The uPAR System as a Potential Therapeutic Target in the Diseased Eye


 ,
,  and
and 
Abstract
:1. Introduction
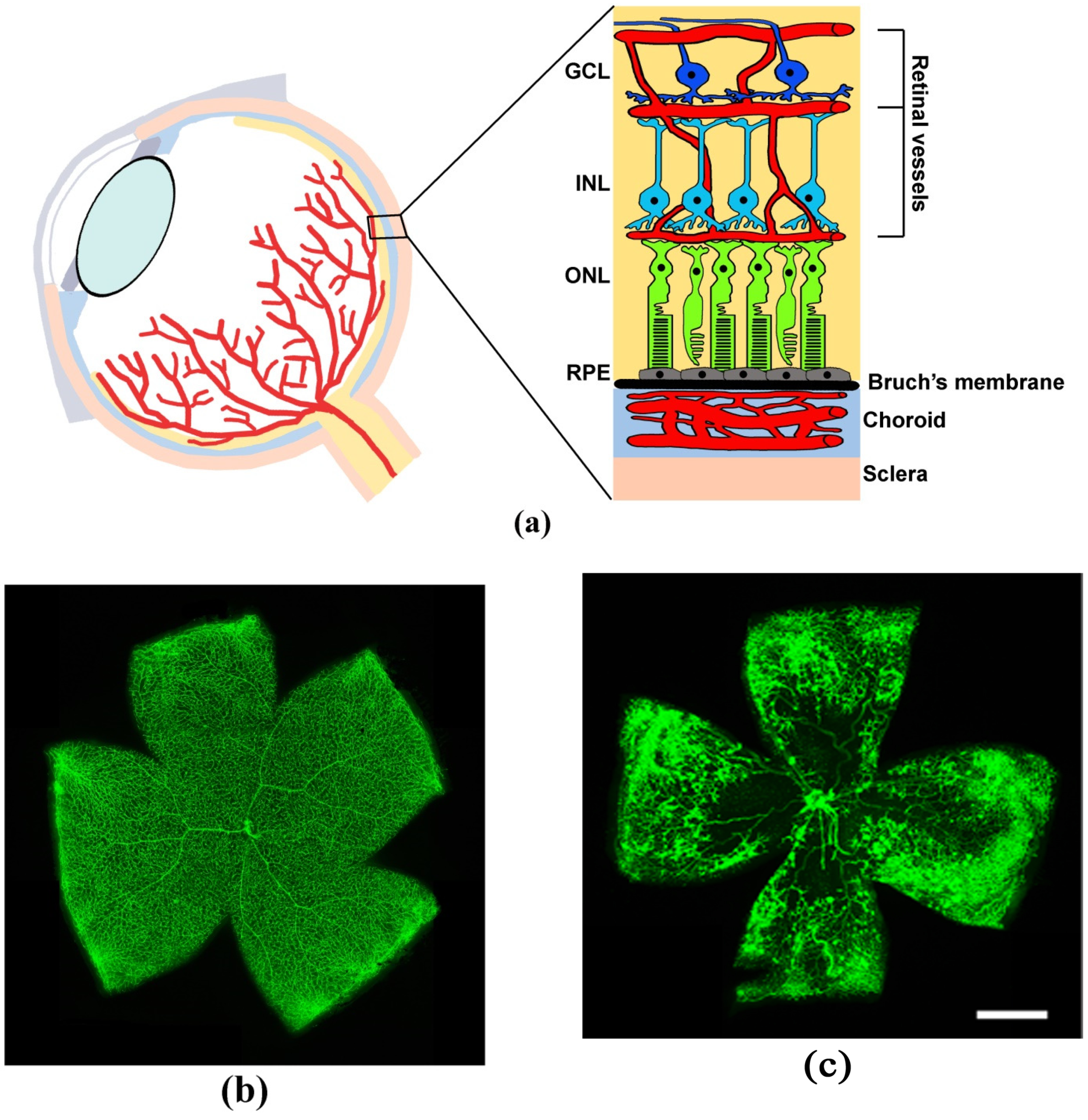
2. Degenerative Diseases of the Retina
3. Animal Models Mimicking Eye Pathologies and Their Potential Value for Developing Novel Treatments
4. The uPAR System
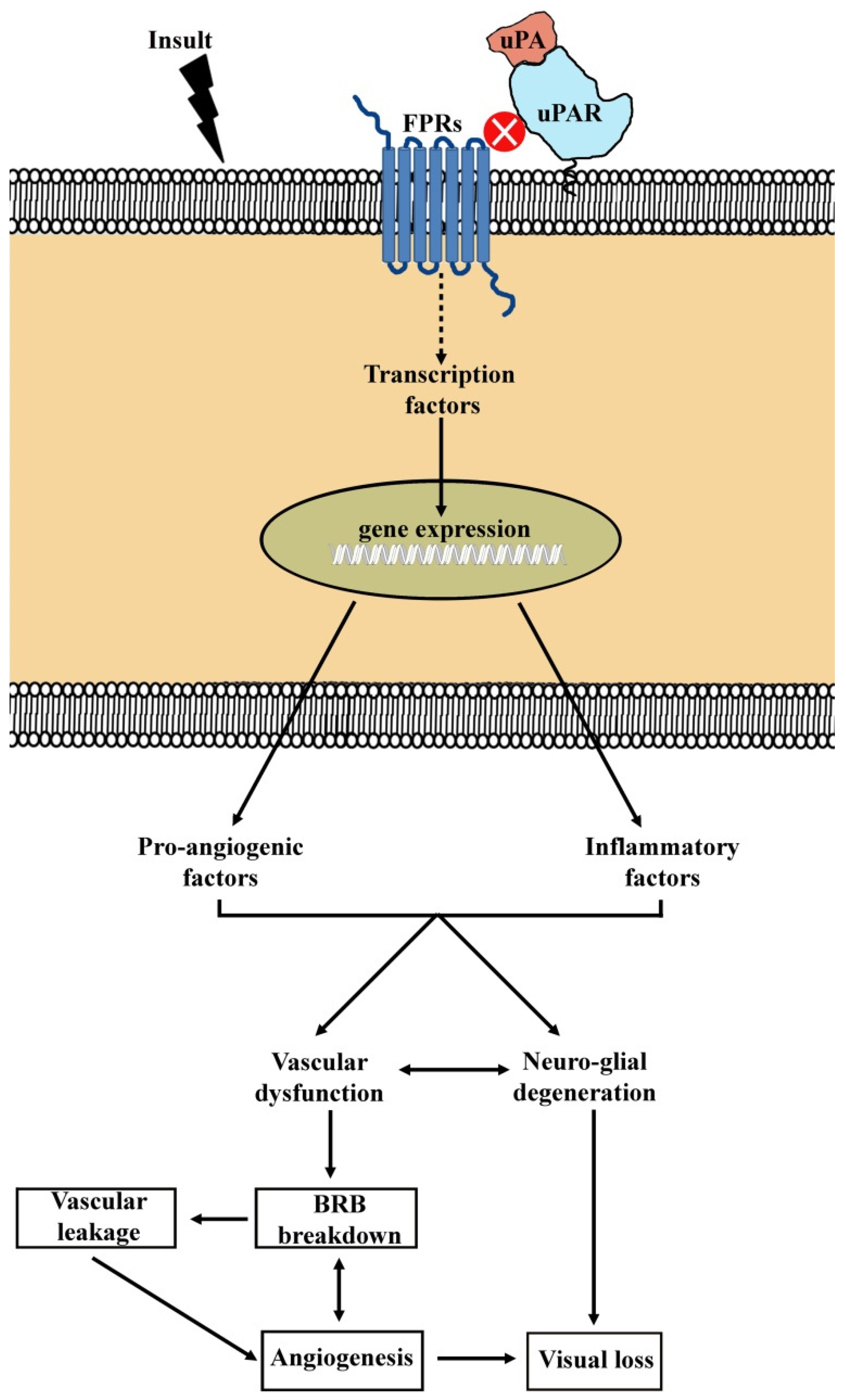
5. uPAR-Co-Receptor Interaction
6. Mechanisms of uPAR Signaling
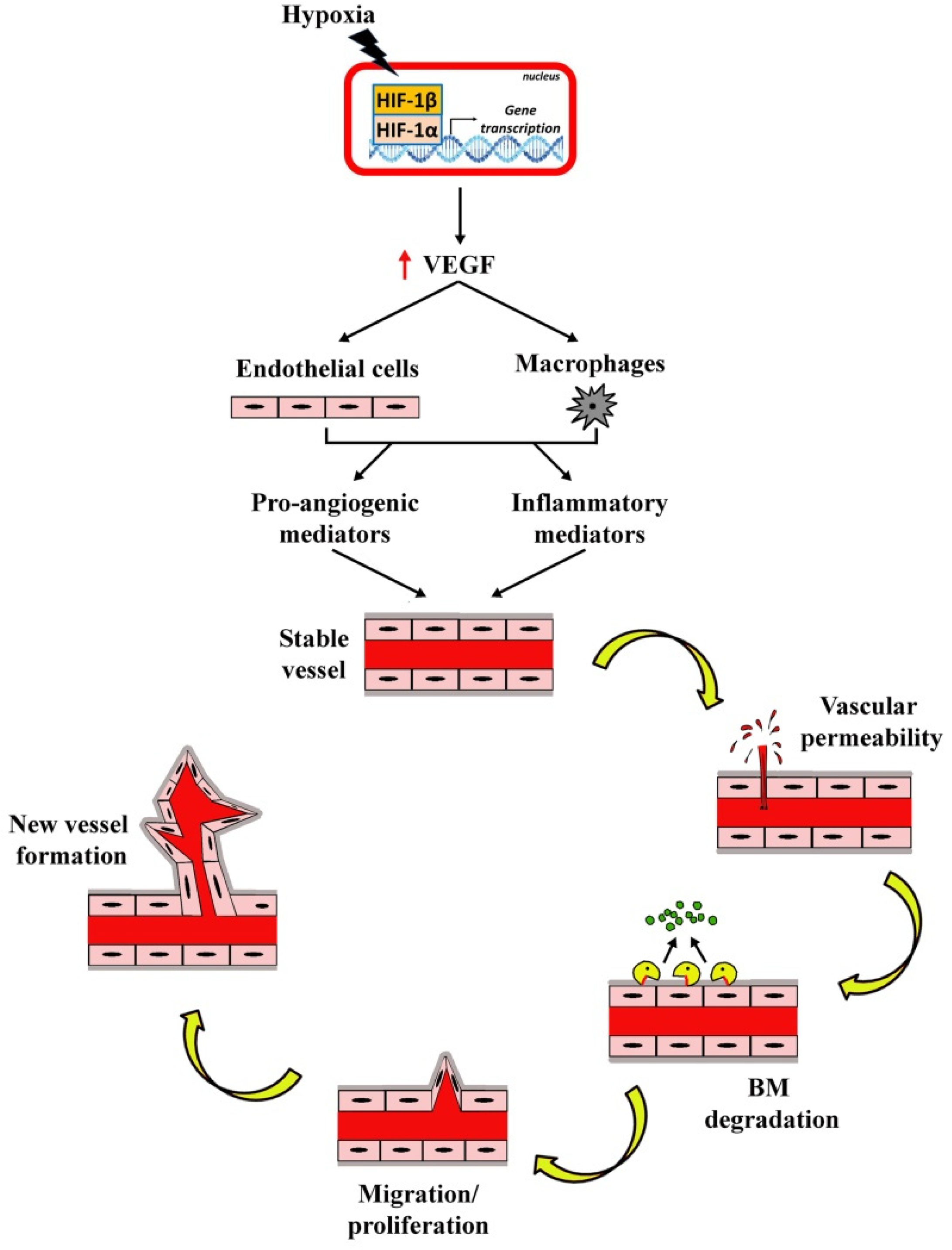
7. The uPAR System in the Diseased Eye
7.1. Ischemic Retinopathies
7.2. Retinitis Pigmentosa
8. Inhibition of the uPAR System
8.1. Pharmacological Approaches to Inhibit the uPAR System
8.2. Genetic Approaches to Inhibit the uPAR System
8.3. Inhibition of uPAR Lateral Partners
9. Conclusion and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ruiz-Ocaña, P.; Espinoza Requena, P.; Alonso-Ojembarrena, A.; Alemany Márquez, P.; Jiménez Carmona, S.; Lechuga-Sancho, A.M. Decreased Retinal Thickness in Type 1 Diabetic Children with Signs of Nonproliferative Diabetic Retinopathy. Int. J. Endocrinol. 2018, 2018, 1078531. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.C.; Holm, M.; Austeng, D.; Morken, T.S.; Zhou, T.E.; Beaudry-Richard, A.; Sierra, E.M.; Dammann, O.; Chemtob, S. Retinopathy of prematurity: Inflammation, choroidal degeneration, and novel promising therapeutic strategies. J. Neuroinflammation 2017, 14, 165. [Google Scholar] [CrossRef] [PubMed]
- Aouiss, A.; Anka Idrissi, D.; Kabine, M.; Zaid, Y. Update of inflammatory proliferative retinopathy: Ischemia, hypoxia and angiogenesis. Curr. Res. Transl. Med. 2019, 67, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.B.; Abe, R.Y.; Zangalli, C.; Sodre, S.L.; Donini, F.A.; Costa, D.C.; Leite, A.; Felix, J.P.; Torigoe, M.; Diniz-Filho, A.; et al. Neovascular glaucoma: A review. Int. J. Retina Vitreous 2016, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Knight, D.K.; Vu, T.T.; Mehta, M.C. Retinitis Pigmentosa: Review of Current Treatment. Int. Ophthalmol. Clin. 2019, 59, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Daruich, A.; Matet, A.; Moulin, A.; Kowalczuk, L.; Nicolas, M.; Sellam, A.; Rothschild, P.R.; Omri, S.; Gélizé, E.; Jonet, L.; et al. Mechanisms of macular edema: Beyond the surface. Prog. Retin. Eye Res. 2018, 63, 20–68. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Harris, A.; Ciulla, T.A.; Siesky, B.; Patel, P.; Belamkar, A.; Mathew, S.; Verticchio Vercellin, A.C. Vascular dysfunction in retinitis pigmentosa. Acta Ophthalmol. 2019. [Google Scholar] [CrossRef]
- Altmann, C.; Schmidt, M.H.H. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int. J. Mol. Sci. 2018, 19, 110. [Google Scholar] [CrossRef]
- Graca, A.B.; Hippert, C.; Pearson, R.A. Müller Glia Reactivity and Development of Gliosis in Response to Pathological Conditions. Adv. Exp. Med. Biol. 2018, 1074, 303–308. [Google Scholar] [CrossRef]
- Jeong, Y.C.; Hwang, Y.H. Etiology and features of eyes with rubeosis iridis among Korean patients: A population-based single center study. PLoS ONE 2016, 11, e0160662. [Google Scholar] [CrossRef]
- Sun, Y.; Smith, L.E.H. Retinal Vasculature in Development and Diseases. Annu. Rev. Vis. Sci. 2018, 4, 101–122. [Google Scholar] [CrossRef]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight. 2017, 2, 93751. [Google Scholar] [CrossRef]
- Rübsam, A.; Parikh, S.; Fort, P.E. Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 942. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef]
- Gardner, T.W.; Davila, J.R. The neurovascular unit and the pathophysiologic basis of diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 1–6. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef]
- Zhang, H.B.; Wang, X.D.; Xu, K.; Li, X.G. The progress of prophylactic treatment in retinopathy of prematurity. Int. J. Ophthalmol. 2018, 11, 858–873. [Google Scholar] [CrossRef]
- Wang, H.; Hartnett, M.E. Regulation of signaling events involved in the pathophysiology of neovascular AMD. Mol. Vis. 2016, 22, 189–202. [Google Scholar]
- Beaujean, O.; Locri, F.; Aronsson, M.; Kvanta, A.; André, H. A novel in vivo model of puncture-induced iris neovascularization. PLoS ONE 2017, 12, e0180235. [Google Scholar] [CrossRef]
- Locri, F.; Aronsson, M.; Beaujean, O.; Kvanta, A.; André, H. Puncture-Induced Iris Neovascularization as a Mouse Model of Rubeosis Iridis. J. Vis. Exp. 2018, 133, e57398. [Google Scholar] [CrossRef]
- Ozawa, Y.; Kurihara, T.; Sasaki, M.; Ban, N.; Yuki, K.; Kubota, S.; Tsubota, K. Neural degeneration in the retina of the streptozotocin-induced type 1 diabetes model. Exp. Diabetes Res. 2011, 2011, 108328. [Google Scholar] [CrossRef]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef]
- Sasase, T. Pathophysiological characteristics of diabetic ocular complications in spontaneously diabetic torii rat. J. Ophthalmol. 2010, 2010, 615641. [Google Scholar] [CrossRef]
