Cancer Stem Cells and Targeting Strategies


Abstract
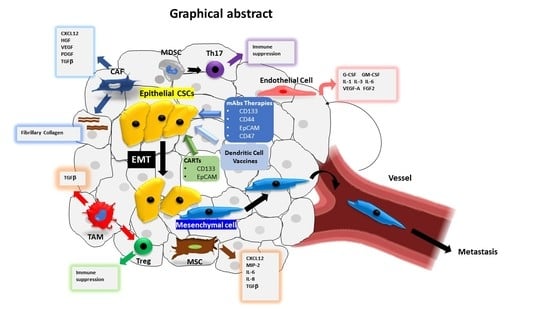
1. Introduction
2. Cancer Stem Cells
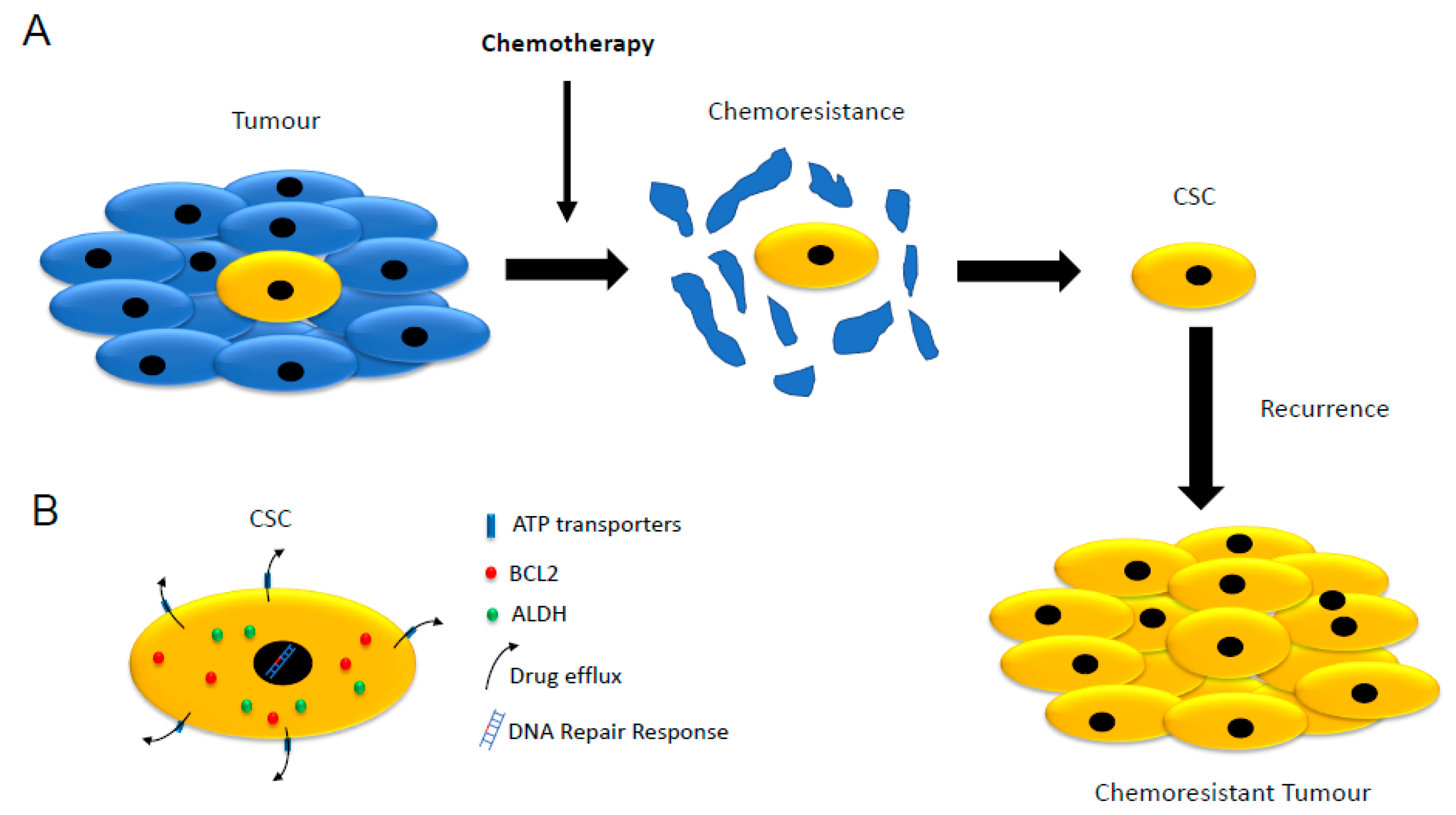
3. Cancer Stemness, Therapy Resistance and Cancer Recurrence
3.1. Low Proliferation Rate
3.2. Expression of ATP-Binding Cassette (ABC) Transporters
3.3. Increased Expression of Aldehyde Dehydrogenases (ALDHs)
3.4. Resistance to Apoptosis
3.5. DNA Repair Response
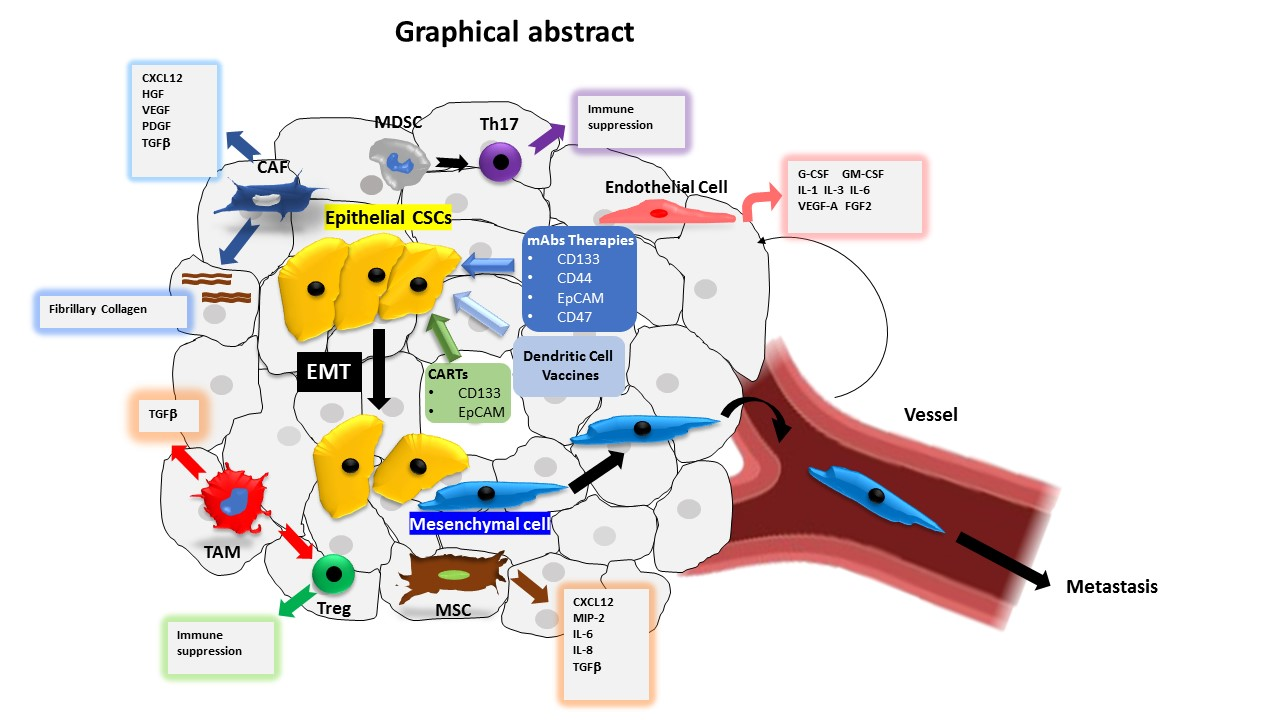
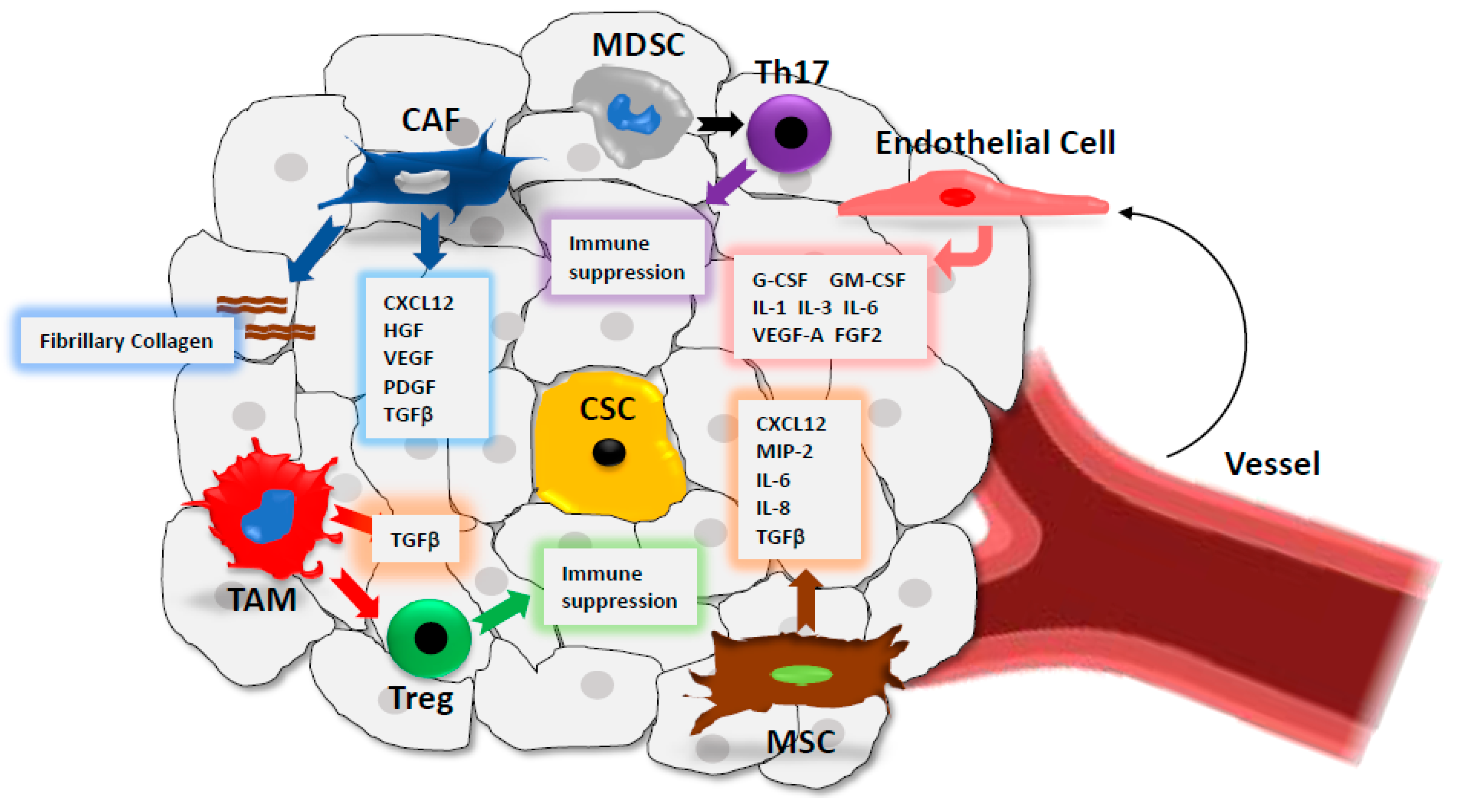
4. CSC Microenvironment
4.1. Cancer-Associated Fibroblasts (CAFs)
4.2. Immune Cells
4.3. Mesenchymal Stem Cells (MSCs)
4.4. Endothelial Cells
5. Cancer Stemness, EMT and Tumour Progression
6. CSC Metabolism
7. Targeting Strategies of CSCs
7.1. Targeting Cell Surface Biomarkers of CSCs
7.1.1. CD133
7.1.2. CD44
7.1.3. EpCAM
7.1.4. CD47
7.2. Dendritic Cell-Based Vaccines
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Global Health Observatory (GHO) data. Available online: https://www.who.int/gho/publications/world_health_statistics/2019/en/ (accessed on 01 July 2019).
