Autophagy Stimulation as a Potential Strategy Against Intestinal Fibrosis

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Heterotopic Transplant Model of Intestinal Fibrosis
2.2. Primary Intestinal Fibroblasts Isolation and Culture
2.3. RNA Extraction and Quantitative PCR
2.4. Western Blot
2.5. Sirius Red Staining
2.6. Statistical Analysis
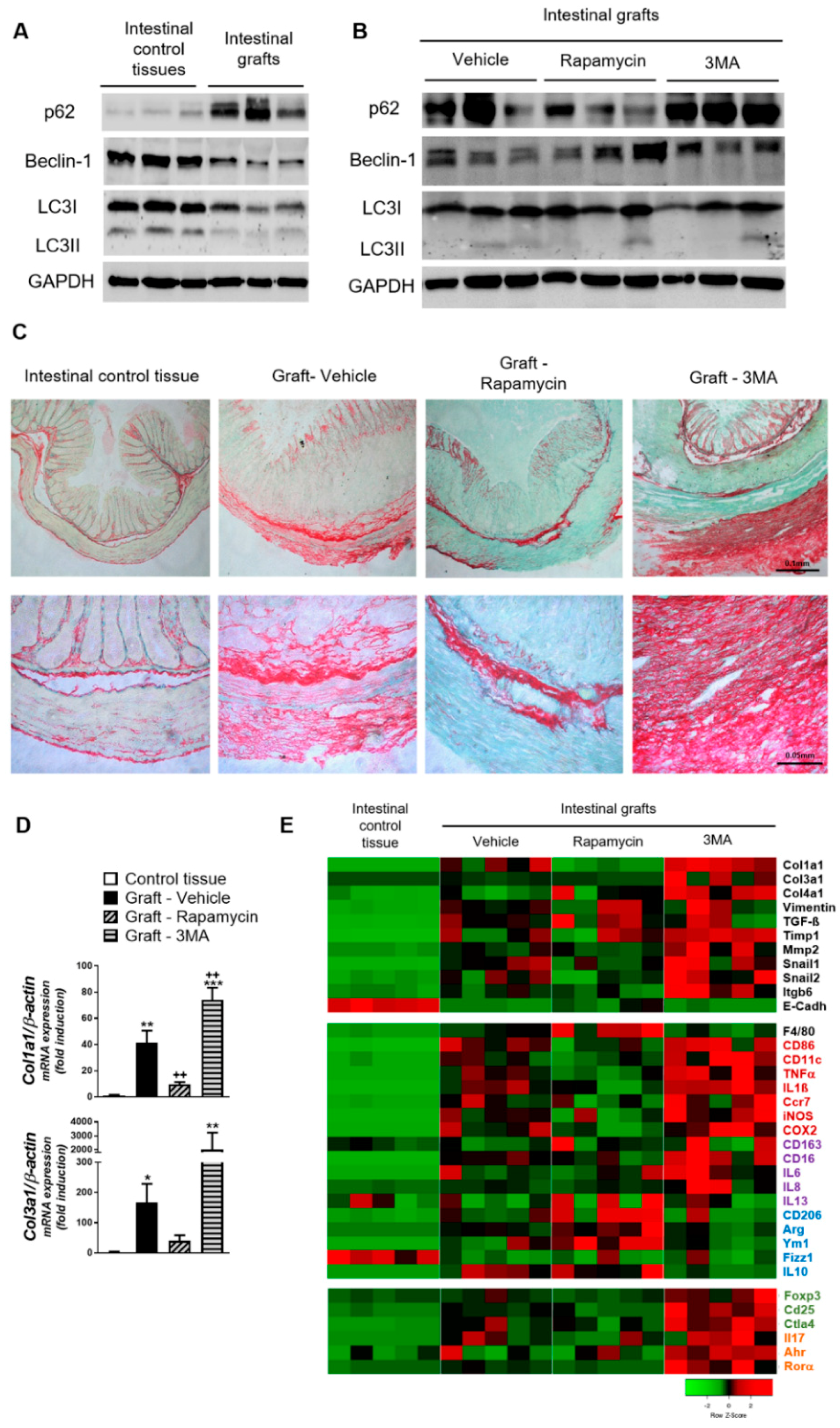
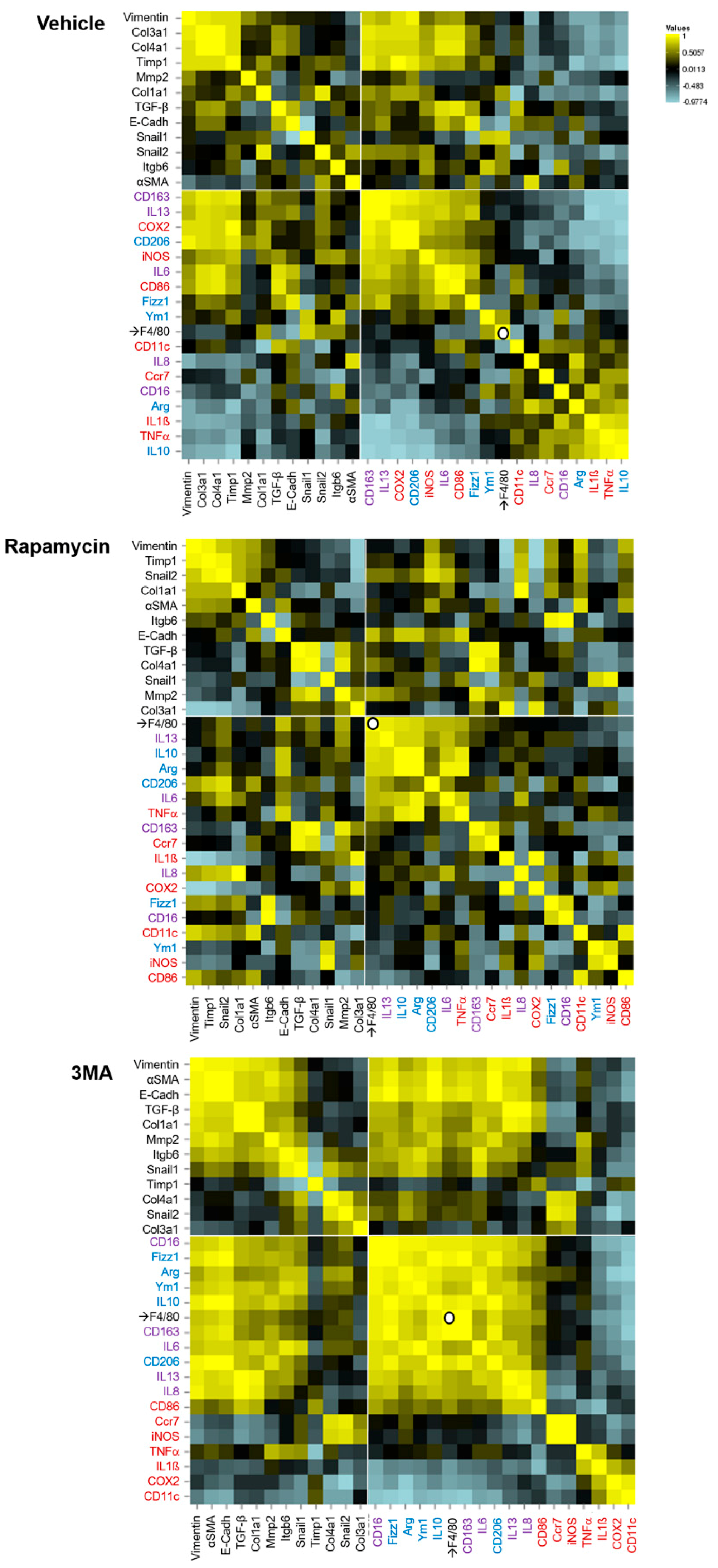
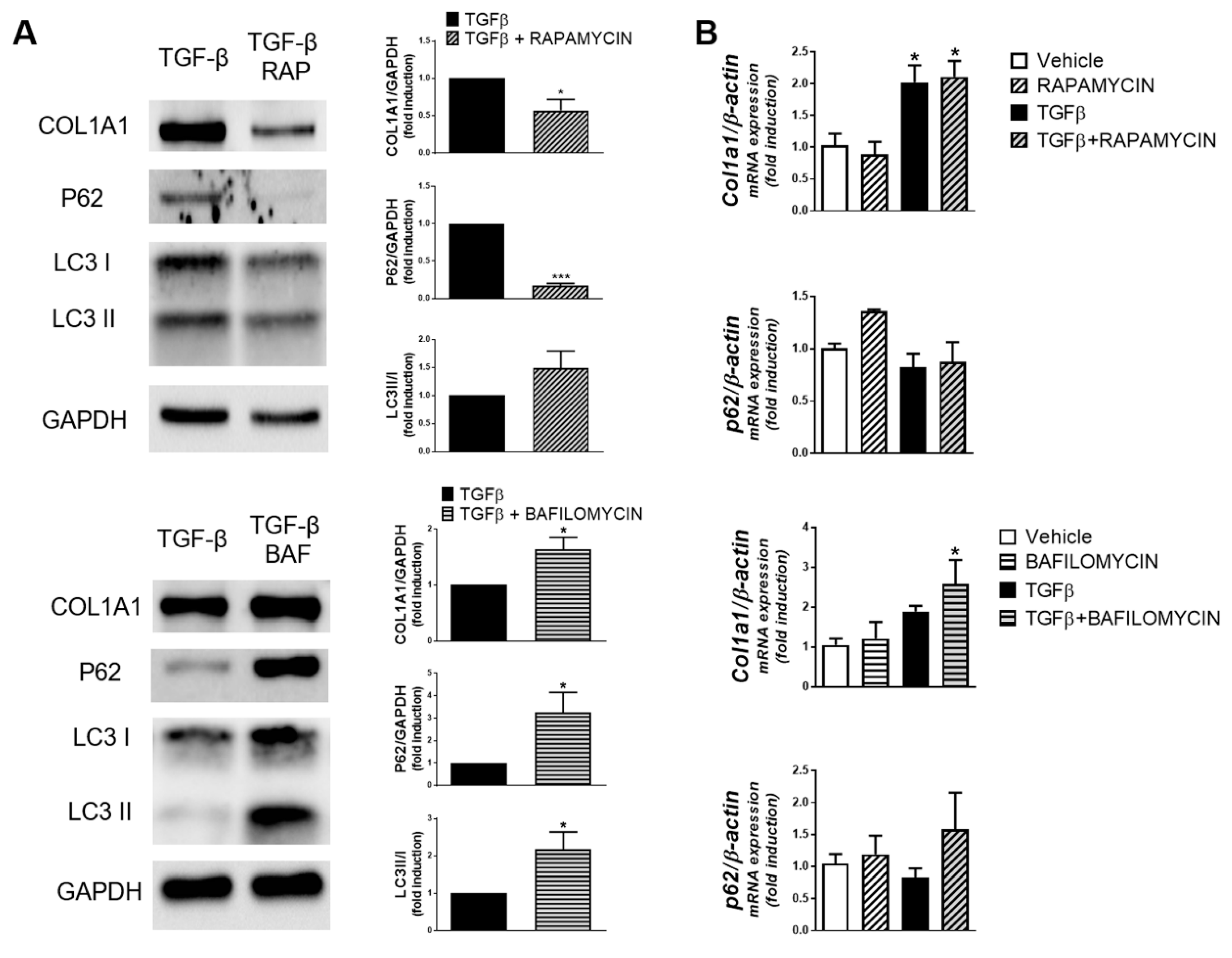
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kim, S.; Eun, H.S.; Jo, E.K. Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells 2019, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Masia, D.; Cosin-Roger, J.; Calatayud, S.; Hernandez, C.; Alos, R.; Hinojosa, J.; Apostolova, N.; Alvarez, A.; Barrachina, M.D. Hypoxic macrophages impair autophagy in epithelial cells through Wnt1: Relevance in IBD. Muc. Immunol. 2014, 7, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Macias-Ceja, D.C.; Cosin-Roger, J.; Ortiz-Masia, D.; Salvador, P.; Hernandez, C.; Esplugues, J.V.; Calatayud, S.; Barrachina, M.D. Stimulation of autophagy prevents intestinal mucosal inflammation and ameliorates murine colitis. Br. J. Pharmacol. 2017, 174, 2501–2511. [Google Scholar] [CrossRef] [PubMed]
