Interplay between Phosphatases and the Anaphase-Promoting Complex/Cyclosome in Mitosis


UCL Cancer Institute, University College London, London WC1E 6DD, UK
*
Authors to whom correspondence should be addressed.
Cells 2019, 8(8), 814; https://doi.org/10.3390/cells8080814
Submission received: 3 July 2019
/
Revised: 25 July 2019
/
Accepted: 1 August 2019
/
Published: 2 August 2019
(This article belongs to the Special Issue Molecular Regulation of Mitosis and Its Role in Disease)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Accurate division of cells into two daughters is a process that is vital to propagation of life. Protein phosphorylation and selective degradation have emerged as two important mechanisms safeguarding the delicate choreography of mitosis. Protein phosphatases catalyze dephosphorylation of thousands of sites on proteins, steering the cells through establishment of the mitotic phase and exit from it. A large E3 ubiquitin ligase, the anaphase-promoting complex/cyclosome (APC/C) becomes active during latter stages of mitosis through G1 and marks hundreds of proteins for destruction. Recent studies have revealed the complex interregulation between these two classes of enzymes. In this review, we highlight the direct and indirect mechanisms by which phosphatases and the APC/C mutually influence each other to ensure accurate spatiotemporal and orderly progression through mitosis, with a particular focus on recent insights and conceptual advances.
1. Introduction
From the outset, the process of cell division might seem mired in extraordinary complexity. Following a series of distinct steps, a eukaryotic cell is able to duplicate its genetic content and partition it among its two daughters along with other organelles with remarkable precision. Decades of research has sought to understand the molecular and physico-chemical underpinnings of the ‘cell division cycle’: from DNA replication, transitory structures such as the mitotic spindle devoted to its distribution, to control systems that monitor the fidelity of these processes. The realization that reversible protein modifications such as phosphorylation and ubiquitylation are at the heart of the cell cycle engine has demystified many of its aspects.
The primary focus of this review is to highlight the roles of two classes of protein modifying enzymes that are central to accurate mitosis and the complex interplay between them: protein phosphatases and the E3 ubiquitin ligase, anaphase-promoting complex/cyclosome (APC/C), with a focus on recent advances. These two modifiers act in concert with numerous mitotic kinases to orchestrate various events of mitosis.
1.1. Protein Phosphatases—Ubiquitous Enzymes that Regulate Cell Physiology and Division
The discovery of phosphatases, then called ‘prosthetic group’-removing or PR enzymes, predated that of kinases [1]. Abundant in rabbit muscle and spleen, the phosphatase contemporaneously referred to as PP1 was shown to remove an inorganic ‘prosthetic group’ from muscle glycogen phosphorylase, converting it to a less active form. A decade later, inorganic phosphate was hinted at [2] and then proven [3] as the identity of the removable prosthetic group. It is now clear that hydroxyl-containing amino acids, i.e., Ser, Thr and Tyr, undergo phosphorylation by protein kinases, while phosphatases catalyze phosphate removal, thereby controlling steady-state levels of this modification.
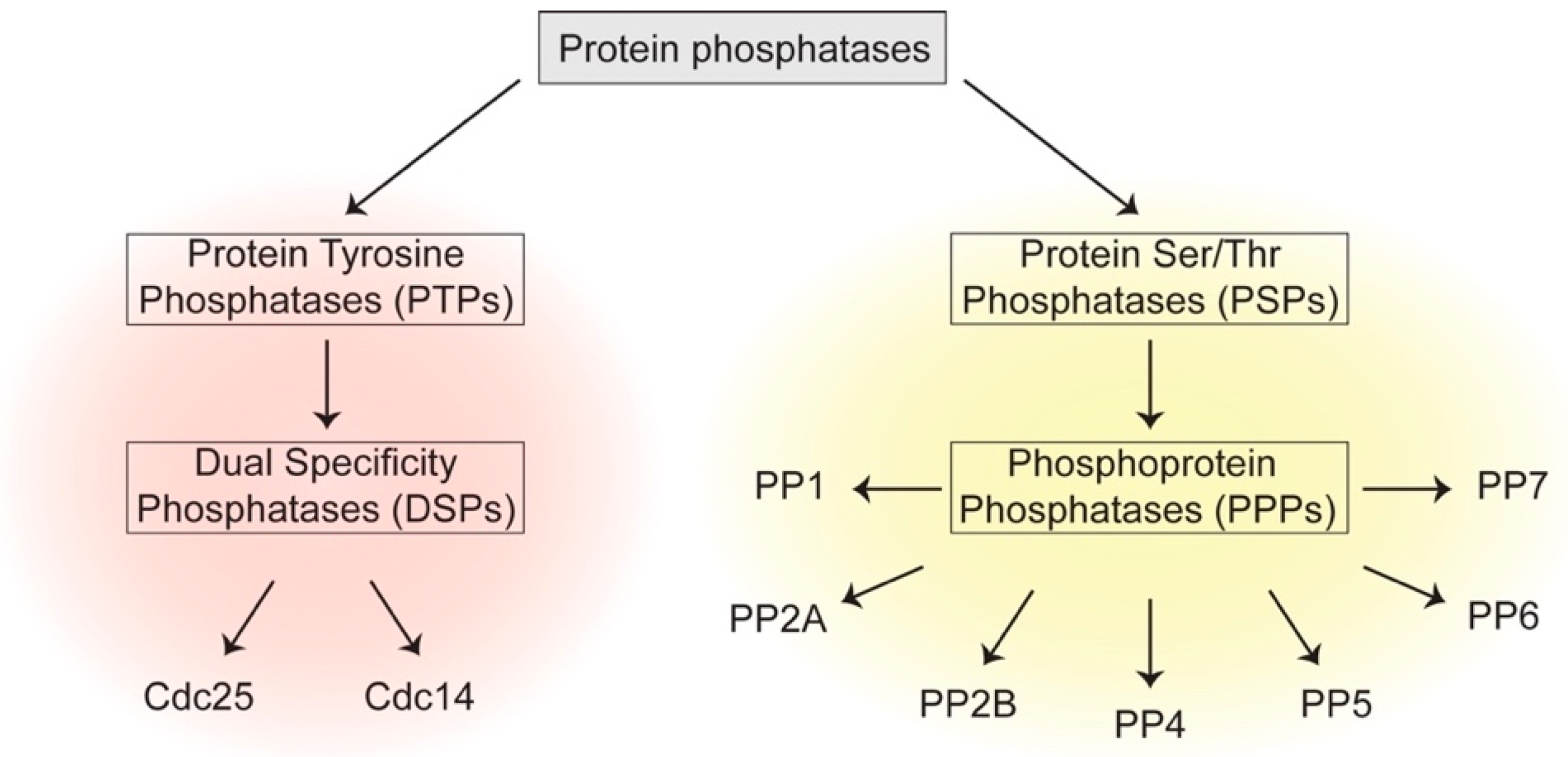
The years following the discovery of PP1 witnessed the identification of many different classes of phosphatases which can be broadly divided into two groups: one is Protein Tyrosine Phosphatases (PTPs), which comprises classical PTPs and Dual Specificity Phosphatases (DSP) that catalyze dephosphorylation of p-Tyr, or p-Tyr, p-Ser and p-Thr residues, respectively [4]. The other family is Protein Serine/Threonine Phosphatases (PSPs) that dephosphorylate p-Ser and p-Thr residues [5] (Figure 1). The human genome contains 189 protein phosphatases, encompassing 10 different protein structural folds and a variety of catalytic mechanisms [6]. This is in contrast to 518 identified human kinases, whose catalytic domains trace back to a single structural fold [7].
