Deregulated miR-29b-3p Correlates with Tissue-Specific Activation of Intrinsic Apoptosis in An Animal Model of Amyotrophic Lateral Sclerosis

 ,
,  and
and 
Abstract
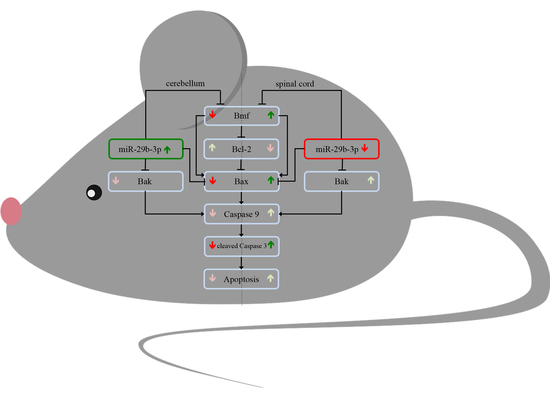
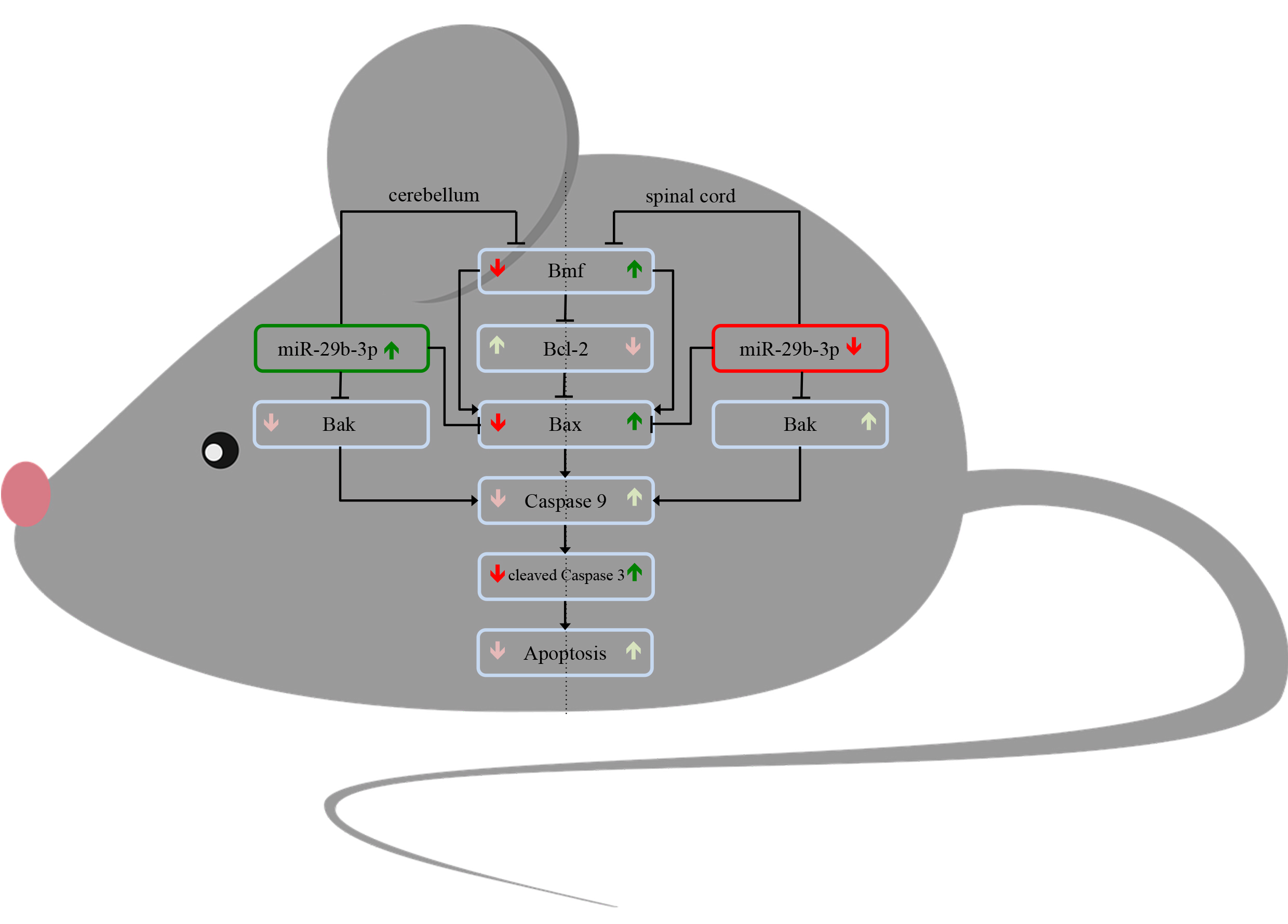{kind=link}
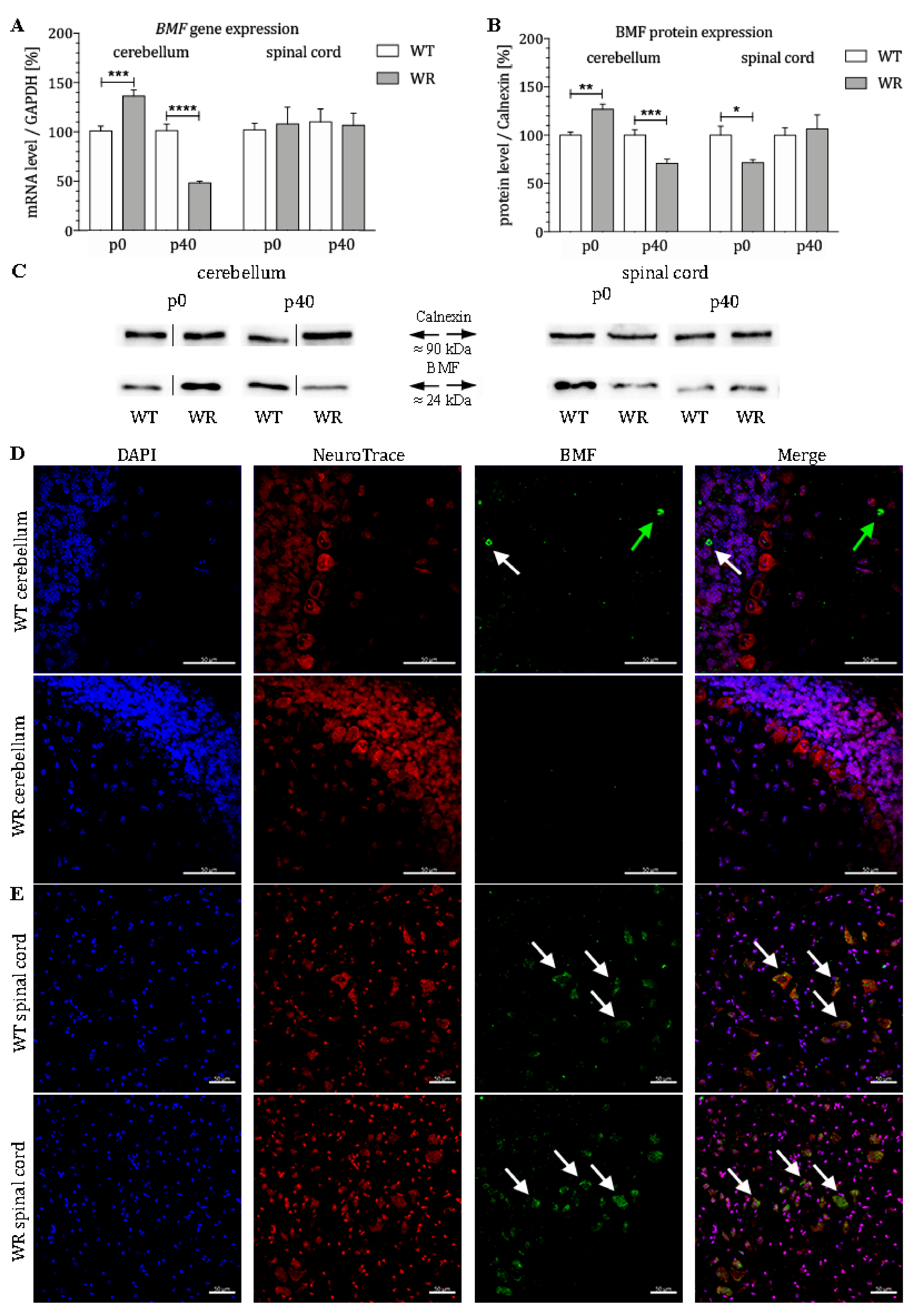{kind=link}
{kind=link}
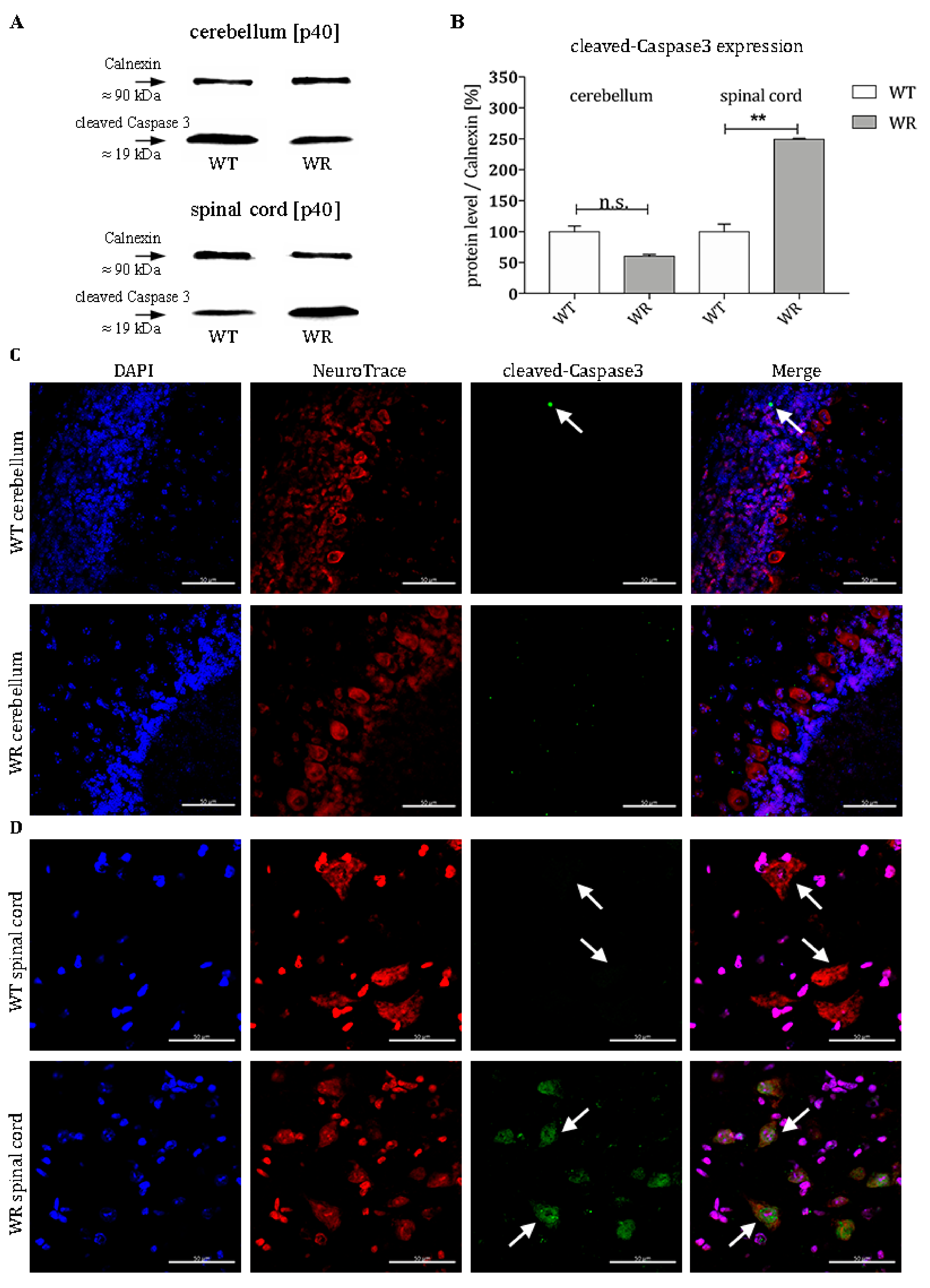{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. RNA Isolation, Reverse Transcription and Quantitative PCR
2.3. In Situ Localization of MicroRNA in Cerebellum and Spinal Cord
2.4. SDS Gel Electrophoresis and Western Blotting
2.5. Immunostaining and Confocal Laser Scanning Microscopy
3. Results
3.1. Differential Expression of the Proapoptotic Bcl-2-Modifying Factor during Different Stable Symptomatic Stages in Cerebellum and Spinal Cord of Wobbler Mice
3.2. Different Tissue-Specific Modulation of Bax Expression
3.3. Increased Level of Cleaved-Caspase 3 in Motor Neurons of Wobbler Spinal Cord
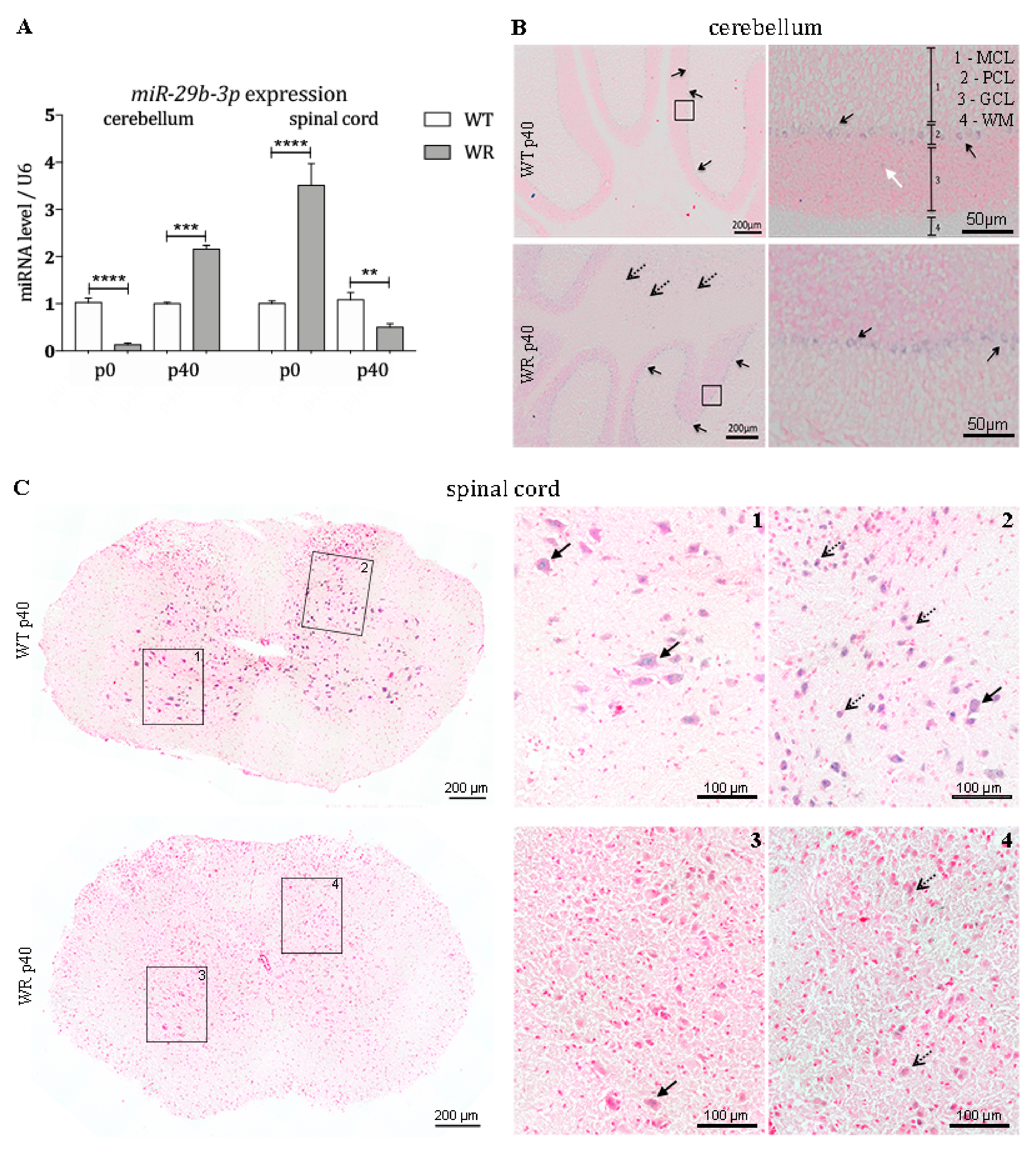
3.4. Differential Expression of miR-29b-3p during Different Stable Symptomatic Stages in Cerebellum and Spinal Cord of Wobbler Mice
3.5. Expression Pattern of miR-29b-3p in Cerebellum and Spinal Cord
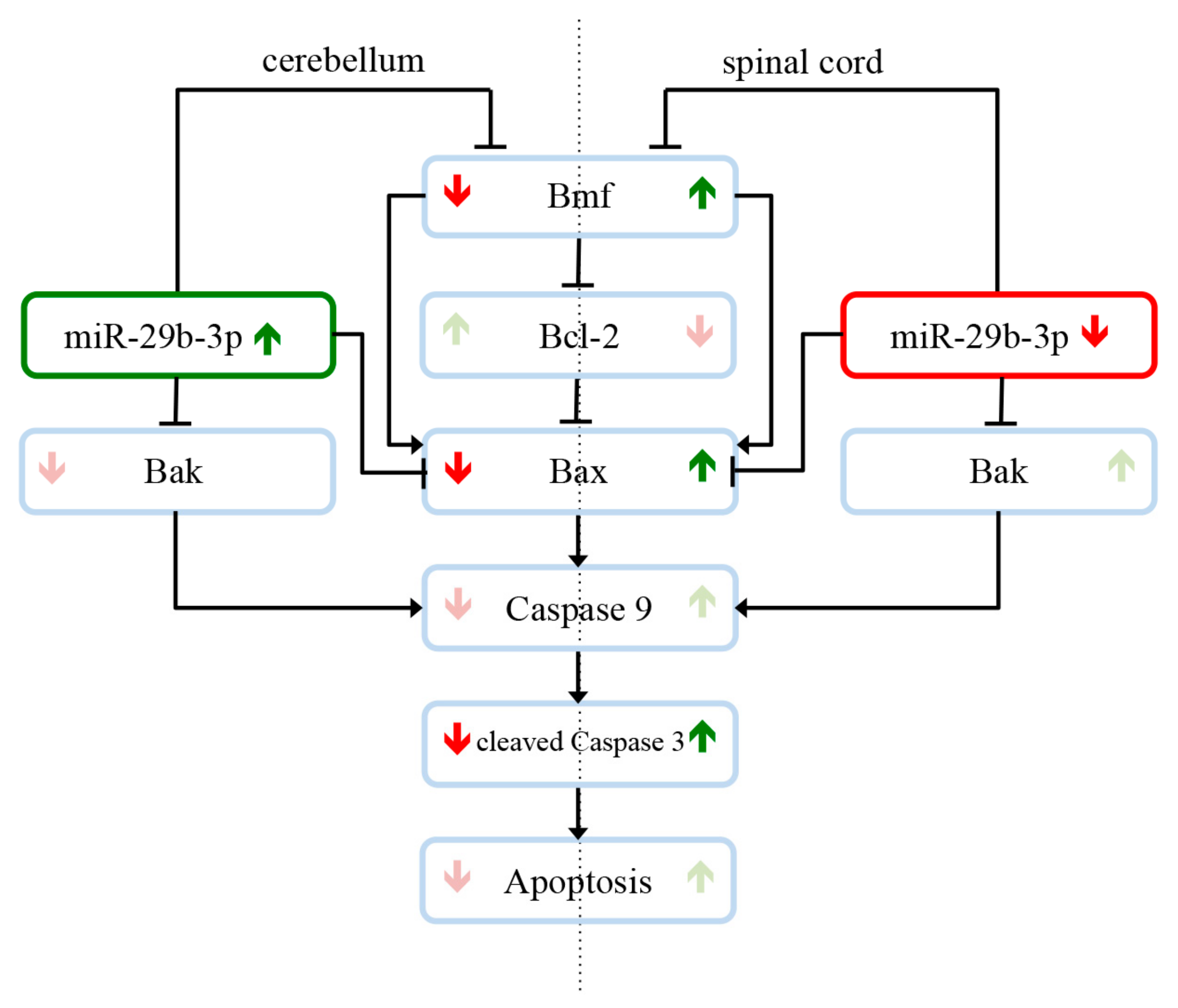
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shaw, P.; Eggett, C. Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J. Neurol. 2001, 247, I17–I27. [Google Scholar] [CrossRef] [PubMed]
- Bucchia, M.; Ramirez, A.; Parente, V.; Simone, C.; Nizzardo, M.; Magri, F.; Dametti, S.; Corti, S. Therapeutic development in amyotrophic lateral sclerosis. Clin. Ther. 2015, 37, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Miller, R.G.; Mitchell, J.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012, 3, CD001447. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Ajroud-Driss, S.; Siddique, T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). BBA-Mol. Basis Dis. 2015, 1852, 679–684. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. New Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Falconer, D.S. Wobbler (wr). Mouse News Lett. 1956, 15, 23. [Google Scholar]
- Schmitt-John, T.; Drepper, C.; Mußmann, A.; Hahn, P.; Kuhlmann, M.; Thiel, C.; Hafner, M.; Lengeling, A.; Heimann, P.; Jones, J.M.; et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat. Genet. 2005, 37, ng1661. [Google Scholar] [CrossRef]
- Pérez-Victoria, J.F.; Abascal-Palacios, G.; Tascón, I.; Kajava, A.; Magadán, J.G.; Pioro, E.P.; Bonifacino, J.S.; Hierro, A. Structural basis for the wobbler mouse neurodegenerative disorder caused by mutation in the Vps54 subunit of the GARP complex. Proc. Natl. Acad. Sci. USA 2010, 107, 12860–12865. [Google Scholar] [CrossRef]
- Fuchs, S.; Resch, K.; Thiel, C.; Ulbrich, M.; Platzer, M.; Jockusch, H.; Schmitt-John, T. Comparative transcription map of the wobbler critical region on mouse chromosome 11 and the homologous region on human chromosome 2p13-14. BMC Genet. 2002, 3, 40. [Google Scholar] [CrossRef]
- Duchen, L.; Strich, S. An hereditary motor neurone disease with progressive denervation of muscle in the mouse: The mutant “wobbler”. J. Neurol. Neurosurg. Psychiatry 1968, 31, 535. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2001, 27, 723–749. [Google Scholar] [CrossRef] [PubMed]
- Achi, E.Y.; Rudnicki, S.A. ALS and frontotemporal dysfunction: A review. Neurol. Res. Int. 2012, 2012, 806306. [Google Scholar] [CrossRef] [PubMed]
- Heimann, P.; Laage, S.; Jockusch, H. Defect of sperm assembly in a neurological mutant of the mouse, wobbler (WR). Differentiation 1991, 47, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2001, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.S. Citron Wobbler mice modeling motor neuron disease display elevated transactive response DNA binding protein. Neuroscience 2009, 158, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, H.; Ferut, A.L.; Kurahashi, K.; McQuarrie, I.G. Impairment of retrograde axonal transport in wobbler mouse motor neuron disease. Muscle Nerve 1990, 13, 121–126. [Google Scholar] [CrossRef]