- Smith, L.E.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111. [Google Scholar]
- Tobe, T.; Ortega, S.; Luna, J.D.; Ozaki, H.; Okamoto, N.; Derevjanik, N.L.; Vinores, S.A.; Basilico, C.; Campochiaro, P.A. Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am. J. Pathol. 1998, 153, 1641–1646. [Google Scholar] [CrossRef]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal degeneration mutants in the mouse. Vision Res. 2002, 42, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Guadagni, V.; Biagioni, M.; Novelli, E.; Aretini, P.; Mazzanti, C.M.; Strettoi, E. Rescuing cones and daylight vision in retinitis pigmentosa mice. FASEB J. 2019. [Google Scholar] [CrossRef]
- Massengill, M.T.; Ahmed, C.M.; Lewin, A.S.; Ildefonso, C.J. Neuroinflammation in Retinitis Pigmentosa, Diabetic Retinopathy, and Age-Related Macular Degeneration: A Minireview. Adv. Exp. Med. Biol. 2018, 1074, 185–191. [Google Scholar] [CrossRef]
- Duran, C.L.; Howell, D.W.; Dave, J.M.; Smith, R.L.; Torrie, M.E.; Essner, J.J.; Bayless, K.J. Molecular Regulation of Sprouting Angiogenesis. Compr. Physiol. 2017, 8, 153–235. [Google Scholar] [CrossRef]
- Ploug, M.; Rønne, E.; Behrendt, N.; Jensen, A.L.; Blasi, F.; Danø, K. Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J. Biol. Chem. 1991, 266, 1926–1933. [Google Scholar]
- Jo, M.; Thomas, K.S.; Marozkina, N.; Amin, T.J.; Silva, C.M.; Parsons, S.J.; Gonias, S.L. Dynamic assembly of the urokinase-type plasminogen activator signaling receptor complex determines the mitogenic activity of urokinase-type plasminogen activator. J. Biol. Chem. 2005, 280, 17449–17457. [Google Scholar] [CrossRef]
- Montuori, N.; Cosimato, V.; Rinaldi, L.; Rea, V.E.; Alfano, D.; Ragno, P. uPAR regulates pericellular proteolysis through a mechanism involving integrins and fMLF-receptors. Thromb. Haemost. 2013, 109, 309–318. [Google Scholar] [CrossRef]
- Bifulco, K.; Longanesi-Cattani, I.; Liguori, E.; Arra, C.; Rea, D.; Masucci, M.T.; De Rosa, M.; Pavone, V.; Stoppelli, M.P.; Carriero, M.V. A urokinase receptor-derived peptide inhibiting VEGFdependent directional migration and vascular sprouting. Mol. Cancer Ther. 2013, 12, 1981–1993. [Google Scholar] [CrossRef]
- Carriero, M.V.; Bifulco, K.; Minopoli, M.; Lista, L.; Maglio, O.; Mele, L.; Di Carluccio, G.; De Rosa, M.; Pavone, V. UPARANT: A urokinase receptor-derived peptide inhibitor of VEGF-driven angiogenesis with enhanced stability and in vitro and in vivo potency. Mol. Cancer Ther. 2014, 13, 1092–1104. [Google Scholar] [CrossRef]
- Eden, G.; Archinti, M.; Furlan, F.; Murphy, R.; Degryse, B. The urokinase receptor interactome. Curr. Pharm. Des. 2011, 17, 1874–1889. [Google Scholar] [CrossRef]
- Madunić, J. The Urokinase Plasminogen Activator System in Human Cancers: An Overview of Its Prognostic and Predictive Role. Thromb. Haemost. 2018, 118, 2020–2036. [Google Scholar] [CrossRef] [Green Version]
- Dinesh, P.; Rasool, M. uPA/uPAR signaling in rheumatoid arthritis: Shedding light on its mechanism of action. Pharmacol. Res. 2018, 134, 31–39. [Google Scholar] [CrossRef]
- Napolitano, F.; Rossi, F.W.; Pesapane, A.; Varricchio, S.; Ilardi, G.; Mascolo, M.; Staibano, S.; Lavecchia, A.; Ragno, P.; Selleri, C.; et al. N-Formyl Peptide Receptors Induce Radical Oxygen Production in Fibroblasts Derived From Systemic Sclerosis by Interacting With a Cleaved Form of Urokinase Receptor. Front. Immunol. 2018, 9, 574. [Google Scholar] [CrossRef] [Green Version]
- Rosetti, F.; Mayadas, T.N. The many faces of Mac-1 in autoimmune disease. Immunol. Rev. 2016, 269, 175–193. [Google Scholar] [CrossRef]
- Rubina, K.A.; Sysoeva, V.Y.; Zagorujko, E.I.; Tsokolaeva, Z.I.; Kurdina, M.I.; Parfyonova, Y.V.; Tkachuk, V.A. Increased expression of uPA, uPAR, and PAI-1 in psoriatic skin and in basal cell carcinomas. Arch. Dermatol. Res. 2017, 309, 433–442. [Google Scholar] [CrossRef]
- Davis, J.; Wagner, M.R.; Zhang, W.; Xu, F.; Van Nostrand, W.E. Amyloid beta-protein stimulates the expression of urokinase-type plasminogen activator (uPA) and its receptor (uPAR) in human cerebrovascular smooth muscle cells. J. Biol. Chem. 2003, 278, 19054–19061. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.F.; Beach, T.G. Increased expression of the urokinase plasminogenactivator receptor in amyloid beta peptide-treated human brain microglia and in AD brains. Brain Res. 2002, 926, 69–79. [Google Scholar] [CrossRef]
- Farris, S.D.; Hu, J.H.; Krishnan, R.; Emery, I.; Chu, T.; Du, L.; Kremen, M.; Dichek, H.L.; Gold, E.; Ramsey, S.A.; et al. Mechanisms of urokinase plasminogen activator (uPA)-mediated atherosclerosis: Role of the uPA receptor and S100A8/A9 proteins. J. Biol. Chem. 2011, 286, 22665–22677. [Google Scholar] [CrossRef]