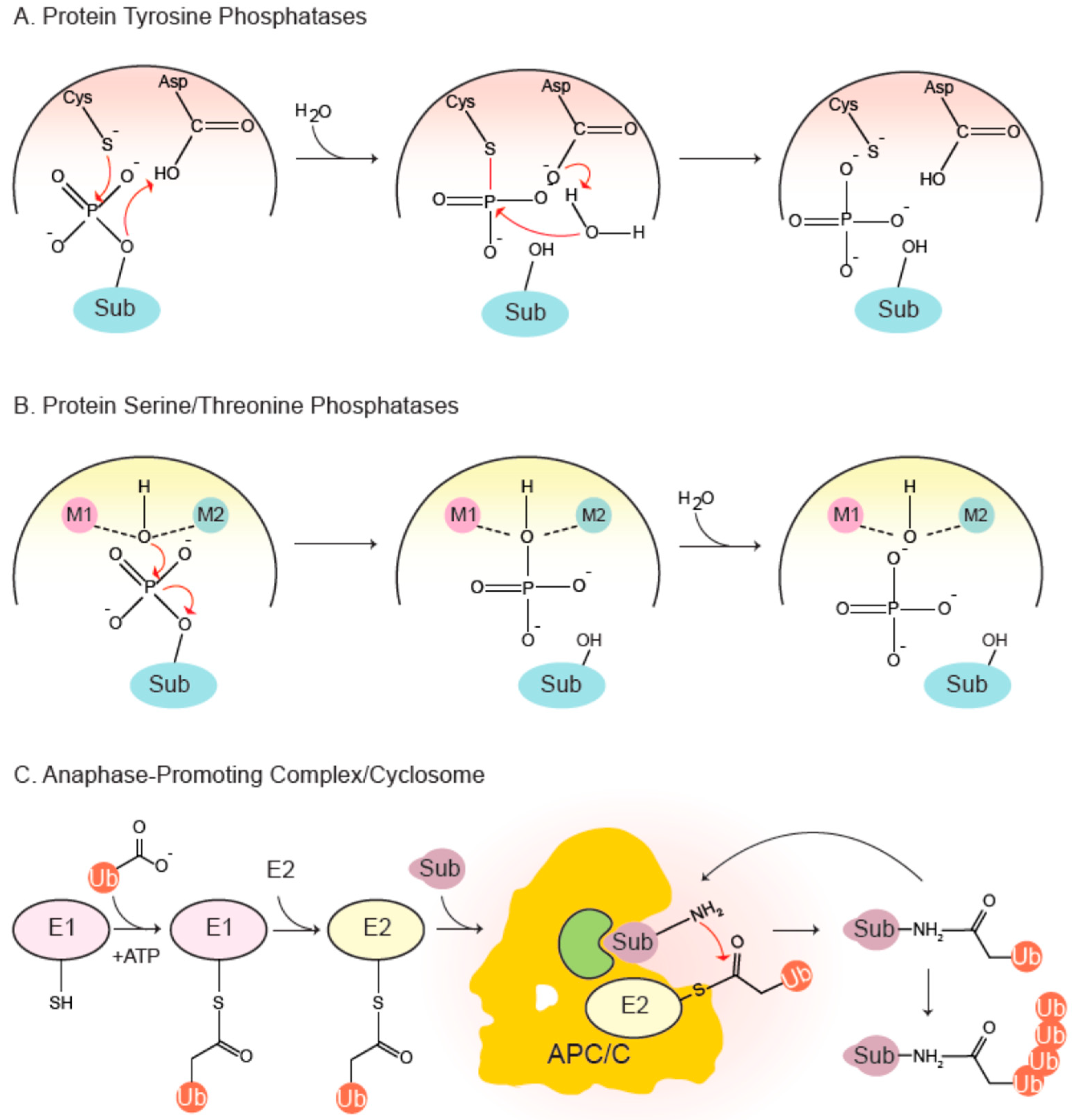
The PTP family is a canopy encompassing diverse phosphatases all of which share the signature motif HCx5R (PTP loop). The sulfur atom of the catalytic cysteine initiates a nucleophilic attack on the phosphorylated substrate (Figure 2), forming a thiophosphate intermediate that is experimentally observable. A conserved aspartate, part of the WPD loop, donates a proton to the dephosphorylated protein, leading to its stabilization and eventual dissociation. Next, a water molecule hydrolyzes the thiophosphate-enzyme intermediate, again mediated by the aspartate, that accepts a proton from the water molecule [4].
The PSP family includes a phosphoprotein phosphatase (PPP) subfamily which comprises of PP1, PP2A, PP2B/calcineurin, PP4, PP5, PP6 and PP7. These phosphatases have been implicated in diverse functions in the cell cycle and beyond. Their catalytic sites adopt similar folds and essentially follow the same mechanism of catalysis (Figure 2). Two divalent metal ions—Fe2+ and Zn2+ in native enzymes, and two Mn2+ ions in recombinant ones—are coordinated by a number of conserved residues involved in metal binding [8,9]. The metal-bound catalytic center holds two water molecules, one of which is activated to mount a nucleophilic attack on the phosphate group of the substrate, thereby leading to its dephosphorylation in a single step [10]. PP1 and PP2A are the most-studied PPPs controlling the cell cycle. PP1 contains surface grooves radiating out from the metal-coordinating center that bind a plethora of degenerate short docking motifs such as RVxF and SILK, to name a few [11]. They mediate interaction of PP1 with hundreds of partner proteins, many of which typically employ multiple docking sites to associate with the phosphatase and conscript it for various functions. Indeed, it has recently been reported that PP1 starts life as a trimer with inhibitor-3 and Sds22 acting as inhibitory chaperones, which are then exchanged for other regulatory proteins [12,13,14]. A restricted set of components constitute the trimeric PP2A holoenzyme: a scaffold A subunit, a catalytic C subunit and a choice of four families of regulatory B subunits, each with multiple isoforms. B55 and B56 families are important in cell division.