- Del Principe, D.; Lista, P.; Malorni, W.; Giammarioli, A.M. Fibroblast autophagy in fibrotic disorders. J. Pathol. 2013, 229, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Hilscher, M.; Hernandez-Gea, V.; Friedman, S.L. Autophagy and mesenchymal cell fibrogenesis. Biochim. Biophys. Acta 2012, 1831, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Macias-Ceja, D.C.; Ortiz-Masia, D.; Salvador, P.; Gisbert-Ferrandiz, L.; Hernandez, C.; Hausmann, M.; Rogler, G.; Esplugues, J.V.; Hinojosa, J.; Alos, R.; et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal. Immunol. 2019, 12, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Fowler, E.V.; Doecke, J.; Simms, L.A.; Zhao, Z.Z.; Webb, P.M.; Hayward, N.K.; Whiteman, D.C.; Florin, T.H.; Montgomery, G.W.; Cavanaugh, J.A.; et al. ATG16L1 T300A shows strong associations with disease subgroups in a large Australian IBD population: further support for significant disease heterogeneity. Am. J. Gastroenterol. 2008, 103, 2519–2526. [Google Scholar] [CrossRef] [PubMed]
- Strisciuglio, C.; Auricchio, R.; Martinelli, M.; Staiano, A.; Giugliano, F.P.; Andreozzi, M.; De Rosa, M.; Giannetti, E.; Gianfrani, C.; Izzo, P.; et al. Autophagy genes variants and paediatric Crohn’s disease phenotype: A single-centre experience. Dig. Liver. Dis. 2014, 46, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Salvador, P.; Macias-Ceja, D.C.; Gisbert-Ferrandiz, L.; Hernandez, C.; Bernardo, D.; Alos, R.; Navarro-Vicente, F.; Esplugues, J.V.; Ortiz-Masia, D.; Barrachina, M.D.; et al. CD16+ Macrophages Mediate Fibrosis in Inflammatory Bowel Disease. J. Crohns Colitis 2018, 12, 589–599. [Google Scholar] [CrossRef]