- Schuliga, M.; Grainge, C.; Westall, G.; Knight, D. The fibrogenic actions of the coagulant and plasminogen activation systems in pulmonary fibrosis. Int. J. Biochem. Cell. Biol. 2018, 97, 108–117. [Google Scholar] [CrossRef]
- Dal Monte, M.; Cammalleri, M.; Pecci, V.; Carmosino, M.; Procino, G.; Pini, A.; De Rosa, M.; Pavone, V.; Svelto, M.; Bagnoli, P. Inhibiting the urokinase-type plasminogen activator receptor system recovers STZ-induced diabetic nephropathy. J. Cell. Mol. Med. 2018, 23, 1034–1049. [Google Scholar] [CrossRef]
- Huang, J.M.; Ren, R.Y.; Bao, Y.; Guo, J.C.; Xiang, W.; Jing, X.Z.; Shi, J.; Zhang, G.X.; Li, L.; Tian, Y.; et al. Ulinastatin Inhibits Osteoclastogenesis and Suppresses Ovariectomy Induced Bone Loss by Downregulating uPAR. Front. Pharmacol. 2018, 9, 1016. [Google Scholar] [CrossRef]
- Huang, W.; Jin, A.; Zhang, J.; Wang, C.; Tsang, L.L.; Cai, Z.; Zhou, X.; Chen, H.; Chan, H.C. Upregulation of CFTR in patients with endometriosis and its involvement in NFκB-uPAR dependent cell migration. Oncotarget 2017, 8, 66951–66959. [Google Scholar] [CrossRef]
- Dande, R.R.; Peev, V.; Altintas, M.M.; Reiser, J. Soluble Urokinase Receptor and the Kidney Response in Diabetes Mellitus. J. Diabetes Res. 2017, 2017, 3232848. [Google Scholar] [CrossRef]
- Desmedt, S.; Desmedt, V.; Delanghe, J.R.; Speeckaert, R.; Speeckaert, M.M. The intriguing role of soluble urokinase receptor in inflammatory diseases. Crit. Rev. Clin. Lab. Sci. 2017, 54, 117–133. [Google Scholar] [CrossRef]
- Scotti, F.; Milani, P.; Setaccioli, M.; Maestroni, S.; Sidenius, N.; De Lorenzi, V.; Massacesi, A.; Bergamini, F.; Zerbini, G. Increased soluble urokinase plasminogen activator receptor (suPAR) levels in neovascular age-related macular degeneration: A role for inflammation in the pathogenesis of the disease? Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 899–903. [Google Scholar] [CrossRef]
- Saylam Kurtipek, G.; Kesli, R.; Tuncez Akyurek, F.; Akyurek, F.; Ataseven, A.; Terzi, Y. Plasma-soluble urokinase plasminogen activator receptor (suPAR) levels in Behçet’s disease and correlation with disease activity. Int. J. Rheum. Dis. 2018, 21, 866–870. [Google Scholar] [CrossRef]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Le Gat, L.; Gogat, K.; Bouquet, C.; Saint-Geniez, M.; Darland, D.; Van Den Berghe, L.; Marchant, D.; Provost, A.; Perricaudet, M.; Menasche, M.; et al. In vivo adenovirus-mediated delivery of a uPA/uPAR antagonist reduces retinal neovascularization in a mouse model of retinopathy. Gene Ther. 2003, 10, 2098–2103. [Google Scholar] [CrossRef] [Green Version]
- Magnussen, S.; Hadler-Olsen, E.; Latysheva, N.; Pirila, E.; Steigen, S.E.; Hanes, R.; Salo, T.; Winberg, J.O.; Uhlin-Hansen, L.; Svineng, G. Tumour microenvironments induce expression of urokinase plasminogen activator receptor (uPAR) and concomitant activation of gelatinolytic enzymes. PLoS ONE 2014, 9, e105929. [Google Scholar] [CrossRef]
- Chintala, S.K. Tissue and urokinase plasminogen activators instigate the degeneration of retinal ganglion cells in a mouse model of glaucoma. Exp. Eye Res. 2016, 143, 17–27. [Google Scholar] [CrossRef]
- Razali, N.; Agarwal, R.; Agarwal, P.; Froemming, G.R.A.; Tripathy, M.; Ismail, N.M. IOP lowering effect of topical trans-resveratrol involves adenosine receptors and TGF-β2 signaling pathways. Eur. J. Pharmacol. 2018, 838, 1–10. [Google Scholar] [CrossRef]
- de Paulis, A.; Montuori, N.; Prevete, N.; Fiorentino, I.; Rossi, F.W.; Visconte, V.; Rossi, G.; Marone, G.; Ragno, P. Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J. Immunol. 2004, 173, 5739–5748. [Google Scholar] [CrossRef]
- Resnati, M.; Pallavicini, I.; Wang, J.M.; Oppenheim, J.; Serhan, C.N.; Romano, M.; Blasi, F. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. USA 2002, 99, 1359–1364. [Google Scholar] [CrossRef] [Green Version]
- Prevete, N.; Liotti, F.; Marone, G.; Melillo, R.M.; de Paulis, A. Formyl peptide receptors at the interface of inflammation, angiogenesis and tumor growth. Pharmacol. Res. 2015, 102, 184–191. [Google Scholar] [CrossRef]
- Weiß, E.; Kretschmer, D. Formyl-Peptide Receptors in Infection, Inflammation, and Cancer. Trends Immunol. 2018, 39, 815–829. [Google Scholar] [CrossRef]
- Filep, J.G.; Sekheri, M.; El Kebir, D. Targeting formyl peptide receptors to facilitate the resolution of inflammation. Eur. J. Pharmacol. 2018, 833, 339–348. [Google Scholar] [CrossRef]
- Yu, Y.; Bao, Z.; Wang, X.; Gong, W.; Chen, H.; Guan, H.; Le, Y.; Su, S.; Chen, K.; Wang, J.M. The G-Protein-Coupled Chemoattractant Receptor Fpr2 Exacerbates High Glucose-Mediated Proinflammatory Responses of Müller Glial Cells. Front. Immunol. 2017, 8, 1852. [Google Scholar] [CrossRef]