- Palmisano, R.; Golfi, P.; Heimann, P.; Shaw, C.; Troakes, C.; Schmitt-John, T.; Bartsch, J.W. Endosomal accumulation of APP in wobbler motor neurons reflects impaired vesicle trafficking: Implications for human motor neuron disease. BMC Neurosci. 2011, 12, 24. [Google Scholar] [CrossRef]
- Moser, J.; Bigini, P.; Schmitt-John, T. The wobbler mouse, an ALS animal model. Mol. Genet. Genomics 2013, 288, 207–229. [Google Scholar] [CrossRef]
- Mezzapesa, D.; Ceccarelli, A.; Dicuonzo, F.; Carella, A.; Caro, D.M.; Lopez, M.; Samarelli, V.; Livrea, P.; Simone, I. Whole-brain and regional brain atrophy in amyotrophic lateral sclerosis. AJNR Am. J. Neuroradiol. 2001, 28, 255–259. [Google Scholar]
- Canu, E.; Agosta, F.; Riva, N.; Sala, S.; Prelle, A.; Caputo, D.; Perini, M.; Comi, G.; Filippi, M. The topography of brain microstructural damage in amyotrophic lateral sclerosis assessed using diffusion tensor MR imaging. AJNR Am. J. Neuroradiol. 2011, 32, 1307–1314. [Google Scholar] [CrossRef]
- Geser, F.; Brandmeir, N.J.; Kwong, L.K.; Martinez-Lage, M.; Elman, L.; McCluskey, L.; Xie, S.X.; Lee, V.; Trojanowski, J.Q. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch. Neurol. (Chicago) 2008, 65, 636–641. [Google Scholar] [CrossRef]
- Troakes, C.; Maekawa, S.; Wijesekera, L.; Rogelj, B.; Siklós, L.; Bell, C.; Smith, B.; Newhouse, S.; Vance, C.; Johnson, L.; et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: Abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology 2012, 32, 505–514. [Google Scholar] [CrossRef]
- Saberi, D.; Ott, B.; Dahlke, C.; Matschke, V.; Schmitt-John, T.; Theiss, C. The spatiotemporal pattern of degeneration in the cerebellum of the wobbler mouse. J. Neuropath. Exp. Neur. 2016, 75, 347–357. [Google Scholar] [CrossRef]
- Prell, T.; Grosskreutz, J. The involvement of the cerebellum in amyotrophic lateral sclerosis. Amyotroph. Lat. Scl. Fr. 2013, 14, 507–515. [Google Scholar] [CrossRef]
- Konrad, C.; Henningsen, H.; Bremer, J.; Mock, B.; Deppe, M.; Buchinger, C.; Turski, P.; Knecht, S.; Brooks, B. Pattern of cortical reorganization in amyotrophic lateral sclerosis: A functional magnetic resonance imaging study. Exp. Brain Res. 2002, 143, 51–56. [Google Scholar] [CrossRef]
- Konrad, C.; Jansen, A.; Henningsen, H.; Sommer, J.; Turski, P.; Brooks, B.; Knecht, S. Subcortical reorganization in amyotrophic lateral sclerosis. Exp. Brain Res. 2006, 172, 361. [Google Scholar] [CrossRef]
- Schoenfeld, M.; Tempelmann, C.; Gaul, C.; Kühnel, G.; Düzel, E.; Hopf, J.-M.; Feistner, H.; Zierz, S.; Heinze, H.-J.; Vielhaber, S. Functional motor compensation in amyotrophic lateral sclerosis. J. Neurol. 2005, 252, 944–952. [Google Scholar] [CrossRef]
- Agosta, F.; Valsasina, P.; Absinta, M.; Riva, N.; Sala, S.; Prelle, A.; Copetti, M.; Comola, M.; Comi, G.; Filippi, M. Sensorimotor functional connectivity changes in amyotrophic lateral sclerosis. Cereb. Cortex 2011, 21, 2291–2298. [Google Scholar] [CrossRef]
- Tessitore, A.; Esposito, F.; Monsurrò, M.R.; Graziano, S.; Panza, D.; Russo, A.; Migliaccio, R.; Conforti, F.L.; Morrone, R.; Quattrone, A.; et al. Subcortical motor plasticity in patients with sporadic ALS: An fMRI study. Brain Res. Bull. 2006, 69, 489–494. [Google Scholar] [CrossRef]
- Cistaro, A.; Valentini, M.; Chiò, A.; Nobili, F.; Calvo, A.; Moglia, C.; Montuschi, A.; Morbelli, S.; Salmaso, D.; Fania, P.; et al. Brain hypermetabolism in amyotrophic lateral sclerosis: A FDG PET study in ALS of spinal and bulbar onset. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 251–259. [Google Scholar] [CrossRef]
- Boillée, S.; Peschanski, M.; Junier, M.-P. The wobbler mouse. Mol. Neurobiol. 2003, 28, 65–106. [Google Scholar] [CrossRef]