The examples of PP1 and PP2A highlight a leitmotif in phosphatase function, including ones controlling cell division: in vivo they tend to function as obligate multimeric enzymes. The catalytic subunit typically associates with one or more additional subunit. These additional subunits can act as scaffolds, regulators or inhibitors, thereby fundamentally altering the properties of the phosphatase. Since early experiments were often performed with the catalytic subunit, phosphatases appeared to show little discrimination towards substrates. This observation, coupled with the relatively few numbers of known phosphatases in comparison to kinases, led to the erroneous yet resilient view that phosphatases are promiscuous housekeeping enzymes working in the background as their kinase counterparts orchestrate major cellular events. In theory, this could indeed serve as an organizational principle in cellular control. For instance, it is well established that oscillating activity of cyclins in complex with cyclin-dependent kinases (Cdks) propels the cell through various transitions by orderly phosphorylation of substrate proteins [15,16], with the tacit assumption that many of these modifications are reversed by background phosphatase activity. Recent research, however, has challenged this view, and now places these enzymes as sophisticated molecular participants. Substrate specificities of phosphatases, imbued by both the active site and docking motif receptors within associated regulatory proteins, are intimately linked to their function [17,18,19,20,21,22,23,24,25,26,27].
1.2. The Multiprotein Complex APC/C is a Master Regulator of the Cell Cycle
In addition to selective phosphorylation, selective degradation of proteins provides a major avenue for control of the cell cycle. A few decades following the discovery of phosphatases, it was found that cells selectively target proteins for degradation by marking them with covalently-attached chains of ubiquitin, a small, heat-stable protein [30,31]. These chains are recognized and irreversibly destroyed by the 26S proteasome. Degradation of mitotic cyclins and Cdk1 inactivation during mitotic exit was then attributed to the ubiquitin-protease system [32], thus marrying together the fields of protein phosphorylation and ubiquitylation in cell cycle control. In fact, ubiquitin-mediated degradation was found to be the impetus for cytological anaphase [33], triggered by destruction of securin [34,35], an inhibitory chaperone for the protease separase. Once released, separase cleaves the kleisin subunit of cohesin rings that hold sister chromatids together [36].
The attachment of ubiquitin, ‘ubiquitylation’, is a multi-step process with sequential actions of three enzymes (E1-E2-E3) [37,38,39]: The C-terminal glycine residue of ubiquitin is first activated by an E1 ubiquitin-activating enzyme in an ATP-dependent process and then covalently attached to the active cysteine residue of the E1 by formation of a thioester-ubiquitin conjugate. The activated ubiquitin is then transferred to the active site cysteine of an E2 ubiquitin-conjugating enzyme. Finally, an E3 ubiquitin ligase coordinates the transfer of ubiquitin to selected substrates by simultaneously binding a ubiquitin-charged E2 and a substrate in a productive manner (Figure 2). Attachment of ubiquitin requires a primary amine within the substrate and typically occurs on lysine residues or the N-terminal initiator methionine. The C-terminal glycine of ubiquitin forms an isopeptide bond with an ε-amine of the substrate. Further ubiquitylation can occur on either the N-terminus or any of the seven lysine residues present within the attached ubiquitin molecule. Polyubiquitylation leads to formation of different types of ubiquitin chains, leading the protein down different trajectories, one of which is irreversible degradation [39].
Deubiquitylating enzymes (DUBs) disassemble ubiquitin chains on substrates, providing another avenue for cellular control and homeostasis. There are about 93 different DUBs in the human genome, compared to about 600 E3 ubiquitin ligases [40,41], drawing parallels with reversible phosphorylation in that there is a discrepancy in numbers of enzymes that add a modification compared to ones that remove it.
The anaphase-promoting complex/cyclosome (APC/C) was found to be the E3 ubiquitin ligase responsible for degradation of Cyclin B during mitosis [42,43,44] by which the master regulatory kinase of mitosis, Cdk1-Cyclin B is switched off, driving cells out of mitosis. The APC/C is a gigantic 1.22 MDa multi-subunit enzyme belonging to the Cullin-RING family of E3 ligases. In vertebrates, it contains 14 highly conserved subunits—Apc1-8, Apc10-13, Apc15 and Apc16—five of which are present as dimers. Apc7 is absent from fission and budding yeast complexes. The APC/C itself is not catalytically active as a ubiquitin ligase. It becomes active only when it associates with a ‘co-activator’ protein belonging to the Cdc20/Fizzy family, such as Cdc20 or Cdh1. It should be noted that co-activators are not stoichiometric components of the APC/C, but their association with the APC/C is essential for its E3 ligase function [45,46,47]. Several organisms also contain meiosis-specific co-activators, such as Ama1 in budding yeast [48], Mfr1/Fzr1, Fzr2 and Fzr3 in fission yeast [49,50], Cort in Drosophila [51], etc. Co-activators not only act as receptors for the substrate protein to be modified, but also act as catalytic activators and induce structural rearrangements essential for E2 binding and ubiquitin transfer [52,53]. Some of the APC/C subunits had already been identified budding yeast genetic screens conducted by Hartwell et al. for temperature sensitive mutations that cause cell cycle arrest at elevated temperatures [54]. Perhaps unsurprisingly, this approach also identified the essential budding yeast cell cycle phosphatase Cdc14.
