J.; Lee, S.Y.; Kim, S.J. TGF-β sensitivity is determined by N-linked glycosylation of the type II TGF-β receptor. Biochem. J. 2012, 445, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.M.; Singh, P.; Varadaraj, A.; Lee, N.Y.; Shah, S.; Flores, H.V.; O’Connell, K.; Mythreye, K. Altering the proteoglycan state of transforming growth factor β type III receptor (TβRIII)/betaglycan modulates canonical Wnt/β-catenin signaling. J. Biol. Chem. 2016, 291, 25716–25728. [Google Scholar] [CrossRef] [PubMed]
- López-Casillas, F.; Cheifetz, S.; Doody, J.; Andres, J.L.; Lane, W.S.; Massagué, J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-β receptor system. Cell 1991, 67, 785–795. [Google Scholar] [CrossRef]
- Esparza-Lopez, J.; Montiel, J.L.; Vilchis-Landeros, M.M.; Okadome, T.; Miyazono, K.; López-Casillas, F.J. Ligand binding and functional properties of betaglycan, a co-receptor of the transforming growth factor-beta superfamily. Specialized binding regions for transforming growth factor-beta and inhibin A. Biol. Chem. 2001, 276, 14588–14596. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [PubMed]
- Velasco, S.; Alvarez-Muñoz, P.; Pericacho, M.; Dijke, P.T.; Bernabéu, C.; López-Novoa, J.M.; Rodríguez-Barbero, A. L- and S-endoglin differentially modulate TGFbeta1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 2008, 121, 913–919. [Google Scholar] [CrossRef]
- Meurer, S.K.; Tihaa, L.; Borkham-Kamphorst, E.; Weiskirchen, R. Expression and functional analysis of endoglin in isolated liver cells and its involvement in fibrogenic Smad signalling. Cell. Signal. 2011, 23, 683–699. [Google Scholar] [CrossRef]
- Meurer, S.K.; Alsamman, M.; Scholten, D.; Weiskirchen, R. Endoglin in liver fibrogenesis: Bridging basic science and clinical practice. World J. Biol. Chem. 2014, 5, 180–203. [Google Scholar] [CrossRef]
- Lee, N.Y.; Blobe, G.C. The interaction of endoglin with beta-arrestin2 regulates transforming growth factor-beta-mediated ERK activation and migration in endothelial cells. J. Biol. Chem. 2007, 282, 21507–21517. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Ray, B.; How, T.; Blobe, G.C. Endoglin promotes transforming growth factor beta-mediated Smad 1/5/8 signaling and inhibits endothelial cell migration through its association with GIPC. J. Biol. Chem. 2008, 283, 32527–32533. [Google Scholar] [CrossRef] [PubMed]
- Paquet, M.E.; Pece-Barbara, N.; Vera, S.; Cymerman, U.; Karabegovic, A.; Shovlin, C.; Letarte, M. Analysis of several endoglin mutants reveals no endogenous mature or secreted protein capable of interfering with normal endoglin function. Hum. Mol. Genet. 2001, 10, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Mallet, C.; Lamribet, K.; Giraudm, S.; Dupuis-Girod, S.; Feige, J.J.; Bailly, S.; Tillet, E. Functional analysis of endoglin mutations from hereditary hemorrhagic telangiectasia type 1 patients reveals different mechanisms for endoglin loss of function. Hum. Mol. Genet. 2015, 24, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Förg, T.; Hafner, M.; Lux, A. Investigation of endoglin wild-type and missense mutant protein heterodimerisation using fluorescence microscopy based IF, BiFC and FRET analyses. PLoS ONE 2014, 9, e102998. [Google Scholar] [CrossRef] [PubMed]
- Ali, B.R.; Ben-Rebeh, I.; John, A.; Akawi, N.A.; Milhem, R.M.; Al-Shehhi, N.A.; Al-Ameri, M.M.; Al-Shamisi, S.A.; Al-Gazali, L. Endoplasmic reticulum quality control is involved in the mechanism of endoglin-mediated hereditary haemorrhagic telangiectasia. PLoS ONE 2011, 6, e26206. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Vara, E.; Tual-Chalot, S.; Botella, L.M.; Arthur, H.M.; Bernabeu, C. Soluble endoglin regulates expression of angiogenesis-related proteins and induction of arteriovenous malformations in a mouse model of hereditary hemorrhagic telangiectasia. Dis. Model. Mech. 2018, 11, dmm034397. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.; Kuiper, P.; Wiercinska, E.; Verspaget, H.W.; Liu, Z.; Pardali, E.; Sier, C.F.; ten Dijke, P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010, 70, 4141–4150. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011, 286, 30034–30046. [Google Scholar] [CrossRef]