- Ott, B.; Dahlke, C.; Meller, K.; Napirei, M.; Schmitt-John, T.; Brand-Saberi, B.; Theiss, C.; Saberi, D. Implementation of a manual for working with wobbler mice and criteria for discontinuation of the experiment. Ann. Anat. 2015, 200, 118–124. [Google Scholar] [CrossRef]
- Martin, L.J. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 2000, 7, 613–622. [Google Scholar] [CrossRef]
- Rohm, M.; May, C.; Marcus, K.; Steinbach, S.; Theis, V.; Theiss, C.; Matschke, V. The microRNA miR-375-3p and the tumor suppressor NDRG2 are involved in sporadic amyotrophic lateral sclerosis. Cell. Physiol. Biochem. 2019, 52, 1412–1426. [Google Scholar]
- Dahlke, C.; Saberi, D.; Ott, B.; Brand-Saberi, B.; Schmitt-John, T.; Theiss, C. Inflammation and neuronal death in the motor cortex of the wobbler mouse, an ALS animal model. J. Neuroinflamm. 2015, 12, 215. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan microRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Gascon, E.; Gao, F.-B. Cause or effect: Misregulation of microRNA pathways in neurodegeneration. Front. Neurosci. 2012, 6, 48. [Google Scholar] [CrossRef]
- Hollins, S.L.; Cairns, M.J. MicroRNA: Small RNA mediators of the brains genomic response to environmental stress. Prog. Neurobiol. 2016, 143, 61–81. [Google Scholar] [CrossRef]
- Rinchetti, P.; Rizzuti, M.; Faravelli, I.; Corti, S. MicroRNA metabolism and dysregulation in amyotrophic lateral sclerosis. Mol. Neurobiol. 2018, 55, 2617–2630. [Google Scholar] [CrossRef]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimer’s Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef]
- Abe, M.; Bonini, N.M. MicroRNAs and neurodegeneration: Role and impact. Trends Cell Biol. 2013, 23, 30–36. [Google Scholar] [CrossRef]
- Maes, O.C.; Chertkow, H.M.; Wang, E.; Schipper, H. MicroRNA: Implications for Alzheimer disease and other human CNS disorders. Curr. Genomics 2009, 10, 154–168. [Google Scholar] [CrossRef]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/β-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; Strooper, B. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef]
- Quinlan, S.; Kenny, A.; Medina, M.; Engel, T.; Jimenez-Mateos, E.M. Chapter seven microRNAs in neurodegenerative diseases. Int. Rev. Cel. Mol. Bio. 2017, 334, 309–343. [Google Scholar]
- Sempere, L.F.; Freemantle, S.; Pitha-Rowe, I.; Moss, E.; Dmitrovsky, E.; Ambros, V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004, 5, R13. [Google Scholar] [CrossRef]
- Rizzuti, M.; Filosa, G.; Melzi, V.; Calandriello, L.; Dioni, L.; Bollati, V.; Bresolin, N.; Comi, G.; Barabino, S.; Nizzardo, M.; et al. MicroRNA expression analysis identifies a subset of downregulated miRNAs in ALS motor neuron progenitors. Sci. Rep. 2018, 8, 10105. [Google Scholar] [CrossRef]
- Li, H.-F.; Wu, Z.-Y. Genotype-phenotype correlations of amyotrophic lateral sclerosis. Transl. Neurodegener. 2016, 5, 3. [Google Scholar] [CrossRef]
- Emde, A.; Eitan, C.; Liou, L.; Libby, R.T.; Rivkin, N.; Magen, I.; Reichenstein, I.; Oppenheim, H.; Eilam, R.; Silvestroni, A.; et al. Dysregulated miRNA biogenesis downstream of cellular stress and ALS-causing mutations: A new mechanism for ALS. Embo. J. 2015, 34, 2633–2651. [Google Scholar] [CrossRef]
- Campos-Melo, D.; Droppelmann, C.A.; He, Z.; Volkening, K.; Strong, M.J. Altered microRNA expression profile in amyotrophic lateral sclerosis: A role in the regulation of NFL mRNA levels. Mol. Brain 2013, 6, 26. [Google Scholar] [CrossRef]
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352. [Google Scholar] [CrossRef]
- Morlando, M.; Modigliani, S.; Torrelli, G.; Rosa, A.; Carlo, V.; Caffarelli, E.; Bozzoni, I. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. Embo. J. 2012, 31, 4502–4510. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Hur, J.; Lunn, S.J.; Paez-Colasante, X.; Bender, D.E.; Yung, R.; Sakowski, S.A.; Feldman, E.L. Expression of microRNAs in human post-mortem amyotrophic lateral sclerosis spinal cords provides insight into disease mechanisms. Mol. Cell Neurosci. 2016, 71, 34–45. [Google Scholar] [CrossRef]