- Chang, J.; Hisamatsu, T.; Shimamura, K.; Yoneno, K.; Adachi, M.; Naruse, H.; Igarashi, T.; Higuchi, H.; Matsuoka, K.; Kitazume, M.T.; et al. Activated hepatic stellate cells mediate the differentiation of macrophages. Hepatol. Res. 2013, 43, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Liaskou, E.; Zimmermann, H.W.; Li, K.K.; Oo, Y.H.; Suresh, S.; Stamataki, Z.; Qureshi, O.; Lalor, P.F.; Shaw, J.; Syn, W.K.; et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology 2013, 57, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; De Salvo, C.; Pizarro, T.T. Central role of IL-17/Th17 immune responses and the gut microbiota in the pathogenesis of intestinal fibrosis. Curr. Opin. Gastroenterol. 2014, 30, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Filidou, E.; Valatas, V.; Drygiannakis, I.; Arvanitidis, K.; Vradelis, S.; Kouklakis, G.; Kolios, G.; Bamias, G. Cytokine Receptor Profiling in Human Colonic Subepithelial Myofibroblasts: A Differential Effect of Th Polarization-Associated Cytokines in Intestinal Fibrosis. Inflamm. Bowel. Dis. 2018, 24, 2224–2241. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Alam, M.M.; Zhao, X.F.; Liao, Y.; Shen, J.; Morgan, S.; Huang, T.; Lee, H.; Lee, E.; Huang, Y. Induction of autophagy in Cx3cr1(+) mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis. Mucosal. Immunol. 