- Chang, X.; Yao, J.; He, Q.; Liu, M.; Duan, T.; Wang, K. Exosomes from women with preeclampsia induced vascular dysfunction by delivering sFlt (soluble Fms-like tyrosine kinase)-1 and sEng (soluble Endoglin) to endothelial cells. Hypertension 2018, 72, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrahim, S.H.; Verma, V.K.; Morton, L.A.; Shah, V.H.; LaRusso, N.F.; Gores, G.J.; Malhi, H. Extracellular vesicles in liver pathobiology: Small particles with big impact. Hepatology 2016, 64, 2219–2233. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Brenner, D.A.; Kisseleva, T. Combatting fibrosis: Exosome-based therapies in the regression of liver fibrosis. Hepatol. Commun. 2018, 3, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Xia, T.; Du, Y.; Liu, J.; Xie, Y.; Zhang, Y.; Guan, F.; Wu, J.; Wang, X.; Shi, C. Exosomes from actviated hepatic stellate cells contain GLUT1 and PKM2: A role for exosomes in metabolic switch of liver nonparenchymal cells. FASEB J. 2019, 33, 8530–8542. [Google Scholar] [CrossRef] [PubMed]
- Meurer, S.K.; Tihaa, L.; Lahme, B.; Gressner, A.M.; Weiskirchen, R. Identification of endoglin in rat hepatic stellate cells: New insights into transforming growth factor β receptor signaling. J. Biol. Chem. 2005, 280, 3078–3887. [Google Scholar] [CrossRef] [PubMed]
- Alsamman, M.; Sterzer, V.; Meurer, S.K.; Sahin, H.; Schaeper, U.; Kuscuoglu, D.; Strnad, P.; Weiskirchen, R.; Trautwein, C.; Scholten, D. Endoglin in human liver disease and murine models of liver fibrosis—A protective factor against liver fibrosis. Liver Int. 2018, 38, 858–867. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Zhou, S.; da Costa, L.T.; Yu, J.; Kinzler, K.W.; Vogelstein, B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 2509–2514. [Google Scholar] [CrossRef]
- Meurer, S.K.; Rizk, M.S.; Tihaa, L.; Weiskirchen, R.; Gressner, A.M. Soluble endoglin modulated TGF-β1 mediated signaling in isolated hepatocytes. Z. Gastroenterol. 2008, 46, P2_36. [Google Scholar] [CrossRef]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef]
- Knowles, B.B.; Howe, C.C.; Aden, D.P. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science 1980, 209, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Meurer, S.K.; Alsamman, M.; Sahin, H.; Wasmuth, H.E.; Kisseleva, T.; Brenner, D.A.; Trautwein, C.; Weiskirchen, R.; Scholten, D. Overexpression of endoglin modulates TGF-β1-signalling pathways in a novel immortalized mouse hepatic stellate cell line. PLoS ONE 2013, 8, e56116. [Google Scholar] [CrossRef] [PubMed]
- Greenwel, P.; Schwartz, M.; Rosas, M.; Peyrol, S.; Grimaud, J.A.; Rojkind, M. Characterization of fat-storing cell lines derived from normal and CCl4-cirrhotic livers. Differences in the production of interleukin-6. Lab. Investig. 1991, 65, 644–653. [Google Scholar] [PubMed]