- Regad, T. Tissue-specific cancer stem cells: Reality or a mirage? Transl. Med. Rep. 2017, 1. [Google Scholar] [CrossRef]
- Regad, T.; Sayers, T.J.; Rees, R.C. Principles of Stem Cell Biology and Cancer: Future Applications and Therapeutics; Wiley-Blackwell: Oxford, UK, 2015. [Google Scholar]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Boil. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Rich, J.N. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 2008, 26, 2839–2845. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer Stem Cells and Chemoresistance: The Smartest Survives the Raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed]
- La Noce, M.; Paino, F.; Mele, L.; Papaccio, G.; Regad, T.; Lombardi, A.; Papaccio, F.; Desiderio, V.; Tirino, V. HDAC2 depletion promotes osteosarcoma’s stemness both in vitro and in vivo: A study on a putative new target for CSCs directed therapy. J. Exp. Clin. Cancer Res. 2018, 37, 296. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.-U.; Miyazaki, H.; Ochiya, T. The role of microRNAs in the regulation of cancer stem cells. Front. Genet. 2014, 4, 295. [Google Scholar] [CrossRef]
- Oskarsson, T.; Batlle, E.; Massagué, J. Metastatic Stem Cells: Sources, Niches, and Vital Pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef]
- Voog, J.; Jones, D.L. Stem Cells and the Niche: A Dynamic Duo. Cell Stem Cell 2010, 6, 103–115. [Google Scholar] [CrossRef]
- Myron, S. Rudolf Virchow. Emerg Infect Dis. 2008, 14, 1480–1481. [Google Scholar]
- Welte, Y.; Adjaye, J.; Lehrach, H.R.; Regenbrecht, C.R. Cancer stem cells in solid tumors: Elusive or illusive? Cell Commun. Signal. 2010, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, D.E.; McCulloch, E.A.; Park, C.H. Mouse Myeloma Tumor Stem Cells: A Primary Cell Culture Assay2. J. Natl. Cancer Inst. 1971, 46, 411–422. [Google Scholar]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Gorain, M.; Kundu, G.; Kundu, G.C. Therapeutic implications of cellular and molecular biology of cancer stem cells in melanoma. Mol. Cancer 2017, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.G.; Miles, A.K.; Ahmad, M.; Buczek, M.E.; Pockley, A.G.; Rees, R.C.; Regad, T. The helicase HAGE prevents interferon-alpha-induced PML expression in ABCB5+ malignant melanoma-initiating cells by promoting the expression of SOCS1. Cell Death Disease 2014, 5, e1061. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef]
- Yeung, T.M.; Gandhi, S.C.; Wilding, J.L.; Muschel, R.; Bodmer, W.F. Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl. Acad. Sci. USA 2010, 107, 3722–3727. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Todaro, M.; de Sousa Mello, F.; Sprick, M.R.; Kemper, K.; Perez Alea, M.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.; El-Rayes, B. Identifying and targeting cancer stem cells in the treatment of gastric cancer. Cancer 2017, 123, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.R. Gastric cancer stem cells: A novel therapeutic target. Cancer Lett. 2013, 338, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Chinn, S.B.; Darr, O.A.; Owen, J.H.; Bellile, E.; McHugh, J.B.; Spector, M.E.; Papagerakis, S.M.; Chepeha, D.B.; Bradford, C.R.; Carey, T.E.; et al. Cancer stem cells: Mediators of tumorigenesis and metastasis in head and neck squamous cell carcinoma. Head Neck 2015, 37, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.; Buckanovich, R.J.; Rueda, B.R. Ovarian cancer stem cells: Working towards the root of stemness. Cancer Lett. 2013, 338, 147–157. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M.; Lepelletier, Y.; et al. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Hou, Y.; Zou, Q.; Ge, R.; Shen, F.; Wang, Y. The critical role of CD133 (+) CD44(+/high) tumor cells in hematogenous metastasis of liver cancers. Cell Res. 2012, 22, 259–272. [Google Scholar] [CrossRef]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.; Lam, C.T.; Poon, R.T.; Fan, S.T. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef]
- Kimura, O.; Takahashi, T.; Ishii, N.; Inoue, Y.; Ueno, Y.; Kogure, T.; Fukushima, K.; Shiina, M.; Yamagiwa, Y.; Kondo, Y.; et al. Characterization of the epithelial cell adhesion molecule (EpCAM)+ cell population in hepatocellular carcinoma cell lines. Cancer Sci. 2010, 101, 2145–2155. [Google Scholar] [CrossRef]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Ohkuma, M.; Kim, H.M.; Akita, H.; Takiuchi, D.; Hatano, H.; Nagano, H.; et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Investig. 2010, 120, 3326–3339. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Q.; Liu, X.; Liu, C.; Liu, R.; Rycaj, K.; Zhang, D.; Liu, B.; Jeter, C.; Calhoun-Davis, T.; et al. Defining a population of stem-like human prostate cancer cells that can generate and propagate castration-resistant prostate cancer (CRPC). Clin. Cancer Res. 2016, 22, 4505–4516. [Google Scholar] [CrossRef] [PubMed]