2019, 12, 612–623. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosin-Roger, J.; Canet, F.; Macias-Ceja, D.C.; Gisbert-Ferrándiz, L.; Ortiz-Masiá, D.; Esplugues, J.V.; Alós, R.; Navarro, F.; Barrachina, M.D.; Calatayud, S. Autophagy Stimulation as a Potential Strategy Against Intestinal Fibrosis. Cells 2019, 8, 1078. https://doi.org/10.3390/cells8091078
Cosin-Roger J, Canet F, Macias-Ceja DC, Gisbert-Ferrándiz L, Ortiz-Masiá D, Esplugues JV, Alós R, Navarro F, Barrachina MD, Calatayud S. Autophagy Stimulation as a Potential Strategy Against Intestinal Fibrosis. Cells. 2019; 8(9):1078. https://doi.org/10.3390/cells8091078
Chicago/Turabian StyleCosin-Roger, Jesus, Francisco Canet, Dulce C. Macias-Ceja, Laura Gisbert-Ferrándiz, Dolores Ortiz-Masiá, Juan V. Esplugues, Rafael Alós, Francisco Navarro, María D. Barrachina, and Sara Calatayud. 2019. "Autophagy Stimulation as a Potential Strategy Against Intestinal Fibrosis" Cells 8, no. 9: 1078. https://doi.org/10.3390/cells8091078
APA StyleCosin-Roger, J., Canet, F., Macias-Ceja, D. C., Gisbert-Ferrándiz, L., Ortiz-Masiá, D., Esplugues, J. V., Alós, R., Navarro, F., Barrachina, M. D., & Calatayud, S. (2019). Autophagy Stimulation as a Potential Strategy Against Intestinal Fibrosis. Cells, 8(9), 1078. https://doi.org/10.3390/cells8091078