- Nawaz, I.M.; Rezzola, S.; Cancarini, A.; Russo, A.; Costagliola, C.; Semeraro, F.; Presta, M. Human vitreous in proliferative diabetic retinopathy: Characterization and translational implications. Prog. Retin. Eye Res. 2019. [Google Scholar] [CrossRef]
- Guerrero, P.A.; McCarty, J.H. Integrins in Vascular Development and Pathology. Adv. Pharmacol. 2018, 81, 129–153. [Google Scholar] [CrossRef]
- Longmate, W.; DiPersio, C.M. Beyond adhesion: Emerging roles for integrins in control of the tumor microenvironment. F1000Res 2017, 6, 1612. [Google Scholar] [CrossRef]
- Ferraris, G.M.; Schulte, C.; Buttiglione, V.; De Lorenzi, V.; Piontini, A.; Galluzzi, M.; Podestà, A.; Madsen, C.D.; Sidenius, N. The interaction between uPAR and vitronectin triggers ligand independent adhesion signalling by integrins. EMBO J. 2014, 33, 2458–2472. [Google Scholar] [CrossRef]
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef]
- Chen, J.; Green, J.; Yurdagul, A., Jr.; Albert, P.; McInnis, M.C.; Orr, A.W. αvβ3 Integrins Mediate Flow-Induced NF-κB Activation, Proinflammatory Gene Expression, and Early Atherogenic Inflammation. Am. J. Pathol. 2015, 185, 2575–2589. [Google Scholar] [CrossRef]
- Lagos-Cabré, R.; Alvarez, A.; Kong, M.; Burgos-Bravo, F.; Cárdenas, A.; Rojas-Mancilla, E.; Pérez-Nuñez, R.; Herrera-Molina, R.; Rojas, F.; Schneider, P.; et al. αVβ3 Integrin regulates astrocyte reactivity. J. Neuroinflammation 2017, 14, 194. [Google Scholar] [CrossRef]
- Bi, J.J.; Yi, L. Effects of integrins and integrin αvβ3 inhibitor on angiogenesis in cerebral ischemic stroke. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2014, 34, 299–305. [Google Scholar] [CrossRef]
- Keasey, M.P.; Jia, C.; Pimentel, L.F.; Sante, R.R.; Lovins, C.; Hagg, T. Blood vitronectin is a major activator of LIF and IL-6 in the brain through integrin-FAK and uPAR signaling. J. Cell Sci. 2018, 131, jcs202580. [Google Scholar] [CrossRef]
- Gonzalez-Salinas, R.; Hernández-Zimbrón, L.F.; Gulias-Cañizo, R.; Sánchez-Vela, M.A.; Ochoa-De La Paz, L.; Zamora, R.; Quiroz-Mercado, H. Current Anti-Integrin Therapy for Ocular Disease. Semin. Ophthalmol. 2018, 33, 634–642. [Google Scholar] [CrossRef]
- Hu, T.T.; Vanhove, M.; Porcu, M.; Van Hove, I.; Van Bergen, T.; Jonckx, B.; Barbeaux, P.; Vermassen, E.; Feyen, J.H.M. The potent small molecule integrin antagonist THR-687 is a promising next-generation therapy for retinal vascular disorders. Exp. Eye Res. 2019, 180, 43–52. [Google Scholar] [CrossRef]
- Barlow, H.R.; Cleaver, O. Building Blood Vessels-One Rho GTPase at a Time. Cells 2019, 8, 545. [Google Scholar] [CrossRef]
- Liu, K.; Fan, J.; Wu, J. Sushi repeat-containing protein X-linked 2 promotes angiogenesis through the urokinase-type plasminogen activator receptor dependent integrin αvβ3/focal adhesion kinase pathways. Drug Discov. Ther. 2017, 11, 212–217. [Google Scholar] [CrossRef]
- Larusch, G.A.; Merkulova, A.; Mahdi, F.; Shariat-Madar, Z.; Sitrin, R.G.; Cines, D.B.; Schmaier, A.H. Domain 2 of uPAR regulates single-chain urokinase-mediated angiogenesis through β1 integrin and VEGFR2. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H305–H320. [Google Scholar] [CrossRef]
- Herkenne, S.; Paques, C.; Nivelles, O.; Lion, M.; Bajou, K.; Pollenus, T.; Fontaine, M.; Carmeliet, P.; Martial, J.A.; Nguyen, N.Q.; et al. The interaction of uPAR with VEGFR2 promotes VEGF-induced angiogenesis. Sci. Signal. 2015, 8, ra117. [Google Scholar] [CrossRef]
- Uhrin, P.; Breuss, J.M. uPAR: A modulator of VEGF-induced angiogenesis. Cell Adh. Migr. 2013, 7, 23–26. [Google Scholar] [CrossRef]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Alizadeh, E.; Mammadzada, P.; André, H. The Different Facades of Retinal and Choroidal Endothelial Cells in Response to Hypoxia. Int. J. Mol. Sci. 2018, 19, 3846. [Google Scholar] [CrossRef]
- Huang, H.; Parlier, R.; Shen, J.K.; Lutty, G.A.; Vinores, S.A. VEGF receptor blockade markedly reduces retinal microglia/macrophage infiltration into laser-induced CNV. PLoS ONE 2013, 8, e71808. [Google Scholar] [CrossRef]
- Kuiper, E.J.; Hughes, J.M.; Van Geest, R.J.; Vogels, I.M.; Goldschmeding, R.; Van Noorden, C.J.; Schlingemann, R.O.; Klaassen, I. Effect of VEGF-A on expression of profibrotic growth factor and extracellular matrix genes in the retina. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4267–4276. [Google Scholar] [CrossRef]
- Cai, W.; Cheng, Y.; Ke, L.; Zhang, P.; Deng, G.; Li, G. Effects of avastin on expression of AQP4 in Müller cells under hypoxia. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2012, 32, 607–612. [Google Scholar] [CrossRef]
- Kurihara, T. Roles of Hypoxia Response in Retinal Development and Pathophysiology. Keio J. Med. 2018, 67, 1–9. [Google Scholar] [CrossRef]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.W.; Ye, S.K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef]