Recent structural studies have illuminated the overall architecture and subunit arrangement of the APC/C holocomplex. It adopts a hollow triangular shape, with the co-activator binding into this nook. Together with the core subunit Apc10, a co-activator forms the substrate recognition module of the enzyme and binds short linear degradation signals, or ‘degrons’ such as the destruction box (D-box) or the KEN-box present within intrinsically unstructured regions of target proteins [32,55,56,57,58]. The catalytic core module is composed of the Cullin-RING subunits Apc2-Apc11, respectively. The rest of the complex is devoted to co-activator recruitment and scaffolding functions, which are thought to coordinate optimal placement of the co-activator, the catalytic module and E2 enzymes for productive catalysis. The tetratricopeptide (TPR) lobe (the substrate recognition sub-complex) consists of the TPR repeat-containing proteins Apc3 (Cdc27), Apc6 (Cdc16), Apc7 (situated at the apex of the APC/C triangle; absent in yeast) and Apc8 (Cdc23) proteins that form homodimers, acting as a scaffold for co-activator binding. On the other hand, the multi-domain subunit Apc1, in conjunction with Apc4 and Apc5, forms a platform that bridges the catalytic module Apc2-Apc11 and the TPR lobe in a catalytically favorable conformation. This geometry also enables cell cycle-controlled binding of the APC/C inhibitors: the Mitotic Checkpoint Complex (MCC), the main effector of a surveillance mechanism called the Spindle Assembly Checkpoint (SAC) and Emi1 (early mitotic inhibitor), a metazoan APC/C inhibitor which is a vertebrate homologue of Rca1 (regulator of cyclin A) [59,60,61,62,63,64].
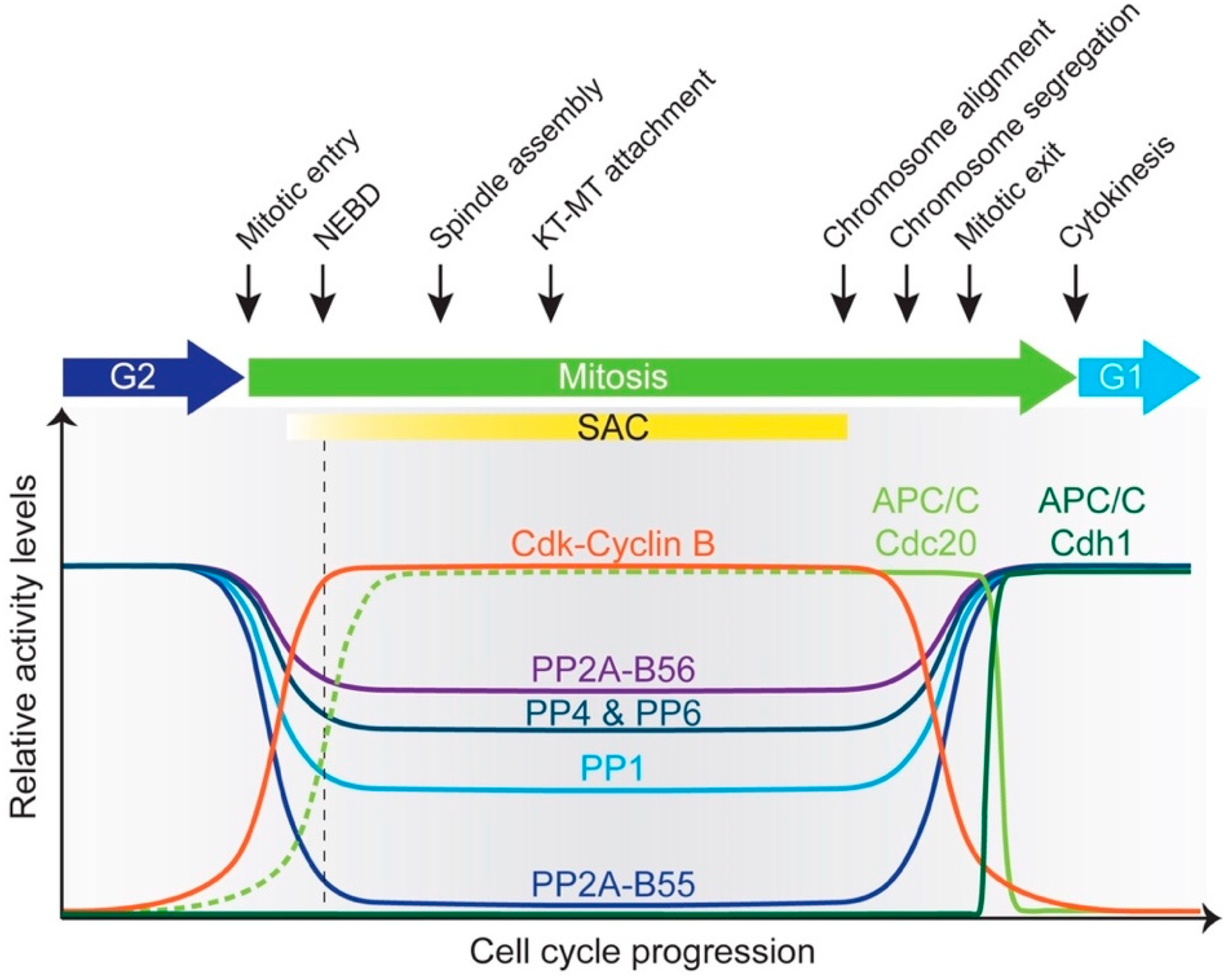
In humans, it has been reported that 170 proteins are ubiquitylated by the APC/C, likely to be an underestimate [65]. Due to its importance in regulating entry into and exit from mitosis, cytokinesis and G1/S transition, APC/C activity is strictly controlled during the cell cycle. During G1, APC/C-Cdh1 activity not only prevents premature entry into S-phase by degrading Cyclins and Skp2, but also regulates replication origin assembly by degrading regulators of DNA replication (e.g., geminin). At G1/S, multiple mechanisms, such as direct Cdh1 phosphorylation, Emi1 inhibitor binding and Cdh1 degradation, collaborate to shut off APC/C-Cdh1 activity and trigger S-phase. Emi1 continues to inhibit APC/C-Cdh1 throughout G2. In mitosis, prior to metaphase, APC/C-Cdc20 activity is restrained by MCC binding until all of the chromosomes are bi-oriented at the metaphase plate. While SAC prevents degradation of most substrates, Cyclin A and Nek2A bypass it by directly binding the APC/C [66,67]. Once the SAC is satisfied, APC/C-Cdc20 is unleashed from inhibition to degrade critical substrates such as Cyclin B and securin, heralding the onset of anaphase [68,69,70,71]. The latter stages of mitotic exit witness Cdh1 activation due to its dephosphorylation by reactivated phosphatases. Cdh1 replaces Cdc20 as the co-activator for the remainder of mitosis and targets the latter for destruction (Figure 3).