- MacDonagh, L.; Gray, S.G.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. Lung cancer stem cells: The root of resistance. Cancer Lett. 2016, 372, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in Cancer: Cancer Stem Cells versus Clonal Evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95, S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Lavi, O.; Hall, M.D.; Gillet, J.-P. Towards a Better Understanding of the Complexity of Cancer Drug Resistance. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 85–102. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M.; et al. A Tumorigenic Subpopulation with Stem Cell Properties in Melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Saikawa. Tumor initiating potential of side population cells in human gastric cancer. Int. J. Oncol. 2009, 34, 1201–1207. [Google Scholar] [CrossRef]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side Population in Human Lung Cancer Cell Lines and Tumors Is Enriched with Stem-like Cancer Cells. Cancer Res. 2007, 67, 4827–4833. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Curtis, S.J.; Sinkevicius, K.W.; Li, D.; Lau, A.N.; Roach, R.R.; Zamponi, R.; Woolfenden, A.E.; Kirsch, D.G.; Wong, K.-K.; Kim, C.F. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell 2010, 7, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, B.; Strife, A.; Fried, J.; Sakai, Y.; Ota, K.; Ohkita, T.; Masuda, R. Studies of cellular proliferation in human leukemia. IV. Behavior of normal hematopoietic cells in 3 adults with acute leukemia given continuous infusions of3 H-thymidine for 8 or 10 days. Cancer 1970, 26, 1–19. [Google Scholar] [CrossRef]
- Moore, N.; Houghton, J.; Lyle, S. Slow-Cycling Therapy-Resistant Cancer Cells. Stem Cells Dev. 2012, 21, 1822–1830. [Google Scholar] [CrossRef]
- Vidal, S.J.; Rodriguez-Bravo, V.; Galsky, M.; Cordon-Cardo, C.; Domingo-Domenech, J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene 2013, 33, 4451–4463. [Google Scholar] [CrossRef]
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer Stem Cells: The Promise and the Potential. Semin. Oncol. 2015, 42, S3–S17. [Google Scholar] [CrossRef]
- Abdullah, L.N.; Chow, E.K.-H. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef]
- Kolenda, J.; Jensen, S.S.; Aaberg-Jessen, C.; Christensen, K.; Andersen, C.; Brünner, N.; Kristensen, B.W. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J. Neuro-Oncol. 2010, 103, 43–58. [Google Scholar] [CrossRef]
- Deeley, R.G.; Cole, S.P. Substrate recognition and transport by multidrug resistance protein 1 (ABCC1). FEBS Lett. 2005, 580, 1103–1111. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Wang, L.-L.; Di, Y.M.; Xue, C.C.; Duan, W.; Li, C.G.; Li, Y. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr. Med. Chem. 2008, 15, 1981–2039. [Google Scholar] [CrossRef]
- Shaffer, B.C.; Gillet, J.-P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug resistance: Still a daunting challenge to the successful treatment of AML. Drug Resist. Updat. 2012, 15, 62–69. [Google Scholar] [CrossRef]
- Xi, G.; Hayes, E.; Lewis, R.; Ichi, S.; Mania-Farnell, B.; Shim, K.; Takao, T.; Allender, E.; Mayanil, C.S.; Tomita, T. CD133 and DNA-PK regulate MDR1 via the PI3K- or Akt-NF-κB pathway in multidrug-resistant glioblastoma cells in vitro. Oncogene 2015, 35, 241–250. [Google Scholar] [CrossRef]
- Huang, B.; Fu, S.J.; Fan, W.Z.; Wang, Z.H.; Chen, Z.B.; Guo, S.J.; Chen, J.X.; Qiu, S.P. PKCε inhibits isolation and stemness of side population cells via the suppression of ABCB1 transporter and PI3K/Akt, MAPK/ERK signaling in renal cell carcinoma cell line 769P. Cancer Lett. 2016, 376, 148–154. [Google Scholar] [CrossRef]
- Johnson, R.A.; Shepard, E.M.; Scotto, K.W. Differential Regulation of MDR1 Transcription by the p53 Family Members. J. Boil. Chem. 2005, 280, 13213–13219. [Google Scholar] [CrossRef]
- Chou, J.-L.; Huang, R.-L.; Shay, J.; Chen, L.-Y.; Lin, S.-J.; Yan, P.S.; Chao, W.-T.; Lai, Y.-H.; Lai, Y.-L.; Chao, T.-K.; et al. Hypermethylation of the TGF-β target, ABCA1 is associated with poor prognosis in ovarian cancer patients. Clin. Epigenet. 2015, 7, 1. [Google Scholar] [CrossRef]
- Sládek, N.E. Human aldehyde dehydrogenases: Potential pathological, pharmacological, and toxicological impact. J. Biochem. Mol. Toxicol. 2003, 17, 7–23. [Google Scholar] [CrossRef]
- Ikawa, M.; Impraim, C.C.; Wang, G.; Yoshida, A. Isolation and characterization of aldehyde dehydrogenase isozymes from usual and atypical human livers. J. Boil. Chem. 1983, 258, 6282–6287. [Google Scholar]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Fitzgerald, T.L.; Rangan, S.; Dobbs, L.; Starr, S.; Sigounas, G. The impact of Aldehyde dehydrogenase 1 expression on prognosis for metastatic colon cancer. J. Surg. Res. 2014, 192, 82–89. [Google Scholar] [CrossRef]
- Vogler, T.; Kriegl, L.; Horst, D.; Engel, J.; Sagebiel, S.; Schäffauer, A.J.; Kirchner, T.; Jung, A. The expression pattern of aldehyde dehydrogenase 1 (ALDH1) is an independent prognostic marker for low survival in colorectal tumors. Exp. Mol. Pathol. 2012, 92, 111–117. [Google Scholar] [CrossRef]
- Deng, L.; Zhou, J.-F.; Sellers, R.S.; Li, J.-F.; Nguyen, A.V.; Wang, Y.; Orlofsky, A.; Liu, Q.; Hume, D.A.; Pollard, J.W.; et al. A Novel Mouse Model of Inflammatory Bowel Disease Links Mammalian Target of Rapamycin-Dependent Hyperproliferation of Colonic Epithelium to Inflammation-Associated Tumorigenesis. Am. J. Pathol. 2010, 176, 952–967. [Google Scholar] [CrossRef]
- Hessman, C.J.; Bubbers, E.J.; Billingsley, K.G.; Herzig, D.O.; Wong, M.H. Loss of expression of the cancer stem cell marker aldehyde dehydrogenase1 correlates with advanced stage colorectal cancer. Am. J. Surg. 2012, 203, 649–653. [Google Scholar] [CrossRef][Green Version]