- Bartolotti, N.; Lazarov, O. CREB signals as PBMC-based biomarkers of cognitive dysfunction: A novel perspective of the brain-immune axis. Brain Behav. Immun. 2019, 78, 9–20. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-κB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)—Key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Haq, S.; Ansari, W.H.; Han, M.M.; Conti, T.F.; Conti, F.F.; Silva, F.Q.; Singh, R.P. Characterization of the Systemic Findings of Patients Undergoing Initiation of Anti-Vascular Endothelial Growth Factor Therapy for Diabetic Macular Edema in Routine Clinical Practice. Ophthalmic Surg. Lasers Imaging Retina 2019, 50, 16–24. [Google Scholar] [CrossRef]
- Iyer, S.; Radwan, A.E.; Hafezi-Moghadam, A.; Malyala, P.; Amiji, M. Long-acting intraocular delivery strategies for biological therapy of age-related macular degeneration. J. Control Release 2019, 296, 140–149. [Google Scholar] [CrossRef]
- Simó, R.; Carrasco, E.; García-Ramírez, M.; Hernández, C. Angiogenic and antiangiogenic factors in proliferative diabetic retinopathy. Curr. Diabetes Rev. 2006, 2, 71–98. [Google Scholar] [CrossRef]
- Rangasamy, S.; Srinivasan, R.; Maestas, J.; McGuire, P.G.; Das, A. A potential role for angiopoietin 2 in the regulation of the blood-retinal barrier in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3784–3791. [Google Scholar] [CrossRef]
- Jin, J.; Zhao, W.C.; Yuan, F. CXCR7/CXCR4/CXCL12 axis regulates the proliferation, migration, survival and tube formation of choroid-retinal endothelial cells. Ophthalmic Res. 2013, 50, 6–12. [Google Scholar] [CrossRef]
- Noda, K.; Nakao, S.; Ishida, S.; Ishibashi, T. Leukocyte adhesion molecules in diabetic retinopathy. J. Ophthalmol. 2012, 2012, 279037. [Google Scholar] [CrossRef]
- Bolinger, M.T.; Antonetti, D.A. Moving Past Anti-VEGF: Novel Therapies for Treating Diabetic Retinopathy. Int. J. Mol. Sci. 2016, 17, 1498. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, V.M.; Chan, C.-C. The role of anti-inflammatory agents in age-related macular degeneration (AMD) treatment. Eye (Lond.) 2011, 25, 127–139. [Google Scholar] [CrossRef]
- Stefánsson, E.; Olafsdottir, O.B.; Eliasdottir, T.S.; Vehmeijer, W.; Einarsdottir, A.B.; Bek, T.; Torp, T.L.; Grauslund, J.; Eysteinsson, T.; Karlsson, R.A.; et al. Retinal oximetry: Metabolic imaging for diseases of the retina and brain. Prog. Retin. Eye Res. 2019, 70, 1–22. [Google Scholar] [CrossRef]
- Olivares-González, L.; Martínez-Fernández de la Cámara, C.; Hervás, D.; Millán, J.M.; Rodrigo, R. HIF-1α stabilization reduces retinal degeneration in a mouse model of retinitis pigmentosa. FASEB J. 2018, 32, 2438–2451. [Google Scholar] [CrossRef]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Clinical evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology 2013, 120, 100–105. [Google Scholar] [CrossRef]
- Noailles, A.; Maneu, V.; Campello, L.; Lax, P.; Cuenca, N. Systemic inflammation induced by lipopolysaccharide aggravates inherited retinal dystrophy. Cell Death Dis. 2018, 9, 350. [Google Scholar] [CrossRef]
- Xu, X.J.; Wang, S.M.; Jin, Y.; Hu, Y.T.; Feng, K.; Ma, Z.Z. Melatonin delays photoreceptor degeneration in a mouse model of autosomal recessive retinitis pigmentosa. J. Pineal Res. 2017, 63, e12428. [Google Scholar] [CrossRef]
- Okamoto, T.; Ozawa, Y.; Kamoshita, M.; Osada, H.; Toda, E.; Kurihara, T.; Nagai, N.; Umezawa, K.; Tsubota, K. The Neuroprotective Effect of Rapamycin as a Modulator of the mTOR-NF-κB Axis during Retinal Inflammation. PLoS ONE 2016, 11, e0146517. [Google Scholar] [CrossRef]
- Montuori, N.; Ragno, P. Role of uPA/uPAR in the modulation of angiogenesis. Chem. Immunol. Allergy 2014, 99, 105–122. [Google Scholar] [CrossRef]
- D’Alessio, S.; Blasi, F. The urokinase receptor as an entertainer of signal transduction. Front. Biosci. (Landmark Ed.) 2009, 14, 4575–4587. [Google Scholar] [CrossRef]
- Malla, R.R.; Gopinath, S.; Gondi, C.S.; Alapati, K.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Cathepsin B and uPAR knockdown inhibits tumor-induced angiogenesis by modulating VEGF expression in glioma. Cancer Gene Ther. 2011, 18, 419–434. [Google Scholar] [CrossRef]
- Motta, C.; Lupo, G.; Rusciano, D.; Olivieri, M.; Lista, L.; De Rosa, M.; Pavone, V.; Anfuso, C.D. Molecular mechanisms mediating antiangiogenic action of the urokinase receptor-derived peptide UPARANT in human retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5723–5735. [Google Scholar] [CrossRef]
- Gao, P.; Niu, N.; Wei, T.; Tozawa, H.; Chen, X.; Zhang, C.; Zhang, J.; Wada, Y.; Kapron, C.M.; Liu, J. The roles of signal transducer and activator of transcription factor 3 in tumor angiogenesis. Oncotarget 2017, 8, 69139–69161. [Google Scholar] [CrossRef] [Green Version]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012, 4, a006049. [Google Scholar] [CrossRef]