- Aulas, A.; Velde, C. Alterations in stress granule dynamics driven by TDP-43 and FUS: A link to pathological inclusions in ALS? Front. Cell Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Correction: Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderweyde, T.; Vanderwyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef]
- Gagliardi, D.; Comi, G.P.; Bresolin, N.; Corti, S. MicroRNAs as regulators of cell death mechanisms in amyotrophic lateral sclerosis. J. Cell Mol. Med. 2019, 23, 1647–1656. [Google Scholar] [CrossRef]
- Modi, P.; Jaiswal, S.; Sharma, P. Regulation of neuronal cell cycle and apoptosis by microRNA 34a. Mol. Cell Biol. 2016, 36, 84–94. [Google Scholar]
- Bhinge, A.; Namboori, S.C.; Bithell, A.; Soldati, C.; Buckley, N.J.; Stanton, L.W. MiR-375 is essential for human spinal motor neuron development and may be involved in motor neuron degeneration. Stem Cells 2016, 34, 124–134. [Google Scholar] [CrossRef]
- Santis, R.; Santini, L.; Colantoni, A.; Peruzzi, G.; de Turris, V.; Alfano, V.; Bozzoni, I.; Rosa, A. FUS mutant human motoneurons display altered transcriptome and microRNA pathways with implications for ALS pathogenesis. Stem Cell Rep. 2017, 9, 1450–1462. [Google Scholar] [CrossRef]
- Willimott, S.; Wagner, S.D. miR-125b and miR-155 Contribute to BCL2 Repression and proliferation in response to CD40 ligand (CD154) in human leukemic B-cells. J. Biol. Chem. 2012, 287, 2608–2617. [Google Scholar] [CrossRef]
- Marcuzzo, S.; Bonanno, S.; Kapetis, D.; Barzago, C.; Cavalcante, P.; D’Alessandro, S.; Mantegazza, R.; Bernasconi, P. Upregulation of neural and cell cycle-related microRNAs in brain of amyotrophic lateral sclerosis mice at late disease stage. Mol. Brain 2015, 8, 5. [Google Scholar] [CrossRef]
- Park, J.-H.; Jang, H.; Lee, I.; Oh, H.; Choi, E.-J.; Rhim, H.; Kang, S. Amyotrophic lateral sclerosis-related mutant superoxide dismutase 1 aggregates inhibit 14-3-3-mediated cell survival by sequestration into the JUNQ compartment. Hum. Mol. Genet. 2017, 26, 3615–3629. [Google Scholar] [CrossRef]
- Parisi, C.; Napoli, G.; Amadio, S.; Spalloni, A.; Apolloni, S.; Longone, P.; Volonté, C. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ. 2016, 23, 531–541. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Thompson, C.B. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell 2002, 109, S97–S107. [Google Scholar] [CrossRef]
- Westphal, D.; Kluck, R.; Dewson, G. Building blocks of the apoptotic pore: How Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014, 21, 196. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Panda, P.; Sinha, N.; Das, D.; Bhutia, S. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef]
- Oltval, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programed cell death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Watson, E.; Whitehead, L.; Adams, R.; Dewson, G.; Coultas, L. Endothelial cell survival during angiogenesis requires the pro-survival protein MCL1. Cell Death Differ. 2016, 23, 1371. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Heinlein, M.; Huang, D.C.; Vaux, D.L. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc. Natl. Acad. Sci. USA 2014, 111, 8512–8517. [Google Scholar] [CrossRef]
- Tripathi, P.; Koss, B.; Opferman, J.; Hildeman, D. Mcl-1 antagonizes Bax/Bak to promote effector CD4+ and CD8+ T-cell responses. Cell Death Differ. 2013, 20, 998. [Google Scholar] [CrossRef]
- Liu, X.-J.; Zheng, X.-P.; Zhang, R.; Guo, Y.-L.; Wang, J.-H. Combinatorial effects of miR-20a and miR-29b on neuronal apoptosis induced by spinal cord injury. Int. J. Clin. Exp. Patho. 2015, 8, 3811–3818. [Google Scholar]