- Kim, M.P.; Fleming, J.B.; Wang, H.; Abbruzzese, J.L.; Choi, W.; Kopetz, S.; McConkey, D.J.; Evans, D.B.; Gallick, G.E. ALDH Activity Selectively Defines an Enhanced Tumor-Initiating Cell Population Relative to CD133 Expression in Human Pancreatic Adenocarcinoma. PLoS ONE 2011, 6, e20636. [Google Scholar] [CrossRef]
- Li, T.; Su, Y.; Mei, Y.; Leng, Q.; Leng, B.; Liu, Z.; Stass, S.A.; Jiang, F. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Investig. 2009, 90, 234–244. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Scadden, D.T. Harnessing the apoptotic programs in cancer stem-like cells. EMBO Rep. 2015, 16, 1084–1098. [Google Scholar] [CrossRef]
- Costello, R.T.; Mallet, F.; Chambost, H.; Sainty, D.; Arnoulet, C.; Gastaut, J.A.; Olive, D. The immunophenotype of minimally differentiated acute myeloid leukemia (AML-M0): Reduced immunogenicity and high frequency of CD34+/CD38− leukemic progenitors. Leukemia 1999, 13, 1513–1518. [Google Scholar] [CrossRef]
- Weller, M.; Frei, K.; Groscurth, P.; Krammer, P.H.; Yonekawa, Y.; Fontana, A. Anti-Fas/APO-1 antibody-mediated apoptosis of cultured human glioma cells. Induction and modulation of sensitivity by cytokines. J. Clin. Investig. 1994, 94, 954–964. [Google Scholar] [CrossRef]
- Frei, K.; Ambar, B.; Adachi, N.; Yonekawa, Y.; Fontana, A. Ex vivo malignant glioma cells are sensitive to Fas (CD95/APO-1) ligand-mediated apoptosis. J. Neuroimmunol. 1998, 87, 105–113. [Google Scholar] [CrossRef]
- Tao, J.; Qiu, B.; Zhang, D.; Wang, Y. Expression levels of Fas/Fas-L mRNA in human brain glioma stem cells. Mol. Med. Rep. 2012, 5, 1202–1206. [Google Scholar] [CrossRef][Green Version]
- Eisele, G.; Roth, P.; Hasenbach, K.; Aulwurm, S.; Wolpert, F.; Tabatabai, G.; Wick, W.; Weller, M. APO010, a synthetic hexameric CD95 ligand, induces human glioma cell death in vitro and in vivo. Neuro-Oncology 2010, 13, 155–164. [Google Scholar] [CrossRef]
- Eisele, G.; Wolpert, F.; Decrey, G.; Weller, M. APO010, a synthetic hexameric CD95 ligand, induces death of human glioblastoma stem-like cells. Anticancer Res. 2013, 33, 3563–3571. [Google Scholar]
- Zobalova, R.; McDermott, L.; Stantic, M.; Prokopova, K.; Dong, L.-F.; Neuzil, J. CD133-positive cells are resistant to TRAIL due to up-regulation of FLIP. Biochem. Biophys. Res. Commun. 2008, 373, 567–571. [Google Scholar] [CrossRef]
- Dong, L.-F.; Freeman, R.; Liu, J.; Zobalova, R.; Marin-Hernandez, A.; Stantic, M.; Rohlena, J.; Valis, K.; Rodriguez-Enriquez, S.; Butcher, B.; et al. Suppression of Tumor Growth In vivo by the Mitocan α-tocopheryl Succinate Requires Respiratory Complex II. Clin. Cancer Res. 2009, 15, 1593–1600. [Google Scholar] [CrossRef]
- Ding, L.; Yuan, C.; Wei, F.; Wang, G.; Zhang, J.; Bellail, A.C.; Zhang, Z.; Olson, J.J.; Hao, C. Cisplatin Restores TRAIL apoptotic pathway in Glioblastoma-Derived Stem Cells through Up-regulation of DR5 and Down-regulation of c-FLIP. Cancer Investig. 2011, 29, 511–520. [Google Scholar] [CrossRef]
- Dutton, A.; Young, L.S.; Murray, P.G. The role of cellular flice inhibitory protein (c-FLIP) in the pathogenesis and treatment of cancer. Expert Opin. Ther. Targets 2006, 10, 27–35. [Google Scholar] [CrossRef]
- Shirley, S.; Micheau, O. Targeting c-FLIP in cancer. Cancer Lett. 2013, 332, 141–150. [Google Scholar] [CrossRef]
- Lauria, F.; Raspadori, D.; Rondelli, D.; Ventura, M.; Fiacchini, M.; Visani, G.; Forconi, F.; Tura, S. High bcl-2 expression in acute myeloid leukemia cells correlates with CD34 positivity and complete remission rate. Leukemia 1997, 11, 2075–2078. [Google Scholar] [CrossRef]
- Campos, L.; Oriol, P.; Sabido, O.; Guyotat, D. Simultaneous Expression of P-Glycoprotein and BCL-2 in Acute Myeloid Leukemia Blast Cells. Leuk. Lymphoma 1997, 27, 119–125. [Google Scholar] [CrossRef]
- Tóthová, E.; Kafková, A.; Stecová, N.; Fricová, M.; Guman, T.; Svorcová, E. Immune-mediated complications during interferon alpha therapy in chronic myelogenous leukemia. Neoplasma 2002, 49, 91–94. [Google Scholar]
- Maugeri-Saccà, M.; Bartucci, M.; De Maria, R. DNA Damage Repair Pathways in Cancer Stem Cells. Mol. Cancer Ther. 2012, 11, 1627–1636. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Bartucci, M.; Svensson, S.; Romania, P.; Dattilo, R.; Patrizii, M.; Signore, M.; Navarra, S.; Lotti, F.; Biffoni, M.; Pilozzi, E.; et al. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2011, 19, 768–778. [Google Scholar] [CrossRef]
- Karimi-Busheri, F.; Rasouli-Nia, A.; Mackey, J.R.; Weinfeld, M. Senescence evasion by MCF-7 human breast tumor-initiating cells. Breast Cancer Res. 2010, 12, R31. [Google Scholar] [CrossRef]
- Morrison, S.J.; Spradling, A.C. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar]
- Rashidi, N.M.; Lo Celso, C. Flying back to the nest: Intravital microscopy reveals how the niche can induce stemness. Intravital 2014, 3, e29653. [Google Scholar] [CrossRef][Green Version]
- Ferraro, F.; Celso, C.L.; Scadden, D. Adult stem cells and their niches. Single Mol. Single Cell Seq. 2010, 695, 155–168. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Balkwill, F.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Investig. 2011, 121, 3804–3809. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Longley, D.; Johnston, P. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Junttila, M.R.; De Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell. Physiol. 2018, 234, 8381–8395. [Google Scholar] [CrossRef]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef]