- Szade, A.; Grochot-Przeczek, A.; Florczyk, U.; Jozkowicz, A.; Dulak, J. Cellular and molecular mechanisms of inflammation-induced angiogenesis. IUBMB Life 2015, 67, 145–159. [Google Scholar] [CrossRef]
- Jaiswal, R.K.; Varshney, A.K.; Yadava, P.K. Diversity and functional evolution of the plasminogen activator system. Biomed. Pharmacother. 2018, 98, 886–898. [Google Scholar] [CrossRef]
- Chu, S.-C.; Yu, C.-C.; Hsu, L.-S.; Chen, K.-S.; Su, M.-Y.; Chen, P.-N. Berberine reverses epithelial-to-mesenchymal transition and inhibits metastasis and tumor-induced angiogenesis in human cervical cancer cells. Mol. Pharmacol. 2014, 86, 609–623. [Google Scholar] [CrossRef]
- Liu, H.L.; Liu, D.; Ding, G.R.; Liao, P.F.; Zhang, J.W. Hypoxia-inducible factor-1α and Wnt/β-catenin signaling pathways promote the invasion of hypoxic gastric cancer cells. Mol. Med. Rep. 2015, 12, 3365–3373. [Google Scholar] [CrossRef]
- Nishi, H.; Sasaki, T.; Nagamitsu, Y.; Terauchi, F.; Nagai, T.; Nagao, T.; Isaka, K. Hypoxia inducible factor-1 mediates upregulation of urokinase-type plasminogen activator receptor gene transcription during hypoxia in cervical cancer cells. Oncol. Rep. 2016, 35, 992–998. [Google Scholar] [CrossRef]
- Wang, Y.; Dang, J.; Wang, H.; Allgayer, H.; Murrell, G.A.; Boyd, D. Identification of a novel nuclear factor-kappaB sequence involved in expression of urokinase-type plasminogen activator receptor. Eur. J. Biochem. 2000, 267, 3248–3254. [Google Scholar] [CrossRef]
- Asuthkar, S.; Gondi, C.S.; Nalla, A.K.; Velpula, K.K.; Gorantla, B.; Rao, J.S. Urokinase-type plasminogen activator receptor (uPAR)-mediated regulation of WNT/β-catenin signaling is enhanced in irradiated medulloblastoma cells. J. Biol. Chem. 2012, 287, 20576–22089. [Google Scholar] [CrossRef]
- McGuire, P.G.; Jones, T.R.; Talarico, N.; Warren, E.; Das, A. The urokinase/urokinase receptor system in retinal neovascularization: Inhibition by A6 suggests a new therapeutic target. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2736–2742. [Google Scholar] [CrossRef]
- Cammalleri, M.; Dal Monte, M.; Locri, F.; Lista, L.; Aronsson, M.; Kvanta, A.; Rusciano, D.; De Rosa, M.; Pavone, V.; André, H.; et al. The urokinase receptor-derived peptide UPARANT mitigates angiogenesis in a mouse model of laser-induced choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2600–2611. [Google Scholar] [CrossRef]
- Das, A.; Boyd, N.; Jones, T.R.; Talarico, N.; McGuire, P.G. Inhibition of choroidal neovascularization by a peptide inhibitor of the urokinase plasminogen activator and receptor system in a mouse model. Arch. Ophthalmol. 2004, 122, 1844–1849. [Google Scholar] [CrossRef]
- Dal Monte, M.; Rezzola, S.; Cammalleri, M.; Belleri, M.; Locri, F.; Morbidelli, L.; Corsini, M.; Paganini, G.; Semeraro, F.; Cancarini, A.; et al. Antiangiogenic effectiveness of the urokinase receptor-derived peptide UPARANT in a model of oxygen-induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2392–2407. [Google Scholar] [CrossRef]
- Prager, G.W.; Breuss, J.M.; Steurer, S.; Mihaly, J.; Binder, B.R. Vascular endothelial growth factor (VEGF) induces rapid prourokinase (pro-uPA) activation on the surface of endothelial cells. Blood 2004, 103, 955–962. [Google Scholar] [CrossRef] [Green Version]
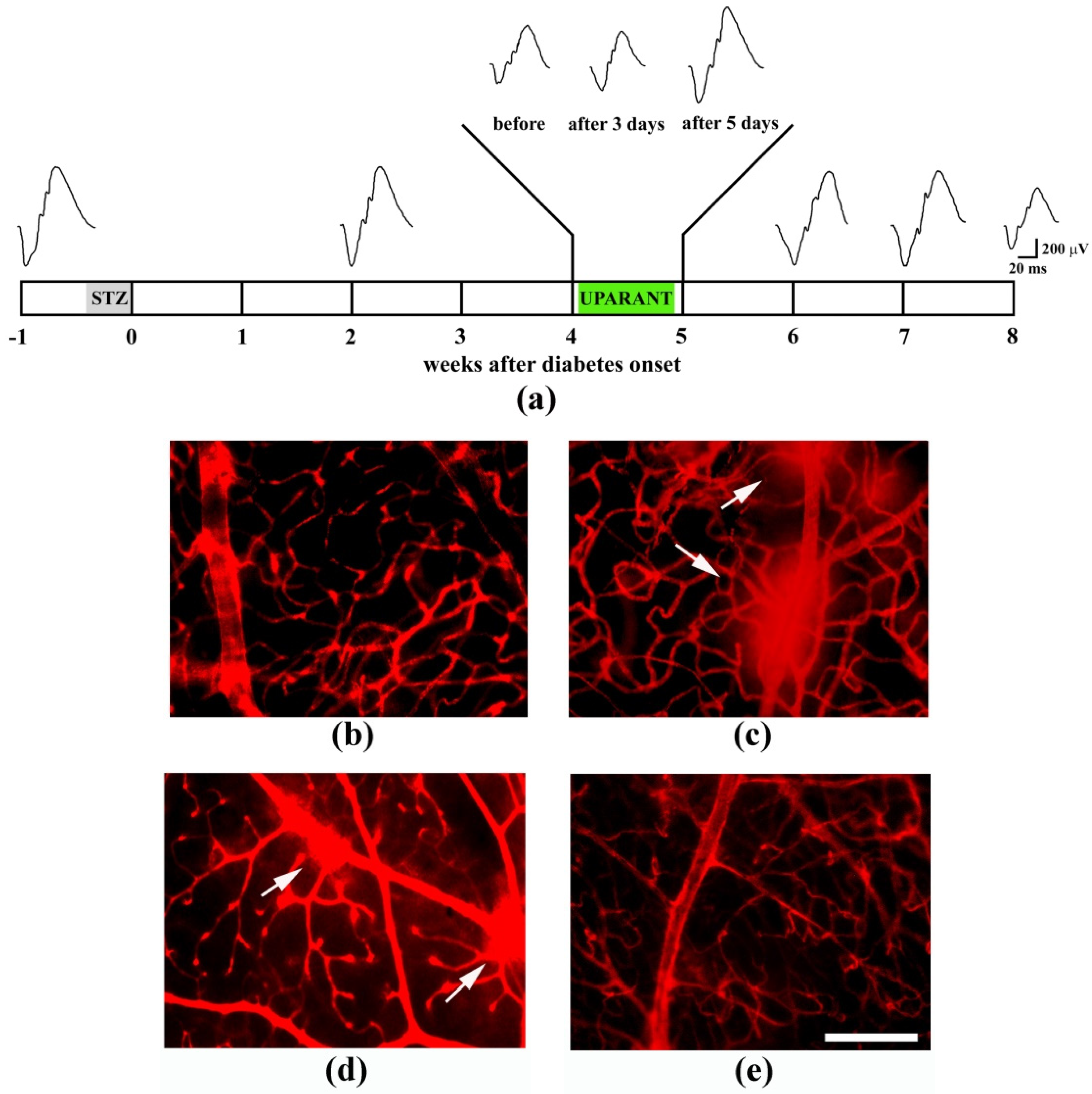
- Cammalleri, M.; Locri, F.; Marsili, S.; Dal Monte, M.; Pisano, C.; Mancinelli, A.; Lista, L.; Rusciano, D.; De Rosa, M.; Pavone, V.; et al. The urokinase receptor-derived peptide UPARANT recovers dysfunctional electroretinogram and blood-retinal barrier leakage in a rat model of diabetes. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3138–3148. [Google Scholar] [CrossRef]
- Cammalleri, M.; Dal Monte, M.; Locri, F.; Marsili, S.; Lista, L.; De Rosa, M.; Pavone, V.; Rusciano, D.; Bagnoli, P. Diabetic retinopathy in the spontaneously diabetic Torii rat: Pathogenetic mechanisms and preventive efficacy of inhibiting the urokinase-type plasminogen activator receptor system. J. Diabetes Res. 2017, 2017, 2904150. [Google Scholar] [CrossRef]
- El-Remessy, A.B.; Franklin, T.; Ghaley, N.; Yang, J.; Brands, M.W.; Caldwell, R.B.; Behzadian, M.A. Diabetes-induced superoxide anion and breakdown of the blood-retinal barrier: Role of the VEGF/uPAR pathway. PLoS ONE 2013, 8, e71868. [Google Scholar] [CrossRef]
- Navaratna, D.; Menicucci, G.; Maestas, J.; Srinivasan, R.; McGuire, P.; Das, A. A peptide inhibitor of the urokinase/urokinase receptor system inhibits alteration of the blood-retinal barrier in diabetes. FASEB J. 2008, 22, 3310–3317. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, J.; Zhu, R.; Zhao, R.; Chen, J.; Jin, J.; Tian, Y.; Su, S.B. The profile of angiogenic factors in vitreous humor of the patients with proliferative diabetic retinopathy. Curr. Mol. Med. 2017, 17, 280–286. [Google Scholar] [CrossRef]