- Stanley, P. Chinese hamster ovary cell mutants with multiple glycosylation defects for production of glycoproteins with minimal carbohydrate heterogeneity. Mol. Cell. Biol. 1989, 9, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Van de Leur, E.; Meurer, S.K.; Buhl, E.M.; Weiskirchen, R. N-glycosylation of Lipocalin 2 is not required for secretion or exosome targeting. Front. Pharmacol. 2018, 9, 426. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Gressner, A.M. Isolation and culture of hepatic stellate cells. Methods Mol. Med. 2005, 117, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, S.; Tag, C.G.; Sauer-Lehnen, S.; Tacke, F.; Weiskirchen, R. Isolation and culture of primary murine hepatic stellate cells. Methods Mol. Biol. 2017, 1627, 165–191. [Google Scholar] [CrossRef] [PubMed]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.; Wiegmans, A.P.; Leimgruber, A.; Möller, A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Kneifel, J.; Weiskirchen, S.; van de Leur, E.; Kunz, D.; Gressner, A.M. Comparative evaluation of gene delivery devices in primary cultures of rat hepatic stellate cells and rat myofibroblasts. BMC Cell Biol. 2000, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G.; Yankelev, H.; Lin, H.Y.; Lodish, H.F. Biosynthesis of the type I and type II TGF-beta receptors. Implications for complex formation. J. Biol. Chem. 1997, 272, 11444–11451. [Google Scholar] [CrossRef] [PubMed]
- Toporsian, M.; Gros, R.; Kabir, M.G.; Vera, S.; Govindaraju, K.; Eidelman, D.H.; Husain, M.; Letarte, M. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ. Res. 2005, 96, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Ilha, M.; Moraes, K.D.S.; Rohden, F.; Martins, L.A.M.; Borojevic, R.; Lenz, G.; Barbé-Tuana, F.; Guma, F.C.R. Exogenous expression of caveolin-1 is sufficient for hepatic stellate cell activation. J. Cell. Biochem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jabalee, J.; Towle, R.; Garnis, C. The role of extracellular vesicles in cancer: Cargo, function, and therapeutic implications. Cells 2018, 7, 93. [Google Scholar] [CrossRef]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Holman, N.S.; Church, R.J.; Nautiyal, M.; Rose, K.A.; Thacker, S.E.; Otieno, M.A.; Wolf, K.K.; LeCluyse, E.; Watkins, P.B.; Mosedale, M. Hepatocyte-derived exosomes promote liver immune tolerance: Possible implications for idiosyncratic drug-induced liver injury. Toxicol. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Zhu, W.; Wang, Y.; Zhao, D.; Wang, X.; Gurley, E.C.; Liang, G.; Chen, W.; Lai, G.; et al. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes hepatic stellate cell activation and cholestatic liver fibrosis. Hepatology 2019. [Google Scholar] [CrossRef]
- Sasaki, R.; Kanda, T.; Yokosuka, O.; Kato, N.; Matsuoka, S.; Moriyama, M. Exosomes and hepatocellular carcinoma: From bench to bedside. Int. J. Mol. Sci. 2019, 20, 1406. [Google Scholar] [CrossRef]
- Chen, L.; Charrier, A.; Zhou, Y.; Chen, R.; Yu, B.; Agarwal, K.; Tsukamoto, H.; Lee, L.J.; Paulaitis, M.E.; Brigstock, D.R. Epigenetic regulation of connective tissue growth factor by MicroRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology 2014, 59, 1118–1129. [Google Scholar] [CrossRef]
- Grange, C.; Tapparo, M.; Collino, F.; Vitillo, L.; Damasco, C.; Deregibus, M.C.; Tetta, C.; Bussolati, B.; Camussi, G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011, 71, 5346–5356. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Gercel-Taylor, C. Exosomes/microvesicles: Mediators of cancer-associated immunosuppressive microenvironments. Semin. Immunopathol. 2011, 33, 441–454. [Google Scholar] [CrossRef]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef]
- Campos, A.; Salomon, C.; Bustos, R.; Díaz, J.; Martínez, S.; Silva, V.; Reyes, C.; Díaz-Valdivia, N.; Varas-Godoy, M.; Lobos-González, L.; et al. Caveolin-1-containing extracellular vesicles transport adhesion proteins and promote malignancy in breast cancer cell lines. Nanomedicine 2018, 13, 2597–2609. [Google Scholar] [CrossRef] [PubMed]
- Ermini, L.; Ausman, J.; Melland-Smith, M.; Yeganeh, B.; Rolfo, A.; Litvack, M.L.; Todros, T.; Letarte, M.; Post, M.; Caniggia, I. A single sphingomyelin species promotes exosomal release of Endoglin into the maternal circulation in preeclampsia. Sci. Rep. 2017, 7, 12172. [Google Scholar] [CrossRef]