- Wang, Y.; Huang, J.; Ma, Y.; Tang, G.; Liu, Y.; Chen, X.; Zhang, Z.; Zeng, L.; Wang, Y.; Ouyang, Y.-B.; et al. MicroRNA-29b is a therapeutic target in cerebral ischemia associated with aquaporin 4. J. Cereb. Blood F. Met. 2015, 35, 1977–1984. [Google Scholar] [CrossRef]
- Akhter, R.; Saleem, S.; Saha, A.; Biswas, S. The proapoptotic protein Bmf co-operates with Bim and Puma in neuron death induced by β-amyloid or NGF deprivation. Mol. Cell Neurosci. 2018, 88, 249–257. [Google Scholar] [CrossRef]
- Hardwick, M.J.; Soane, L. Multiple functions of BCL-2 family proteins. Csh. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef]
- Hsu, Y.-T.; Wolter, K.G.; Youle, R.J. Cytosol-to-membrane redistribution of Bax and Bcl-XL during apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3668–3672. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef]
- Buytaert, E.; Callewaert, G.; Vandenheede, J.; Agostinis, P. Deficiency in apoptotic effectors BAX and BAK reveals an autophagic cell death pathway initiated by photodamage to the endoplasmic reticulum. Autophagy 2006, 2, 238–240. [Google Scholar] [CrossRef]
- Huang, Z.; Lu, L.; Jiang, T.; Zhang, S.; Shen, Y.; Zheng, Z.; Zhao, A.; Gao, R.; Li, R.; Zhou, S.; et al. miR-29b affects neurocyte apoptosis by targeting MCL-1 during cerebral ischemia/reperfusion injury. Exp. Ther. Med. 2018, 16, 3399–3404. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Cuconati, A.; Mukherjee, C.; Perez, D.; White, E. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Gene Dev. 2003, 17, 2922–2932. [Google Scholar] [CrossRef]
- Kole, A.J.; Swahari, V.; Hammond, S.M.; Deshmukh, M. miR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Gene Dev. 2011, 25, 125–130. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC T method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ekegren, T.; Grundström, E.; Lindholm, D.; Aquilonius, S.-M. Upregulation of Bax protein and increased DNA degradation in ALS spinal cord motor neurons. Acta Neurol. Scand. 1999, 100, 317–321. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Leigh, N.P. Amyotrophic lateral sclerosis. Orphanet. J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef]
- Grad, L.I.; Rouleau, G.A.; Ravits, J.; Cashman, N.R. Clinical spectrum of amyotrophic lateral sclerosis (ALS). Csh. Perspect. Med. 2017, 7, a024117. [Google Scholar] [CrossRef]
- Delgado, M.; Tesfaigzi, Y. BH3-only proteins, Bmf and Bim, in autophagy. Cell Cycle 2013, 12, 3453–3454. [Google Scholar] [CrossRef]
- Hockings, C.; Anwari, K.; Ninnis, R.; Brouwer, J.; O’Hely, M.; Evangelista, M.; Hinds, M.; Czabotar, P.; Lee, E.; Fairlie, W.; et al. Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax. Cell Death Dis. 2015, 6, e1735. [Google Scholar] [CrossRef]
- Puthalakath, H.; Villunger, A.; O’Reilly, L.A.; Beaumont, J.G.; Coultas, L.; Cheney, R.E.; Huang, D.C.; Strasser, A. Bmf: A proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 2001, 293, 1829–1832. [Google Scholar] [CrossRef]
- Schmelzle, T.; Mailleux, A.A.; Overholtzer, M.; Carroll, J.S.; Solimini, N.L.; Lightcap, E.S.; Veiby, O.P.; Brugge, J.S. Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 3787–3792. [Google Scholar] [CrossRef]
- Hausmann, M.; Leucht, K.; Ploner, C.; Kiessling, S.; Villunger, A.; Becker, H.; Hofmann, C.; Falk, W.; Krebs, M.; Kellermeier, S.; et al. BCL-2 modifying factor (BMF) is a central regulator of anoikis in human intestinal epithelial cells. J. Biol. Chem. 2011, 286, 26533–26540. [Google Scholar] [CrossRef]
- Ramjaun, A.; Tomlinson, S.; Eddaoudi, A.; Downward, J. Upregulation of two BH3-only proteins, Bmf and Bim, during TGFβ-induced apoptosis. Oncogene 2007, 26, 1209852. [Google Scholar] [CrossRef]
- Lau, G.J.; Godin, N.; Maachi, H.; Lo, C.-S.; Wu, S.-J.; Zhu, J.-X.; Brezniceanu, M.-L.; Chénier, I.; Fragasso-Marquis, J.; Lattouf, J.-B.; et al. Bcl-2–modifying factor induces renal proximal tubular cell apoptosis in diabetic mice. Diabetes 2012, 61, 474–484. [Google Scholar] [CrossRef]