- Locri, F.; Dal Monte, M.; Aronsson, M.; Cammalleri, M.; De Rosa, M.; Pavone, V.; Kvanta, A.; Bagnoli, P.; André, H. UPARANT is an effective antiangiogenic agent in a mouse model of rubeosis iridis. J. Mol. Med. (Berl.) 2019. [Google Scholar] [CrossRef]
- Shi, H.; Carion, T.W.; Jiang, Y.; Steinle, J.J.; Berger, E.A. VIP protects human retinal microvascular endothelial cells against high glucose-induced increases in TNF-α and enhances RvD1. Prostaglandins Other Lipid Mediat. 2016, 123, 28–32. [Google Scholar] [CrossRef]
- Zhang, X.G.; Hui, Y.N.; Huang, X.F.; Du, H.J.; Zhou, J.; Ma, J.X. Activation of formyl peptide receptor-1 enhances restitution of human retinal pigment epithelial cell monolayer under electric fields. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3160–3165. [Google Scholar] [CrossRef]
- Gardner, P.J.; Yazid, S.; Ribeiro, J.; Ali, R.R.; Dick, A.D. Augmenting endogenous levels of retinal annexin A1 suppresses uveitis in mice. Transl. Vis. Sci. Technol. 2017, 6, 10. [Google Scholar] [CrossRef]
- Liang, X.Y.; Chen, L.J.; Ng, T.K.; Tuo, J.; Gao, J.L.; Tam, P.O.; Lai, T.Y.; Chan, C.C.; Pang, C.P. FPR1 interacts with CFH, HTRA1 and smoking in exudative age-related macular degeneration and polypoidal choroidal vasculopathy. Eye (Lond.) 2014, 28, 1502–1510. [Google Scholar] [CrossRef] [Green Version]
- Rezzola, S.; Corsini, M.; Chiodelli, P.; Cancarini, A.; Nawaz, I.M.; Coltrini, D.; Mitola, S.; Ronca, R.; Belleri, M.; Lista, L.; et al. Inflammation and N-formyl peptide receptors mediate the angiogenic activity of human vitreous humour in proliferative diabetic retinopathy. Diabetologia 2017, 60, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Sui, A.; Zhong, Y.; Demetriades, A.M.; Shen, J.; Su, T.; Yao, Y.; Gao, Y.; Zhu, Y.; Shen, X.; Xie, B. ATN-161 as an integrin α5β1 antagonist depresses ocular neovascularization by promoting new vascular endothelial cell apoptosis. Med. Sci. Monit. 2018, 24, 5860–5873. [Google Scholar] [CrossRef]
- Roy, S.; Bae, E.; Amin, S.; Kim, D. Extracellular matrix, gap junctions, and retinal vascular homeostasis in diabetic retinopathy. Exp. Eye Res. 2015, 133, 58–68. [Google Scholar] [CrossRef]
- Friedlander, M.; Brooks, P.C.; Shaffer, R.W.; Kincaid, C.M.; Varner, J.A.; Cheresh, D.A. Definition of two angiogenic pathways by distinct alpha v integrins. Science 1995, 270, 1500–1502. [Google Scholar] [CrossRef]
- Friedlander, M.; Theesfeld, C.L.; Sugita, M.; Fruttiger, M.; Thomas, M.A.; Chang, S.; Cheresh, D.A. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc. Natl. Acad. Sci. USA 1996, 93, 9764–9769. [Google Scholar] [CrossRef]
- Takagi, H.; Suzuma, K.; Otani, A.; Oh, H.; Koyama, S.; Ohashi, H.; Watanabe, D.; Ojima, T.; Suganami, E.; Honda, Y. Role of vitronectin receptor-type integrins and osteopontin in ischemia-induced retinal neovascularization. Jpn. J. Ophthalmol. 2002, 46, 270–278. [Google Scholar] [CrossRef]
- Witmer, A.N.; van Blijswijk, B.C.; van Noorden, C.J.; Vrensen, G.F.; Schlingemann, R.O. In vivo angiogenic phenotype of endothelial cells and pericytes induced by vascular endothelial growth factor-A. J. Histochem. Cytochem. 2004, 52, 39–52. [Google Scholar] [CrossRef]
- Wilkinson-Berka, J.L.; Jones, D.; Taylor, G.; Jaworski, K.; Kelly, D.J.; Ludbrook, S.B.; Willette, R.N.; Kumar, S.; Gilbert, R.E. SB-267268, a nonpeptidic antagonist of alpha(v)beta3 and alpha(v)beta5 integrins, reduces angiogenesis and VEGF expression in a mouse model of retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1600–1605. [Google Scholar] [CrossRef]
- Yoshida, T.; Gong, J.; Xu, Z.; Wei, Y.; Duh, E.J. Inhibition of pathological retinal angiogenesis by the integrin αvβ3 antagonist tetraiodothyroacetic acid (tetrac). Exp. Eye Res. 2012, 94, 41–48. [Google Scholar] [CrossRef]
- Silva, R.L.E.; Kanan, Y.; Mirando, A.C.; Kim, J.; Shmueli, R.B.; Lorenc, V.E.; Fortmann, S.D.; Sciamanna, J.; Pandey, N.B.; Green, J.J.; et al. Tyrosine kinase blocking collagen IV-derived peptide suppresses ocular neovascularization and vascular leakage. Sci. Transl. Med. 2017, 9, eaai8030. [Google Scholar] [CrossRef]
- Ning, A.; Cui, J.; Maberley, D.; Ma, P.; Matsubara, J. Expression of integrins in human proliferative diabetic retinopathy membranes. Can. J. Ophthalmol. 2008, 43, 683–688. [Google Scholar] [CrossRef] [Green Version]
- Stepanova, V.; Jayaraman, P.S.; Zaitsev, S.V.; Lebedeva, T.; Bdeir, K.; Kershaw, R.; Holman, K.R.; Parfyonova, Y.V.; Semina, E.V.; Beloglazova, I.B.; et al. Urokinase-type Plasminogen Activator (uPA) promotes angiogenesis by attenuating proline-rich homeodomain protein (PRH) transcription factor activity and de-repressing vascular endothelial growth factor (VEGF) receptor expression. J. Biol. Chem. 2016, 291, 15029–15045. [Google Scholar] [CrossRef]