Unidirectional and orderly transitions through cell cycle phases are critical to our initial question of how cells divide in an error-free manner. Concerning mitotic exit, studies in both yeast and vertebrates show that APC/C-mediated proteolysis and dephosphorylation work in concert to ensure irreversible transition through this phase and establish a biochemically G1 state [72,73].
The APC/C and phosphatases exhibit considerable crosstalk during mitosis; they influence each other directly and indirectly. Broadly, APC/C activation during mitosis requires phosphorylation and is opposed by phosphatase action. Association of co-activators with the APC/C, however, requires them to be dephosphorylated by phosphatases. Phosphatases are also essential to silence the SAC and further promote APC/C activation. Active APC/C catalyzes mitotic cyclin degradation (inactivation of Cdk1) which then leads to phosphatase reactivation. These observations outline the interlinked nature of an extensive network of phosphorylation and ubiquitylation throughout M-phase and will be explored in greater detail in the following sections. Readers interested in detailed kinase and phosphatase crosstalk governing kinetochore-microtubule attachments and the SAC that are beyond the scope of this review are pointed to recent excellent comprehensive reviews [74,75,76,77,78].
2. G2/M Transition
2.1. Cdc25 Promotes Cdk1-Cyclin B Activation and Mitotic Entry
As mentioned previously, a quantitative increase in Cdk activity from G1 to M-phase triggers sequential passage through the cell cycle. Transition from G2 to M-phase requires an explosive, switch-like activation of Cdk1-Cyclin B. During G2, although the synthesis of Cyclin B increases due to transcriptional activation, concomitant increase in associated kinase activity requires removal of two inhibitory phosphates on the Cdk1 subunit: p-Thr14 and p-Tyr15. Phosphorylation of these residues prevents catalysis by causing ATP misalignment [79], and is performed by the inhibitory kinases Wee1 and Myt1. The dual-specificity PTP Cdc25 dephosphorylates these residues. Cdk1-Cyclin B catalyzes its own activation by phosphorylating both Wee1 and Cdc25 to opposite effects: inactivating the former but activating the latter, thus generating double negative and positive feedback loops. In addition, PP2A has long been known as a negative regulator of Cdk1 activation [80,81,82,83]. These activities underlie a bistable switch leading to swift Cdk1 activation and irreversible mitotic commitment [84,85], although studies in Xenopus indicate that preceding Cdk1-Cyclin A-mediated phosphorylation sets in motion mitotic commitment [86].
In humans, three Cdc25 paralogues exist; Cdc25A, Cdc25B and Cdc25C and perform distinct but overlapping functions in Cdk activation. Active Cdk1-Cyclin B first emerges at centrosomes, catalyzed by Cdc25B. In fact, centrosome splitting proceeds a few minutes after emergence of active kinase in unperturbed cells [87]. In a spatial positive feedback loop, partially activated cytoplasmic pool of Cdk1-Cyclin B then translocates into the nucleus driven by phosphorylation of the Cyclin N-terminus, where it eventually promotes nuclear envelope breakdown [88,89,90]. Nuclear pools of Cdc25A and Cdc25C promote further activation of the kinase.
The activity and subcellular localization of these phosphatases are themselves modified through phosphoregulation and proteolysis. For instance, phosphorylation of Cdc25C by Cdk1 and Polo-like kinase (Plk) increases its activity. Additionally, Cdc25C phosphorylation by a number of kinases at a 14-3-3 consensus sequence leads to its cytoplasmic retention and catalytic inactivation by these adaptor proteins [91,92]. An initial Cdk1-mediated phosphorylation allows PP1 binding to Cdc25C via its N-terminal RVxF motif. In a phosphatase relay, PP1 dephosphorylates the 14-3-3 docking site within the protein, thereby relieving Cdc25C from inhibition to dephosphorylate Cdk1 [93,94]. At the end of mitosis, Cdc25A is degraded by APC/C-Cdh1, thus further promoting Cdk1 inactivation [95].
2.2. Phosphatases are Downregulated at Mitotic Entry
Thousands of proteins are phosphorylated during mitosis by Cdk1, Plk, Aurora A and Aurora B, to name a few. They drive the dramatic cytological changes one associates with this phase: reorganization of cortical cytoskeleton to induce cell rounding and dissolution of the nuclear envelope in vertebrates, compaction of chromosomes to aid segregation and formation of a spindle structure to capture and biorient chromosomes at the metaphase plate. A coherent forward step towards this phase would dictate that opposing phosphatase activity be dampened, swinging the balance towards phosphorylation. Downregulation of many phosphatases occurs at this juncture through direct Cdk1-dependent phosphorylation or binding of various small, heat-stable inhibitors (see below).
Increasing Cdk1 kinase levels phosphorylate the highly conserved AGC kinase Greatwall. The two phosphorylated residues lie within the activation loop of the kinase domain and produce a partially activated enzyme. Intramolecular autophosphorylation of its C-terminal tail leads fully active Greatwall by stabilization of its active conformation [96,97].
Active Greatwall phosphorylates two small, heat-stable, intrinsically unstructured proteins: ENSA (α-endosulfine) and Arpp19 (cAMP-regulated phosphoprotein 19) [98,99]. Phosphorylation occurs at a Ser residue within a highly conserved motif present in both proteins and transforms them into potent inhibitors of PP2A-B55 holocomplexes. Specificity for the B55-bound form of PP2A arises as the proteins bind at the interface of B55 and the catalytic subunit, occluding the active site [100].
In keeping with a paradigm often governing enzyme-inhibitor interactions, PP2A-B55 itself is able to dephosphorylate p-ENSA/p-Arpp19, albeit very slowly, with a kcat two-three orders of magnitude lower than that of a generic PP2A-B55 substrate. PP2A-B55—p-ENSA Km is about four orders of magnitude lower than that for a phosphosubstrate, indicating very tight binding of the inhibitor. Strong inhibitor binding coupled with low rate of catalysis has been termed ‘inhibition by unfair competition’ [101]. This implies that as long as p-ENSA is replenished by active Greatwall, the mitotic state will be preserved through phosphatase inhibition. In summary, Cdk1 phosphorylation of Greatwall leads to its activation and phosphorylation of ENSA/Arpp19—inhibitors of PP2A-B55.