- Imai, K.; Adachi, M.; Kawamura, R.; Zhang, Y. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 2005, 13, 129. [Google Scholar]
- Zhang, Y.; Adachi, M.; Kawamura, R.; Zou, H.; Imai, K.; Hareyama, M.; Shinomura, Y. Bmf contributes to histone deacetylase inhibitor-mediated enhancing effects on apoptosis after ionizing radiation. Apoptosis 2006, 11, 1349–1357. [Google Scholar] [CrossRef]
- Lossi, L.; Castagna, C.; Merighi, A. Caspase-3 mediated cell death in the normal development of the mammalian cerebellum. Int. J. Mol. Sci. 2018, 19, 3999. [Google Scholar] [CrossRef]
- Clarke, P.; Posada, A.; Primi, M.; Castagné, V. Neuronal death in the central nervous system during development. Biomed. Pharmacother. 1998, 52, 356–362. [Google Scholar] [CrossRef]
- Oppenheim, R.; Flavell, R.; Vinsant, S.; Prevette, D.; Kuan, C.; Rakic, P. Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J. Neurosci. 2001, 21, 4752–4760. [Google Scholar] [CrossRef]
- Zanjani, H.; Vogel, M.; Delhaye-Bouchaud, N.; Martinou, J.; Mariani, J. Increased inferior olivary neuron and cerebellar granule cell numbers in transgenic mice overexpressing the human Bcl-2 gene. J. Neurobiol. 1997, 32, 502–516. [Google Scholar] [CrossRef]
- Fan, D.; Grooms, S.; Araneda, R.; Johnson, A.; Dobrenis, K.; Kessler, J.; Zukin, R. AMPA receptor protein expression and function in astrocytes cultured from hippocampus. J. Neurosci. Res. 2001, 57, 557–571. [Google Scholar] [CrossRef]
- Baader, S.L.; Sanlioglu, S.; Berrebi, A.S.; Parker-Thornburg, J.; Oberdick, J. Ectopic overexpression of Engrailed-2 in cerebellar purkinje cells causes restricted cell loss and retarded external germinal layer development at lobule junctions. J. Neurosci. 1998, 18, 1763–1773. [Google Scholar] [CrossRef]
- Goswami, J.; Martin, L.A.; Goldowitz, D.; Beitz, A.J.; Feddersen, R.M. Enhanced Purkinje cell survival but compromised cerebellar function in targeted antiapoptotic protein transgenic mice. Mol. Cell Neurosci. 2005, 29, 202–221. [Google Scholar] [CrossRef]
- Jankowski, J.; Miething, A.; Schilling, K.; Oberdick, J.; Baader, S. Cell death as a regulator of cerebellar histogenesis and compartmentation. Cerebellum 2011, 10, 373–392. [Google Scholar] [CrossRef]
- Jankowski, J.; Miething, A.; Schilling, K.; Baader, S.L. Physiological Purkinje cell death is spatiotemporally organized in the developing mouse cerebellum. Cerebellum 2009, 8, 277–290. [Google Scholar] [CrossRef]
- Jung, A.; Kim, T.; Rhyu, I.; Kim, H.; Lee, Y.; Vinsant, S.; Oppenheim, R.W.; Sun, W. Misplacement of Purkinje cells during postnatal development in Bax knock-out mice: A novel role for programmed cell death in the nervous system? J. Neurosci. 2008, 28, 2941–2948. [Google Scholar] [CrossRef]
- Heckroth, J.A.; Goldowitz, D.; Eisenman, L.M. Purkinje cell reduction in the reeler mutant mouse: A quantitative immunohistochemical study. J. Comp. Neurol. 1989, 279, 546–555. [Google Scholar] [CrossRef]
- Croci, L.; Chung, S.-H.; Masserdotti, G.; Gianola, S.; Bizzoca, A.; Gennarini, G.; Corradi, A.; Rossi, F.; Hawkes, R.; Consalez, G.G. A key role for the HLH transcription factor EBF2COE2,O/E-3 in Purkinje neuron migration and cerebellar cortical topography. Development 2006, 133, 2719–2729. [Google Scholar] [CrossRef]
- Hashimoto, K.; Kano, M. Postnatal development and synapse elimination of climbing fiber to Purkinje cell projection in the cerebellum. Neurosci. Res. 2005, 53, 221–228. [Google Scholar] [CrossRef]
- Lowrie, M.B.; Vrbová, G. Dependence of postnatal motoneurones on their targets: Review and hypothesis. Trends Neurosci. 1992, 15, 80–84. [Google Scholar] [CrossRef]