- Batista, B.S.; Eng, W.S.; Pilobello, K.T.; Hendricks-Muñoz, K.D.; Mahal, L.K. Identification of a conserved glycan signature for microvesicles. J. Proteome Res. 2011, 10, 4624–4633. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Tsang, K.S.; Wang, Y.; Chan, J.C.; Xu, G.; Gao, W.Q. Unfolded protein response is required for the definitive endodermal specification of mouse embryonic stem cells via Smad2 and β-catenin signaling. J. Biol. Chem. 2014, 289, 26290–26301. [Google Scholar] [CrossRef]
- Eguchi, A.; Feldstein, A.E. Extracellular vesicles in non-alcoholic and alcoholic fatty liver diseases. Liver Res. 2018, 2, 30–34. [Google Scholar] [CrossRef]
- Cai, S.; Cheng, X.; Pan, X.; Li, J. Emerging role of exosomes in liver physiology and pathology. Hepatol. Res. 2017, 47, 194–203. [Google Scholar] [CrossRef]
- Han, W.; Duan, Z. Roles of exosomes in liver metastases: Novel diagnosis and treatment choices. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Cat. No. | Clonality | Supplier/Reference | Species * | Dilution |
|---|---|---|---|---|---|
| Primary antibodies | |||||
| α-smooth muscle actin (α-SMA) (ASM-1) | CBL-171 | mono (m) | Millipore, Merck, Darmstadt, Germany | h, m, r, b, ch, eq | 1:2000 |
| β-actin | A5441 | mono (m) | Sigma, Taufkirchen, Germany | h, m, r, gp, can, hm, f, p, car, c, rb, sh, b | 1:10,000 |
| Alix (1A12) | sc-43540 | mono (m) | Santa Cruz, Santa Cruz, CA, USA | h, m, r | 1:1000 |
| ALK5 (V-22) | sc-398 | poly (rb) | Santa Cruz | h, m, r | 1:500 |
| Caveolin-1 | CS-3238 | poly (rb) | Cell Signaling Technology, Frankfurt am Main, Germany | h, m, r, ha, z, b, p | 1:1000 |
| CD81 (B-11) | sc-166029 | mono (m) | Santa Cruz | h, m, r | 1:1000 |
| CTGF | sc-14939 | poly (g) | Santa Cruz | h, m, r | 1:1000 |
| Endoglin | AF1320 | poly (g) | R&D Systems, Wiesbaden, Germany | m | 1:2000 |
| MMP-14 | AB38971 | poly (rb) | Abcam, Cambridge, UK | h, m, p | 1:1000 |
| PPabE2 | NA | poly (rb) | [19] | r | 1:2000 |
| pSmad2 | CS-3101 | poly (rb) | Cell Signaling Technology | h, m, r | 1:1000 |
| TGF-β1 | CS-3711 | poly (rb) | Cell Signaling Technology | h, m, r | 1:500 |
| TGF-βRII (L-21) | sc-400 | poly (rb) | Santa Cruz | h, m, r | 1:500 |
| TGF-βRIII | AF242PB | poly (g) | R&D Systems | h | 1:1000 |
| Secondary antibodies | |||||
| IgG-HRP | sc-2004 | NA | Santa Cruz | rb | 1:5000 |
| IgG-HRP | sc-2005 | NA | Santa Cruz | m | 1:5000 |
| IgG-HRP | sc-2056 | NA | Santa Cruz | g | 1:5000 |
| Cell Type | ||||||
|---|---|---|---|---|---|---|
| Immortalized Cell Lines | Primary Cells | |||||
| Surface Marker | HepG2 | LX-2 | Col-GFP | Rat HSC | Mouse HSC | Rat pMF ** |
| CD81 | −/− * | + | + | + | + | + |
| Alix | + | + | + | + | + | + |
| Caveolin-1 | −/− | + | + | + | - | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meurer, S.; Wimmer, A.E.; van de Leur, E.; Weiskirchen, R. Endoglin Trafficking/Exosomal Targeting in Liver Cells Depends on N-Glycosylation. Cells 2019, 8, 997. https://doi.org/10.3390/cells8090997
Meurer S, Wimmer AE, van de Leur E, Weiskirchen R. Endoglin Trafficking/Exosomal Targeting in Liver Cells Depends on N-Glycosylation. Cells. 2019; 8(9):997. https://doi.org/10.3390/cells8090997
Chicago/Turabian StyleMeurer, Steffen, Almut Elisabeth Wimmer, Eddy van de Leur, and Ralf Weiskirchen. 2019. "Endoglin Trafficking/Exosomal Targeting in Liver Cells Depends on N-Glycosylation" Cells 8, no. 9: 997. https://doi.org/10.3390/cells8090997
APA StyleMeurer, S., Wimmer, A. E., van de Leur, E., & Weiskirchen, R. (2019). Endoglin Trafficking/Exosomal Targeting in Liver Cells Depends on N-Glycosylation. Cells, 8(9), 997. https://doi.org/10.3390/cells8090997