PP2A-B56 is present at kinetochores, where it phosphoregulates kinetochore-microtubule interactions, and is opposed by Aurora B. Phosphorylation of another small, heat-stable protein Bod1 recruited by Ndc80 causes it to inhibit PP2A-B56 complexes. This inhibition is proposed to maintain the correct balance of PP2A-B56 and Aurora B activities [102,103], although detailed mechanisms remain elusive.
Direct phosphorylation of PP2A subunits has also been reported to regulate enzyme activity by affecting assembly. Human PP2A catalytic subunit phosphorylation at its C-terminal tail at Thr307 prevented binding of B55 [104]. A recent study reported that phosphorylation of Thr174 of the budding yeast B55 by mitotic Cdk led to inhibition of its phosphatase activity [105]. Phosphorylation of the equivalent site Ser167 on human B55α, possibly by Cdk1-Cyclin B, inhibits holoenzyme assembly [106].
Cdk1 also directly phosphorylates PP1 near its C-terminus at Thr320 to inhibit its activity down to 30–40% of its peak levels [107,108,109]. Protein Kinase A phosphorylates Inhibitor-1 (l-1), a small heat-stable protein inhibitor, which then specifically inhibits PP1 [110]. In fact, inhibition by I-1 was often used to discriminate between PP1 and PP2A enzymes isolated from tissues in early studies [111]. Importantly, PP1 phosphorylation and I-1 inhibition are thought to act in concert to suppress its phosphatase activity. In an analogous paradigm to PP2A-B55 inhibition, PP1 itself removes these inhibitory phosphates, albeit slowly [110].
As previously mentioned, PP1 binding proteins often employ one or more short docking motifs to interact with the enzyme. Of its 200 known interactors, at least 90% employ the RVxF motif [11]. When the ‘x’ within the motif is a Ser or a Thr, it conforms to a basophilic motif preferred by the mitotic kinase Aurora B [112]. Indeed, Aurora B-mediated phosphorylation of these motifs prevents PP1 holoenzyme assembly during mitosis [113], representing another mode of PP1 inhibition.
PP6 is a phosphatase belonging to the PPP superfamily that also forms a heterotrimeric enzyme. It dephosphorylates the regulatory T-loop of Aurora A thus inhibiting its activity at centrosomes [114]. During mitosis, Cdk1 phosphorylation of the PP6 regulatory subunit creates a docking site for Plk1, which then phosphorylates the phosphatase complex at multiple sites and inhibits its activity through unknown mechanisms. Thus, PP6-mediated signaling is temporally inhibited to ensure a preponderance of Aurora A activity [115].
Another ubiquitous phosphatase of the PPP family, PP4, forms heterodimeric or heterotrimeric complexes in vivo [116,117], presumably leading to different substrate specificities or subcellular localization. For instance, Drosophila PP4 is targeted to centromeres via interaction of its regulatory subunit R3 with the centromere protein CENP-C, where it regulates centromeric integrity during mitosis [118]. It has also been implicated in mitotic centrosome function in Drosophila [119], C. elegans [120] and humans [121]. At the G2/M transition, rising Cdk1-Cyclin B activity leads to phosphorylation of a PP4 complex at centrosomes to inhibit its activity. As PP4 opposes γ-tubulin phosphorylation, its inhibition enables formation of the mitotic spindle [121].
During metaphase/anaphase, APC/C-mediated degradation of Cyclin B and various mitotic kinases sets in motion cascade of events that lead to reactivation of phosphatases (Figure 3). Thus, the APC/C is essential for phosphatase function during mitotic exit.
It is important to note that while there is a general swing towards kinase activation, some phosphatase activity is preserved throughout mitosis. Indeed, it is essential for mitotic progression. As is discussed in the next few sections, PP1 and PP2A-B56 activity at kinetochores plays an important role in stabilizing amphitelic kinetochore-microtubule linkages and silencing the SAC. These phosphatases have also been implicated in dephosphorylating the APC/C co-activator Cdc20 and initiating anaphase [122,123,124]. It is possible that association with certain structures within the cells precludes inhibitor binding, just as microtubule binding protects spindle assembly factors from APC/C-mediated turnover [125]. Another possibility is that these phosphatases are locally reactivated. How these complexes evade inhibition or achieve local reactivation is an important question for the future.
3. Building a Metaphase Cell
3.1. PP1 and PP2A Phosphatases Regulate Kinetochore Capture by Microtubules
Successful execution of chromosome segregation requires capture of kinetochore by microtubules emanating from spindle poles or centrosomes at the opposite ends of the cells. This ensures that each daughter cell gets one copy of the genome. Until all sister chromatids have correctly attached, kinetochores activate the SAC and hold cells in metaphase by inhibiting the APC/C. Thus, phosphatase mediated error correction and SAC inactivation are intimately linked to APC/C activation. In turn, APC/C degrades many proteins involved in these processes. Kinetochore capture is a stochastic process; many rounds of incorrect attachments precede mature, end-on attachments [126,127,128]. It is imperative that only bioriented chromosomes attached to the spindle be selectively stabilized.
How does the cell recognize correct attachments? On a molecular level, this occurs through warring activities of kinases and phosphatases at the centromere and kinetochore. Central to error correction is the kinase Aurora B, which is recruited to inner centromeres in two different ways. First, the mitotic Haspin kinase phosphorylates histone H3 at Thr3, which directly binds the Aurora B-containing Chromosome Passenger Complex (CPC) subunit Survivin [129]. Second, Bub1, a mitotic kinase, phosphorylates H2A at Thr120 at centromeres, which then leads to the recruitment of shugoshin-CPC [130]. Centromeric shugoshin contributes to another pool of Aurora B [131]. Upon autophosphorylation of its T-loop, possibly in trans, active Aurora B phosphorylates several kinetochore proteins and severs incorrect interactions with microtubules, thus giving them a chance to pursue correct ones. Among other proteins, the outer kinetochore subunit Ndc80 is the focal point of Aurora B kinase activity. Phosphorylation of its N-terminus reduces its affinity for microtubules and disfavors the connection [132]. Aurora B also phosphorylates the kinetochore localized Ska complex in metazoans, which is capable of efficiently binding curved ends of depolymerizing microtubule protofilaments. Thus, by helping kinetochores hold on to different microtubule architectures and states, the Ska complex enables ‘end tracking’. Phosphorylation reduces Ska-Ndc80 binding, dampening its affinity for the spindle microtubules [133,134]. Targets of Aurora B are also modulated by other kinases such as Cdk1, Plk1 and Mps1 in this endeavor [74].