- Cabarcas, S.M.; Mathews, L.A.; Farrar, W.L. The cancer stem cell niche-there goes the neighborhood? Int. J. Cancer 2011, 129, 2315–2327. [Google Scholar] [CrossRef]
- Buczek, M.E.; Miles, A.K.; Green, W.; Johnson, C.; Boocock, D.J.; Pockley, A.G.; Rees, R.C.; Hulman, G.; Van Schalkwyk, G.; Parkinson, R.; et al. Cytoplasmic PML promotes TGF-β-associated epithelial–mesenchymal transition and invasion in prostate cancer. Oncogene 2015, 35, 3465–3475. [Google Scholar] [CrossRef]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-β signaling and epithelial–mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 31. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Corcoran, K.E.; Trzaska, K.A.; Fernandes, H.; Bryan, M.; Taborga, M.; Srinivas, V.; Packman, K.; Patel, P.S.; Rameshwar, P. Mesenchymal Stem Cells in Early Entry of Breast Cancer into Bone Marrow. PLoS ONE 2008, 3, e2563. [Google Scholar] [CrossRef]
- Nabha, S.M.; Dos Santos, E.B.; Yamamoto, H.A.; Belizi, A.; Dong, Z.; Meng, H.; Saliganan, A.; Sabbota, A.; Bonfil, R.D.; Cher, M.L. Bone marrow stromal cells enhance prostate cancer cell invasion through type I collagen in an MMP-12 dependent manner. Int. J. Cancer 2008, 122, 2482–2490. [Google Scholar] [CrossRef]
- Duda, D.G.; Duyverman, A.M.M.J.; Kohno, M.; Snuderl, M.; Steller, E.J.A.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef]
- Li, W.; Zhou, Y.; Yang, J.; Zhang, X.; Zhang, H.; Zhang, T.; Zhao, S.; Zheng, P.; Huo, J.; Wu, H. Gastric cancer-derived mesenchymal stem cells prompt gastric cancer progression through secretion of interleukin-8. J. Exp. Clin. Cancer Res. 2015, 34, 52. [Google Scholar] [CrossRef]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal Stem Cell Transition to Tumor-Associated Fibroblasts Contributes to Fibrovascular Network Expansion and Tumor Progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma Associated Fibroblast Like Differentiation of Human Mesenchymal Stem Cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef]
- Peng, Y.; Li, Z.; Li, Z. GRP78 secreted by tumor cells stimulates differentiation of bone marrow mesenchymal stem cells to cancer-associated fibroblasts. Biochem. Biophys. Res. Commun. 2013, 440, 558–563. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272. [Google Scholar] [CrossRef]
- Fessler, E.; Borovski, T.; Medema, J.P. Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol. Cancer 2015, 14, 157. [Google Scholar] [CrossRef]
- Pirtskhalaishvili, G.; Nelson, J.B. Endothelium-derived factors as paracrine mediators of prostate cancer progression. Prostate 2000, 44, 77–87. [Google Scholar] [CrossRef]
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 2010, 10, 138–146. [Google Scholar] [CrossRef]
- Grünert, S.; Jechlinger, M.; Beug, H.; Gruenert, S. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev. Mol. Cell Boil. 2003, 4, 657–665. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Boil. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta signalling in context. Nature reviews. Mol. Cell Boil. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Boil. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Fendrich, V.; Waldmann, J.; Esni, F.; Ramaswamy, A.; Mullendore, M.; Buchholz, M.; Maitra, A.; Feldmann, G. Snail and Sonic Hedgehog activation in neuroendocrine tumors of the ileum. Endoc.-Relat. Cancer 2007, 14, 865–874. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, L.; He, C.-S.; Xu, F.; Liu, J.-L.; Hu, Z.-H.; Zhao, L.-P.; Tian, Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell. Biochem. 2012, 113, 150–1513. [Google Scholar] [CrossRef]
- Shirakihara, T.; Horiguchi, K.; Miyazawa, K.; Ehata, S.; Shibata, T.; Morita, I.; Miyazono, K.; Saitoh, M. TGF-β regulates isoform switching of FGF receptors and epithelial–mesenchymal transition. EMBO J. 2011, 30, 783–795. [Google Scholar] [CrossRef]
- Uttamsingh, S.; Bao, X.; Nguyen, K.T.; Bhanot, M.; Gong, J.; Chan, J.L.-K.; Liu, F.; Chu, T.T.; Wang, L.-H. Synergistic effect between EGF and TGF-β1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene 2007, 27, 2626–2634. [Google Scholar] [CrossRef]
- Wendt, M.K.; Smith, J.A.; Schiemann, W.P. Transforming Growth Factor-β-Induced Epithelial-Mesenchymal Transition Facilitates Epidermal Growth Factor-Dependent Breast Cancer Progression. Oncogene 2010, 29, 6485–6498. [Google Scholar] [CrossRef]
- Snyder, V.; Reed-Newman, T.C.; Arnold, L.; Thomas, S.M.; Anant, S. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front. Oncol. 2018, 8, 203. [Google Scholar] [CrossRef]
- Emmink, B.L.; Verheem, A.; Van Houdt, W.J.; Steller, E.J.; Govaert, K.M.; Pham, T.V.; Piersma, S.R.; Rinkes, I.H.B.; Jimenez, C.R.; Kranenburg, O. The secretome of colon cancer stem cells contains drug-metabolizing enzymes. J. Proteom. 2013, 91, 84–96. [Google Scholar] [CrossRef]
- Hammoudi, N.; Ahmed, K.B.R.; Garcia-Prieto, C.; Huang, P. Metabolic alterations in cancer cells and therapeutic implications. Chin. J. Cancer 2011, 30, 508–525. [Google Scholar] [CrossRef]
- Ye, X.-Q.; Li, Q.; Wang, G.-H.; Sun, F.-F.; Huang, G.-J.; Bian, X.-W.; Yu, S.-C.; Qian, G.-S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Kheir, T.B.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef]