- Herculano-Houzel, S.; Mota, B.; Lent, R. Cellular scaling rules for rodent brains. Proc. Natl. Acad. Sci. USA 2006, 103, 12138–12143. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Anilkumar, U.; Chen, G.; Ramírez-Peinado, S.; Galindo-Moreno, J.; Muñoz-Pinedo, C.; Prehn, J. Analysis of BH3-only proteins upregulated in response to oxygen/glucose deprivation in cortical neurons identifies Bmf but not Noxa as potential mediator of neuronal injury. Cell Death Dis. 2014, 5, e1456. [Google Scholar] [CrossRef]
- Jing, X.; Yang, J.; Jiang, L.; Chen, J.; Wang, H. MicroRNA-29b regulates the mitochondria-dependent apoptotic pathway by targeting Bax in doxorubicin cardiotoxicity. Cell Physiol. Biochem. 2018, 48, 692–704. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Bio. 2013, 15, 49–63. [Google Scholar] [CrossRef]
- Ouyang, Y.; Xu, L.; Lu, Y.; Sun, X.; Yue, S.; Xiong, X.; Giffard, R.G. Astrocyte-enriched miR-29a targets PUMA and reduces neuronal vulnerability to forebrain ischemia. Glia 2013, 61, 1784–1794. [Google Scholar] [CrossRef]
- Hwang, H.-W.; Wentzel, E.A.; Mendell, J.T. A Hexanucleotide element directs microRNA nuclear import. Science 2007, 315, 97–100. [Google Scholar] [CrossRef]
- Smirnova, L.; Gräfe, A.; Seiler, A.; Schumacher, S.; Nitsch, R.; Wulczyn, G.F. Regulation of miRNA expression during neural cell specification. Eur. J. Neurosci. 2005, 21, 1469–1477. [Google Scholar] [CrossRef]
- Park, S.-Y.; Lee, J.; Ha, M.; Nam, J.-W.; Kim, N.V. miR-29 miRNAs activate p53 by targeting p85α and CDC42. Nat. Struct. Mol. Bio. 2008, 16, nsmb1533. [Google Scholar] [CrossRef]
- Kwon, J.J.; Factora, T.D.; Dey, S.; Kota, J. Systematic review of miR-29 in cancer. Mol Ther. 2018, 12, 173–194. [Google Scholar] [CrossRef]
- Cushing, L.; Kuang, P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lü, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Resp. Cell Mol. 2011, 45, 287–294. [Google Scholar] [CrossRef]
- Yang, W.-M.; Jeong, H.-J.; Park, S.-Y.; Lee, W. Induction of miR-29a by saturated fatty acids impairs insulin signaling and glucose uptake through translational repression of IRS-1 in myocytes. Febs. Lett. 2014, 588, 2170–2176. [Google Scholar] [CrossRef]
- Zhang, X.; Gong, X.; Han, S.; Zhang, Y. MiR-29b protects dorsal root ganglia neurons from diabetic rat. Cell Biochem. Biophys. 2014, 70, 1105–1111. [Google Scholar] [CrossRef]
- Jauhari, A.; Singh, T.; Yadav, S. Expression of miR-145 and its target proteins are regulated by miR-29b in differentiated neurons. Mol. Neurobiol. 2001, 55, 8978–8990. [Google Scholar] [CrossRef]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma exosomal miRNAs in persons with and without Alzheimer disease: Altered expression and prospects for biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef]
- Jiao, J.; Herl, L.D.; Farese, R.V.; Gao, F.-B. MicroRNA-29b regulates the expression level of human progranulin, a secreted glycoprotein implicated in frontotemporal dementia. PLoS ONE 2010, 5, e10551. [Google Scholar] [CrossRef]
- Roshan, R.; Shridhar, S.; Sarangdhar, M.A.; Banik, A.; Chawla, M.; Garg, M.; Singh, V.; Pillai, B. Brain-specific knockdown of miR-29 results in neuronal cell death and ataxia in mice. RNA 2014, 20, 1287–1297. [Google Scholar] [CrossRef]
- Papadopoulou, A.S.; Serneels, L.; Achsel, T.; Mandemakers, W.; Callaerts-Vegh, Z.; Dooley, J.; Lau, P.; Ayoubi, T.; Radaelli, E.; Spinazzi, M.; et al. Deficiency of the miR-29a/b-1 cluster leads to ataxic features and cerebellar alterations in mice. Neurobiol. Dis. 2015, 73, 275–288. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, Y.; Zhang, S.; Jiang, T.-P.; Chen, L.; Liu, J.; Zhou, S. MicroRNA-29b alleviates oxygen and glucose deprivation/reperfusion-induced injury via inhibition of the p53-dependent apoptosis pathway in N2a neuroblastoma cells. Exp. Ther. Med. 2018, 15, 67–74. [Google Scholar] [CrossRef]
- Wei, R.; Zhang, R.; Li, H.; Li, H.; Zhang, S.; Xie, Y.; Li, S.; Chen, F. MiR-29 targets PUMA to suppress oxygen and glucose deprivation/reperfusion (OGD/R)-induced cell death in hippocampal neurons. Curr. Neurovasc. Res. 2018, 15, 47–54. [Google Scholar] [CrossRef]