A collaboration between PP1 and PP2A-B56 phosphatases, recruited through many mechanisms, leads to direct dephosphorylation of Aurora B substrates to strengthen kinetochore-microtubule interactions. PP2A-B56 directly binds the outer kinetochore, and serves to stabilize initial microtubule-kinetochore contacts, enabling them to form mature, end-on attachments [135]. B56-bound complex is drafted to an unattached outer kinetochore by directly binding the spindle checkpoint protein BubR1 via a LxxIxE motif. To give an example of the complex cross-talk at these regulatory foci, Cdk1 and Plk1 positively regulate PP2A-B56 binding in this context by phosphorylating residues within and downstream of the motif and enhancing B56 binding [24,25,26,27]. This population of the phosphatase opposes Aurora B phosphorylation of the PP1-binding RVxF motif in the outer kinetochore protein Knl1 and allows its recruitment [136]. PP1 can also be delivered to the site of action by binding to plus end-directed motors such as CENP-E. Analogous to its Knl1 binding, PP1 interaction with CENP-E is blocked by Aurora A- (at spindle poles) and B-mediated phosphorylation of a threonine within its RVxF motif. Once dephosphorylated, PP1-CENP-E stabilizes mature attachments [137]. Finally, the Ska complex can also directly engage PP1 [138].
As previously mentioned, Cyclin A is degraded during prometaphase by APC/C action, even though the spindle checkpoint is active. Timely degradation of Cyclin A at this juncture is essential for formation of stable kinetochore-microtubule (KT-MT) attachments [139]. The question remains of how Cyclin A evades the SAC. As kinase activity rises in mitosis, APC/C subunits are phosphorylated. The Cdk accessory subunit Cks1/2, in the form of Cyclin A-Cdk-Cks, independently recruits Cyclin A to the APC/C by binding phosphorylated residues within the latter and leads to its ubiquitylation [67,140]. Thus, APC/C action in prometaphase enables correct KT-MT attachments by limiting Cyclin A-associated kinase activity.
Once correctly attached, the two sister chromatids are pulled to opposite poles of the cell, thereby generating tension that is thought to place the kinetochore proteins outside the zone of influence of inner centromere localized Aurora B, more than 100 nm away [141]. Here, phosphatase activity predominates and preferentially stabilizes these attachments [142], through direct dephosphorylation of seminal targets such as Knl1 and Ndc80.
During mitotic exit, BubR1 (Mad3 in yeast, worms and plants) and Bub1 are targeted for degradation by the APC/C [143,144], shutting down PP2A-B56 recruitment and SAC signaling (discussed below). PP1, now in complex with Repo-Man, nips Aurora B signaling at the bud by dephosphorylating H3 p-Thr3 [145,146]. Aurora B is also physically evicted from the inner centromere to the central spindle. Dephosphorylation of budding yeast CPC subunit Sli15 (INCENP) by Cdc14 precipitates this event [147]. In humans, this role is performed by PP2A-B55 [21]. Finally, Aurora kinases are degraded by APC/C action in late mitosis, thus ensuring that their phosphosignaling is temporally constrained within the cell.
3.2. The Spindle Assembly Checkpoint Inhibits APC/C Activity in Metaphase and is Regulated by Phosphatases
Until all kinetochores are correctly captured and bioriented at the metaphases plate, the APC/C activity is held in check by a highly potent, diffusible complex that acts as a stoichiometric inhibitor—the Mitotic Checkpoint Complex (MCC). Even one unattached kinetochore can sequentially recruit an array of proteins to form the MCC, holding the entire cell in metaphase and safeguarding genome segregation.
The MCC is a complex composed of Mad2, BubR1, Cdc20 and Bub3. At unattached kinetochores, the Ndc80 complex, unfettered by microtubule binding, instead recruits the kinase Mps1 and drives a phospho-dependent signaling cascade. By virtue of being present at the outer kinetochore, Mps1 phosphorylates various proteins, including the MELT motifs of Knl1. This is the most upstream event in the MCC formation. As seen during error correction, Knl1 again acts as a scaffold and recruits Bub1-Bub3 complexes via its phosphorylated MELT motifs, which then recruit BubR1-Bub3 complexes. At this juncture, Mps1 directly phosphorylates Bub1 and leads to Mad1-Mad2 heterodimer binding [78]. Next, Mad1 phosphorylation by Mps1 stimulates its catalytic activity, and leads to conversion of open-Mad2 (o-Mad2), to the MCC-assembly competent closed-Mad2 (c-Mad2) by structural remodeling of the protein [148]. This process is critical to rapid MCC assembly and therefore instantaneous checkpoint response within the cell. Termed a ‘template-assisted mechanism’, Mad1-c-Mad2 heterodimers catalyze conversion of cytosolic o-Mad2 proteins to their closed conformers at unattached kinetochores. Importantly, spontaneous Mad2 conversion is disfavored outside the context of kinetochores as the protein faces a large energy barrier [149,150]. C-Mad2 is the structurally relevant form of the MCC. By capturing Cdc20 via its N-terminal KILR motif, it leads to MCC formation. MCC binds APC/C complexes already containing a bound Cdc20 co-activator and inhibits their activity [151]. Cdk1 and Plk1 kinases impinge at various stages throughout this process, for instance by directly phosphorylating Mps1 to enhance its kinase activity [74,152,153].