- Cammalleri, M.; Dal Monte, M.; Locri, F.; Pecci, V.; De Rosa, M.; Pavone, V.; Bagnoli, P. The urokinase-type plasminogen activator system as drug target in retinitis pigmentosa: New preclinical evidence in the rd10 mouse model. J. Cell. Mol. Med. 2019, 23, 5176–5192. [Google Scholar] [CrossRef]
- Zhao, L.; Zabel, M.K.; Wang, X.; Ma, W.; Shah, P.; Fariss, R.N.; Qian, H.; Parkhurst, C.N.; Gan, W.B.; Wong, W.T. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 2015, 7, 1179–1197. [Google Scholar] [CrossRef]
- Song, H.; Vijayasarathy, C.; Zeng, Y.; Marangoni, D.; Bush, R.A.; Wu, Z.; Sieving, P.A. NADPH Oxidase Contributes to Photoreceptor Degeneration in Constitutively Active RAC1 Mice. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2864–2875. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.J.; Freeman, W.R.; Azen, S.P.; Flaxel, C.J.; Labree, L.D.; Cheng, L.; Wills, M.; Jones, T.R. Effect of a novel octapeptide urokinase fragment, A6, on experimental choroidal neovascularization in the monkey. Retina 2006, 26, 202–209. [Google Scholar] [CrossRef]
- Colombo, E.S.; Menicucci, G.; McGuire, P.G.; Das, A. Hepatocyte growth factor/scatter factor promotes retinal angiogenesis through increased urokinase expression. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1793–1800. [Google Scholar] [CrossRef]
- Bifulco, K.; Longanesi-Cattani, I.; Gargiulo, L.; Maglio, O.; Cataldi, M.; De Rosa, M.; Stoppelli, M.P.; Pavone, V.; Carriero, M.V. An urokinase receptor antagonist that inhibits cell migration by blocking the formyl peptide receptor. FEBS Lett. 2008, 582, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Bifulco, K.; Longanesi-Cattani, I.; Franco, P.; Pavone, V.; Mugione, P.; Di Carluccio, G.; Masucci, M.T.; Arra, C.; Pirozzi, G.; Stoppelli, M.P.; et al. Single amino acid substitutions in the chemotactic sequence of urokinase receptor modulate cell migration and invasion. PLoS ONE 2012, 7, e44806. [Google Scholar] [CrossRef]
- Papadopoulos, Z. Aflibercept: A review of its effect on the treatment of exudative age-related macular degeneration. Eur. J. Ophthalmol. 2019, 29, 368–378. [Google Scholar] [CrossRef]
- Lulli, M.; Cammalleri, M.; Granucci, I.; Witort, E.; Bono, S.; Di Gesualdo, F.; Lupia, A.; Loffredo, R.; Casini, G.; Dal Monte, M.; et al. In vitro and in vivo inhibition of proangiogenic retinal phenotype by an antisense oligonucleotide downregulating uPAR expression. Biochem. Biophys. Res. Commun. 2017, 490, 977–983. [Google Scholar] [CrossRef]
- Rakic, J.M.; Lambert, V.; Munaut, C.; Bajou, K.; Peyrollier, K.; Alvarez-Gonzalez, M.L.; Carmeliet, P.; Foidart, J.M.; Noël, A. Mice without uPA, tPA, or plasminogen genes are resistant to experimental choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1732–1739. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Pathological Signs | References |
|---|---|---|
| Cancer | Angiogenesis, tumor cell proliferation, motility and metastasis | for Ref., see [36] |
| Rheumatoid arthritis | Angiogenesis and inflammation | for Ref., see [37] |
| Systemic sclerosis | Oxidative stress | [38] |
| Lupus erythematosus | Inflammation | for Ref., see [39] |
| Psoriasis | Cell proliferation and invasion | [40] |
| Alzheimer disease | Inflammation, oxidative stress and altered blood brain barrier | [41,42] |
| Coronary artery disease | Inflammation, atherosclerosis and aortic dilation | [43] |
| Pulmonary fibrosis | Inflammation and fibrosis | for Ref., see [44] |
| Kidney disease | Inflammation, altered vascular permeability, impaired glomerular filtration and fibrosis | for Ref., see [45] |
| Bone destructive disease | Inflammation | [46] |
| Endometriosis | Angiogenesis, inflammation and cell proliferation | [47] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cammalleri, M.; Dal Monte, M.; Pavone, V.; De Rosa, M.; Rusciano, D.; Bagnoli, P. The uPAR System as a Potential Therapeutic Target in the Diseased Eye. Cells 2019, 8, 925. https://doi.org/10.3390/cells8080925
Cammalleri M, Dal Monte M, Pavone V, De Rosa M, Rusciano D, Bagnoli P. The uPAR System as a Potential Therapeutic Target in the Diseased Eye. Cells. 2019; 8(8):925. https://doi.org/10.3390/cells8080925
Chicago/Turabian StyleCammalleri, Maurizio, Massimo Dal Monte, Vincenzo Pavone, Mario De Rosa, Dario Rusciano, and Paola Bagnoli. 2019. "The uPAR System as a Potential Therapeutic Target in the Diseased Eye" Cells 8, no. 8: 925. https://doi.org/10.3390/cells8080925
APA StyleCammalleri, M., Dal Monte, M., Pavone, V., De Rosa, M., Rusciano, D., & Bagnoli, P. (2019). The uPAR System as a Potential Therapeutic Target in the Diseased Eye. Cells, 8(8), 925. https://doi.org/10.3390/cells8080925