- Khanna, S.; Rink, C.; Ghoorkhanian, R.; Gnyawali, S.; Heigel, M.; Wijesinghe, D.S.; Chalfant, C.E.; Chan, Y.; Banerjee, J.; Huang, Y.; et al. Loss of miR-29b following acute ischemic stroke contributes to neural cell death and infarct size. J. Cereb. Blood F. Met. 2013, 33, 1197–1206. [Google Scholar] [CrossRef]
- Fuller-Carter, P.I.; Carter, K.W.; Anderson, D.; Harvey, A.R.; Giles, K.M.; Rodger, J. Integrated analyses of zebrafish miRNA and mRNA expression profiles identify miR-29b and miR-223 as potential regulators of optic nerve regeneration. BMC Genomics 2015, 16, 591. [Google Scholar] [CrossRef]
- Jones, M.; Lal, A. MicroRNAs, wild-type and mutant p53: More questions than answers. RNA Biol. 2014, 9, 781–791. [Google Scholar] [CrossRef]
- Eve, D.J.; Dennis, J.S.; Citron, B.A. Transcription factor p53 in degenerating spinal cords. Brain Res. 2007, 1150, 174–181. [Google Scholar] [CrossRef]
- Ranganathan, S.; Bowser, R. p53 and cell cycle proteins participate in spinal motor neuron cell death in ALS. Open Pathol. J. 2010, 4, 11–22. [Google Scholar] [CrossRef]
- Nolan, K.; Walter, F.; Tuffy, L.P.; Poeschel, S.; Gallagher, R.; Haunsberger, S.; Bray, I.; Stallings, R.L.; Concannon, C.G.; Prehn, J.H. Endoplasmic reticulum stress-mediated upregulation of miR-29a enhances sensitivity to neuronal apoptosis. Eur. J. Neurosci. 2016, 43, 640–652. [Google Scholar] [CrossRef]
- Yin, K.-J.; Deng, Z.; Huang, H.; Hamblin, M.; Xie, C.; Zhang, J.; Chen, E.Y. miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol. Dis. 2010, 38, 17–26. [Google Scholar] [CrossRef]
- Moon, J.; Xu, L.; Giffard, R.G. Inhibition of microRNA-181 reduces forebrain ischemia-induced neuronal loss. J. Cereb. Blood F. Met. 2013, 33, 1976–1982. [Google Scholar] [CrossRef]
- Ripa, R.; Dolfi, L.; Terrigno, M.; Pandolfini, L.; Savino, A.; Arcucci, V.; Groth, M.; Tozzini, E.; Baumgart, M.; Cellerino, A. MicroRNA miR-29 controls a compensatory response to limit neuronal iron accumulation during adult life and aging. BMC Biol. 2017, 15, 9. [Google Scholar] [CrossRef]
- Pandi, G.; Nakka, V.P.; Dharap, A.; Roopra, A.; Vemuganti, R. MicroRNA miR-29c downregulation leading to de-repression of its target DNA methyltransferase 3a promotes ischemic brain damage. PLoS ONE 2013, 8, e58039. [Google Scholar] [CrossRef]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, B.M.; Lamon, S.; Léger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef]
- Catapano, F.; Domingos, J.; Perry, M.; Ricotti, V.; Phillips, L.; Servais, L.; Seferian, A.; de Groot, I.; Krom, Y.D.; Niks, E.H.; et al. Downregulation of miRNA-29, -23 and -21 in urine of Duchenne muscular dystrophy patients. Epigenomics 2018, 10. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klatt, C.L.; Theis, V.; Hahn, S.; Theiss, C.; Matschke, V. Deregulated miR-29b-3p Correlates with Tissue-Specific Activation of Intrinsic Apoptosis in An Animal Model of Amyotrophic Lateral Sclerosis. Cells 2019, 8, 1077. https://doi.org/10.3390/cells8091077
Klatt CL, Theis V, Hahn S, Theiss C, Matschke V. Deregulated miR-29b-3p Correlates with Tissue-Specific Activation of Intrinsic Apoptosis in An Animal Model of Amyotrophic Lateral Sclerosis. Cells. 2019; 8(9):1077. https://doi.org/10.3390/cells8091077
Chicago/Turabian StyleKlatt, Christina L., Verena Theis, Stephan Hahn, Carsten Theiss, and Veronika Matschke. 2019. "Deregulated miR-29b-3p Correlates with Tissue-Specific Activation of Intrinsic Apoptosis in An Animal Model of Amyotrophic Lateral Sclerosis" Cells 8, no. 9: 1077. https://doi.org/10.3390/cells8091077
APA StyleKlatt, C. L., Theis, V., Hahn, S., Theiss, C., & Matschke, V. (2019). Deregulated miR-29b-3p Correlates with Tissue-Specific Activation of Intrinsic Apoptosis in An Animal Model of Amyotrophic Lateral Sclerosis. Cells, 8(9), 1077. https://doi.org/10.3390/cells8091077