- De Luca, A.; Fiorillo, M.; Peiris-Pagés, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Reue, K.; Frohnen, P.; Chan, M.; Alhiyari, Y.; Dratver, M.B.; Pajonk, F. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res. Treat. 2014, 146, 525–534. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Yin, A.H.; Miraglia, S.; Zanjani, E.D.; Almeida-Porada, G.; Ogawa, M.; Leary, A.G.; Olweus, J.; Kearney, J.; Buck, D.W. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997, 90, 5002–5012. [Google Scholar]
- Feng, K.-C.; Guo, Y.-L.; Liu, Y.; Dai, H.-R.; Wang, Y.; Lv, H.-Y.; Huang, J.-H.; Yang, Q.-M.; Han, W.-D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Boil. 2017, 5, 18. [Google Scholar] [CrossRef]
- Vey, N.; Delaunay, J.; Martinelli, G.; Fiedler, W.; Raffoux, E.; Prebet, T.; Gomez-Roca, C.; Papayannidis, C.; Kebenko, M.; Paschka, P.; et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 2016, 7, 32532–32542. [Google Scholar] [CrossRef]
- Macdonald, J.; Henri, J.; Roy, K.; Hays, E.; Bauer, M.; Veedu, R.N.; Pouliot, N.; Shigdar, S. EpCAM immunotherapy versus specific targeted delivery of drugs. Cancers 2018, 10, 19. [Google Scholar] [CrossRef]
- Ragnhammar, P.; Fagerberg, J.; Frödin, J.-E.; Hjelm, A.-L.; Lindemalm, C.; Magnusson, I.; Masucci, G.; Mellstedt, H. Effect of monoclonal antibody 17-1A and gm-CSF in patients with advanced colorectal carcinoma—long-lasting, complete remissions can be induced. Int. J. Cancer 1993, 53, 751–758. [Google Scholar] [CrossRef]
- Riethmüller, G.; Schneider-Gädicke, E.; Schlimok, G.; Schmiegel, W.; Raab, R.; Höffken, K.; Gruber, R.; Pichlmaier, H.; Hirche, H.; Pichlmayr, R. Randomised trial of monoclonal antibody for adjuvant therapy of resected Dukes’ C colorectal carcinoma. German Cancer Aid 17-1A Study Group. Lancet 1994, 343, 1177–1183. [Google Scholar] [CrossRef]
- Schmoll, H.-J.; Wittig, B.; Arnold, D.; Riera-Knorrenschild, J.; Nitsche, D.; Kroening, H.; Mayer, F.; Andel, J.; Ziebermayr, R.; Scheithauer, W. Maintenance treatment with the immunomodulator MGN1703, a Toll-like receptor 9 (TLR9) agonist, in patients with metastatic colorectal carcinoma and disease control after chemotherapy: A randomised, double-blind, placebo-controlled trial. J. Cancer Res. Clin. Oncol. 2014, 140, 1615–1624. [Google Scholar] [CrossRef]
- Fields, A.L.; Keller, A.; Schwartzberg, L.; Bernard, S.; Kardinal, C.; Cohen, A.; Schulz, J.; Eisenberg, P.; Forster, J.; Wissel, P. Adjuvant therapy with the monoclonal antibody edrecolomab plus fluorouracil-based therapy does not improve overall survival of patients with stage III colon cancer. J. Clin. Oncol. 2009, 27, 1941–1947. [Google Scholar] [CrossRef]
- Münz, M.; Murr, A.; Kvesic, M.; Rau, D.; Mangold, S.; Pflanz, S.; Lumsden, J.; Volkland, J.; Fagerberg, J.; Riethmüller, G.; et al. Side-by-side analysis of five clinically tested anti-EpCAM monoclonal antibodies. Cancer Cell Int. 2010, 10, 44. [Google Scholar] [CrossRef]
- Schmidt, M.; Scheulen, M.E.; Dittrich, C.; Obrist, P.; Marschner, N.; Dirix, L.; Schmidt, M.; Ruttinger, D.; Schuler, M.; Reinhardt, C.; et al. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann. Oncol. Off. J. of Eur. Soc. Med. Oncol. 2010, 21, 275–282. [Google Scholar] [CrossRef]
- Gao, A.-G.; Lindberg, F.P.; Finn, M.B.; Blystone, S.D.; Brown, E.J.; Frazier, W.A. Integrin-associated protein is a receptor for the C-terminal domain of thrombospondin. J. Boil. Chem. 1996, 271, 21–24. [Google Scholar] [CrossRef]
- Lindberg, F.P.; Bullard, D.C.; Caver, T.E.; Gresham, H.D.; Beaudet, A.L.; Brown, E.J. Decreased resistance to bacterial infection and granulocyte defects in IAP-Deficient mice. Science 1996, 274, 795–798. [Google Scholar] [CrossRef]
- Miyashita, M.; Ohnishi, H.; Okazawa, H.; Tomonaga, H.; Hayashi, A.; Fujimoto, T.-T.; Furuya, N.; Matozaki, T. Promotion of neurite and filopodium formation by CD47: Roles of integrins, rac, and Cdc42. Mol. Boil. Cell 2004, 15, 3950–3963. [Google Scholar] [CrossRef]
- Liu, Y.; Merlin, D.; Burst, S.L.; Pochet, M.; Madara, J.L.; Parkos, C.A. The role of CD47 in neutrophil transmigration: Increased rate of migration correlates with increased cell surface expression of cd47. J. Boil. Chem. 2001, 276, 40156–40166. [Google Scholar] [CrossRef]
- Reinhold, M.I.; Lindberg, F.P.; Kersh, G.J.; Allen, P.M.; Brown, E.J. Costimulation of T cell activation by integrin-associated protein (CD47) is an adhesion-dependent, CD28-independent signaling pathway. J. Exp. Med. 1997, 185, 1–11. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; Van Rooijen, N.; Weissman, I.L. CD47 is up-regulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, J.-C.; Lee, J.-J.; Kim, S.; Lee, S.G.; Park, S.-W.; Sung, M.W.; Heo, D.S. Association of CD47 with natural killer cell-mediated cytotoxicity of head-and-neck squamous cell carcinoma lines. Tumor Boil. 2008, 29, 28–34. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef]
- Chan, K.S.; Espinosa, I.; Chao, M.; Wong, D.; Ailles, L.; Diehn, M.; Gill, H.; Presti, J., Jr.; Chang, H.Y.; van de Rijn, M.; et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14016–14021. [Google Scholar] [CrossRef]
- Manna, P.P.; Frazier, W.A. CD47 mediates killing of breast tumor cells via Gi-dependent inhibition of protein kinase A. Cancer Res. 2004, 64, 1026–1036. [Google Scholar] [CrossRef]
- Schulenburg, A.; Blatt, K.; Cerny-Reiterer, S.; Sadovnik, I.; Herrmann, H.; Marian, B.; Grunt, T.W.; Zielinski, C.C.; Valent, P. Cancer stem cells in basic science and in translational oncology: Can we translate into clinical application? J. Hematol. Oncol. 2015, 8, 16. [Google Scholar] [CrossRef]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 2015, 8, 130. [Google Scholar] [CrossRef]
- Fang, X.; Chen, C.; Xia, F.; Yu, Z.; Zhang, Y.; Zhang, F.; Gu, H.; Wan, J.; Zhang, X.; Weng, W.; et al. CD274 promotes cell cycle entry of leukemia-initiating cells through JNK/Cyclin D2 signaling. J. Hematol. Oncol. 2016, 9, 124. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, J.; Kong, X.; Li, E.; Liu, Y.; Du, X.; Kang, Z.; Tang, Y.; Kuang, Y.; Yang, Z.; et al. CD47 promotes tumor invasion and metastasis in non-small cell lung cancer. Sci. Rep. 2016, 6, 29719. [Google Scholar] [CrossRef]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef]
- Liu, X.; Kwon, H.; Li, Z.; Fu, Y.-X. Is CD47 an innate immune checkpoint for tumor evasion? J. Hematol. Oncol. 2017, 10, 12. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 blockade by Hu5F9-G4 and rituximab in non-hodgkin’s lymphoma. New Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Huber, A.; Dammeijer, F.; Aerts, J.G.J.V.; Vroman, H. Current state of dendritic cell-based immunotherapy: Opportunities for in vitro antigen loading of different DC subsets? Front. Immunol. 2018, 9, 2804. [Google Scholar] [CrossRef]
- Van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; De Vries, I.J.M.; Bol, K.F. Dendritic cell cancer therapy: Vaccinating the right patient at the right time. Front. Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef]
- Lin, P.-C.; Hsieh, H.-Y.; Chu, P.-C.; Chen, C.S. Therapeutic opportunities of targeting histone deacetylase isoforms to eradicate cancer stem cells. Int. J. Mol. Sci. 2018, 19, 1939. [Google Scholar] [CrossRef]
- Mele, L.; Paino, F.; Papaccio, F.; Regad, T.; Boocock, D.; Stiuso, P.; Lombardi, A.; Liccardo, D.; Aquino, G.; Barbieri, A.; et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis. 2018, 9, 572. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Tumour | Biomarkers | References |
|---|---|---|
| Breast cancer | CD44+/CD24−/low/ALDH+ | [16] |
| Prostate cancer | CD44+/a2b1+/ALDH+ | [34] |
| Melanoma | ABCB5+ | [17] |
| CD20+ | [18] | |
| CD271+ | ||
| Glioblastoma | CD133+ | [20] |
| Colon cancer | CD133+/CD44+/ALDH+ | [21] |
| EpCAM+/CD44+/CD166+ | [22] | |
| CD44+/CD24+ | [23] | |
| CD133+/CD24+ | [24] | |
| Lung cancer | CD133+ | |
| CD44+ | ||
| ALDH+ | [35,47] | |
| CD117+ | ||
| Gastric cancer | CD133+ | |
| CD44+/CD24+ | [25] | |
| CD90+ | [26] | |
| CD44+/CD54+ | ||
| Head and neck cancer | CD44+/ALDH+ | [27] |
| CD44+/CD66- | ||
| Ovarian cancer | CD133+ | [28] |
| CD44+ | ||
| ALDH+ | ||
| CD117+ | ||
| Pancreatic cancer | CD133+/CD44+/CD24+/ESA+ | [29] |
| Liver cancer | CD133+/CD44+ | [30] |
| CD90+ | [31] | |
| EpCAM+ | [32] | |
| CD13+ | [33] |
| Target | Tumour Type | Therapy | Clinical Trial/Reports | Results |
|---|---|---|---|---|
| CD133 | Advanced cholangiosarcoma | Cocktail CD133 CAR-T and CART-EGFR | NCT02541370 (Phase I and II) | Toxicity |
| Glioblastoma multiforme | ICT-121 DC vaccine | NCT02049489 (Phase I) | Not published | |
| CD44 | Refractory Acute Myeloid Leukemia | Monoclonal antibody (RG7356) | 146 | Limited clinical activity |
| EpCAM | Metastatic colorectal cancer | Edrecolomab (Monoclonal antibody) | 151 | Limited response |
| 150 | Not statistically significant | |||
| 3622W94 (Monoclonal antibody) | 152 | Toxicity | ||
| ING-1 (Monoclonal antibody) | Toxicity | |||
| Adecatumumab (Monoclonal antibody) | 153 | Favourable response in patients with highest expression | ||
| Various types of tumours | EPCAM-targeted CAR-T cells | NCT02729493 (N/A) | Ongoing | |
| NCT02725125 (N/A) | Ongoing | |||
| NCT02915445 (Phase I) | Ongoing | |||
| NCT03013712 (Phase I and II) | Ongoing | |||
| CD47 | Various solid tumours and hematological malignancies | Hu5F9-G4 (Monoclonal antibody) | NCT02216409 (Phase I) | Not statistically significant |
| NCT02678338 (Phase I) | Not published | |||
| Combination Hu5F9-G4 and Cetuximab | NCT02953782 (Phase I and II) | Ongoing | ||
| Combination Hu5F9-G4 and Retuximab | NCT02953509 (Phase I and II) | Ongoing | ||
| CC-90002 (Monoclonal antibody) | NCT02641002 (Phase I) | Ongoing | ||
| Combination CC-90002 and Retuximab | NCT02367196 (Phase I) | Ongoing | ||
| Autogenic glioma stem-like cells (A2B5+) | Glioblastoma multiforme | Dendritic cell-based vaccine | NCT01567202 (Phase II) | Ongoing |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbato, L.; Bocchetti, M.; Di Biase, A.; Regad, T. Cancer Stem Cells and Targeting Strategies. Cells 2019, 8, 926. https://doi.org/10.3390/cells8080926
Barbato L, Bocchetti M, Di Biase A, Regad T. Cancer Stem Cells and Targeting Strategies. Cells. 2019; 8(8):926. https://doi.org/10.3390/cells8080926
Chicago/Turabian StyleBarbato, Luisa, Marco Bocchetti, Anna Di Biase, and Tarik Regad. 2019. "Cancer Stem Cells and Targeting Strategies" Cells 8, no. 8: 926. https://doi.org/10.3390/cells8080926
APA StyleBarbato, L., Bocchetti, M., Di Biase, A., & Regad, T. (2019). Cancer Stem Cells and Targeting Strategies. Cells, 8(8), 926. https://doi.org/10.3390/cells8080926