It was initially thought that the MCC functions by sequestering Cdc20 away from the APC/C. Recently, however, it has become clear that the MCC employs a multitude of mechanisms to target numerous aspects of APC/C function to obstruct its activity. Cryo-electron microscopy structures have revealed molecular details of APC/C-MCC interactions [61,64]. The hollow of the APC/C is occupied by the MCC complex and its own molecule of Cdc20. MCC binding causes the APC/C-bound Cdc20 to move away from Apc10, thus disrupting D-box binding site within the holocomplex. BubR1, by virtue of its various inhibitory degron motifs (D-box, KEN and the ABBA motifs), mounts a multi-pronged attack on the two Cdc20 molecules within the complex [56,61,64]. It caps all available degron-binding sites within Cdc20, hobbling any potential for substrate recruitment. In some conformations, BubR1 also precludes binding of the initiating E2 UbcH10 by occupying its binding site within the catalytic module at the base of the enzyme. In a different state however, possibly regulated by the Apc15 subunit of the enzyme, the MCC rotates away and vacates the initiator E2 binding site. How the APC/C switches between the two conformations is unclear. Subsequent UbcH10 binding to the second conformer allows ubiquitylation and degradation of the Cdc20 molecule within the MCC, thereby leading to MCC turnover [61,64]. Thus, APC/C-bound MCC is a dynamic, self-limiting inhibitor with an inbuilt ‘diffuse’ function. Cdc20 turnover is important for cells to recover from a SAC arrest and avoid cohesion fatigue. Cdc20 levels are replenished by synthesis during mitosis [154]; as long as kinetochores remain unattached, the MCC will continue to inhibit the E3 ligase. This process is continuously antagonized by the DUB Usp44 to maintain a fine balance of Cdc20 levels [155,156]. Another protein antagonizes MCC assembly: p31comet adopts a Mad2-like fold, and extracts c-Mad2 from its complex with Mad1 and from within the MCC by directly dimerizing with it [157,158]. p31comet also acts as an adaptor protein and delivers c-Mad2 to the AAA+ ATPase TRIP13, which then catalyzes its conversion to inactive o-Mad2 by ATP hydrolysis and inactivates the MCC [159]. Phosphorylation has been reported to regulate p31comet activity in Xenopus and humans [160,161,162]. However, the details of the molecular mechanism are unclear, as is the nature of phosphatases that regulate this process.
Finally, in addition to MCC dissociation, further SAC signaling must also be extinguished at the source to ensure timely anaphase entry. Indeed, kinetochores proteins are in a state of flux due to presence of both kinases (Aurora B and Mps1) and recruited phosphatases (PP2A-B56-BubR1 and PP1-Knl1), which enables quick responsiveness of the system to changes in the input. Once the Ndc80 complex is bound to kinetochores, Mps1 can no longer bind and initiate its signaling cascade and MCC formation [163]. Direct dephosphorylation of Mps1 substrates is also required to prevent any potential for further MCC formation and is achieved by PP1 and PP2A phosphatases. PP1, recruited to outer kinetochores in a number of ways, is thought to take the lead in Mps1 substrate dephosphorylation [74]. PP1-Knl1 directly dephosphorylates Knl1-MELT motifs to halt any further binding of Bub1. PP2A-B56 bound to BubR1 is also thought to dephosphorylate Bub1, in addition to its role in recruiting PP1 by dephosphorylating its docking motifs within Knl1 [164,165,166]. As the same phosphatases regulate both error correction and SAC silencing, their interplay, in particular together with Aurora B and Mps1 kinases is complex. However, it is vital to understand how the antagonistic activities are integrated [74].
These molecular interactions underlie the highly responsive nature of MCC-mediated inhibition and explain how cells commence Cyclin B degradation as soon as the last chromosome is aligned [167].
3.3. Constructing and Segregating Mitotic Chromosomes
One of the hallmarks of the mitotic phase is the appearance of discrete, rod-shaped ‘condensed’ chromosomes, a prerequisite for accurate chromosome segregation to daughter cells. This is mediated by actions of two different SMC (structural maintenance of chromosome) protein complexes, condensin and cohesin [168,169]. They govern sister chromatid resolution, chromosome compaction and segregation, and are subject to regulation by kinases, phosphatases and the APC/C.
Condensin I and condensin II are ring-shaped multisubunit complexes that perform distinct functions in chromosome organisation and segregation in mitosis. In vitro reconstitution of mitotic chromatids has revealed that Cdk1 phosphorylation of condensin I is the sole mitosis-specific modification required for chromatid reconstitution [170]. Nonetheless, actions of Cdk1, Plk1 and Aurora B are also implicated in chromatin targeting of distinct condensin complexes and stimulating their activity [171,172,173,174,175,176]. In budding yeast, Cdc14 dephosphorylates the Smc4 subunit of condensin during mitotic exit [177]. In higher organisms, PP2A has been implicated in regulating condensin [178]. Additionally, PP6 has also been shown to positively regulate condensin I complexes by opposing inhibitory phosphorylation by casein kinase 2 [115,179].
Cohesin is a ring-shaped proteinaceous complex that holds together duplicated sister chromatids by physically entrapping them. Cell cycle-dependent acetylation of one of the cohesin subunits serves to ‘lock’ the ring in both yeast and vertebrates. Sororin, a vertebrate-specific protein is specifically recruited to acetylated cohesin. It protects cohesin from the ‘anti-establishment’ activity of a negative regulator, Wapl. Following entry into mitosis, vertebrate cells witness a removal of majority of cohesin complexes due to extensive phosphorylation of both Sororin and Wapl catalyzed by Cdk1, Plk1 and Aurora B. This occurs concomitant to condensin activity. Sororin phosphorylation destabilizes its interaction with cohesin, allowing Wapl activity to predominate [180,181]. Centromeric cohesin is preserved until anaphase through recruitment of PP2A-B56, which antagonizes these phosphorylation events and holds sister chromatids together until anaphases commences [182,183]. A mitosis-specific signaling cascade ensures the presence of PP2A-B56 complexes where their action is needed. As previously mentioned, Bub1 recruits Sgo1 by phosphorylating histone H2A at Thr120 [130]. Sgo1 directly binds PP2A-B56 through contacts with both the catalytic and regulatory subunits [184]. Bub1 degradation during anaphase turns off this pathway [185]; however, the identity of the phosphatase that resets H2A p-Thr120 is unknown.
Anaphase onset liberates separase by APC/C-Cdc20-mediated degradation of its inhibitor, securin. Centromeric cohesin is cleaved by this protease and sister chromatids commence their movement to opposite poles of the cell. A collaboration with PP2A-B56 phosphatase ensures that this step ensues in a switch-like fashion and precedes later events such as cytokinesis, which can damage any lagging chromosomes. CaMKII phosphorylates securin and makes it more amenable to APC/C-mediated degradation by increasing its degron-APC/C affinity. Separase-bound securin, however, is kept in a hypophosphorylated state by recruitment of PP2A-B56. Thus, free securin is targeted by the APC/C before separase-bound securin, ensuring that liberated separase is not re-inhibited by remaining securin molecules. Once activated, separase self-cleavage precludes PP2A binding and therefore re-inhibition by securin [186,187,188].
In budding yeast, securin degradation and decisive anaphase onset is also under phosphatase control, albeit with different underlying molecular details. Phosphorylation of yeast securin by Cdk1 impedes its degradation. As small levels of securin are degraded by the APC/C, Cdc14 is liberated, which dephosphorylates securin, promoting its degradation. Thus, a positive feedback loop is generated that leads to switch-like anaphase onset [189].
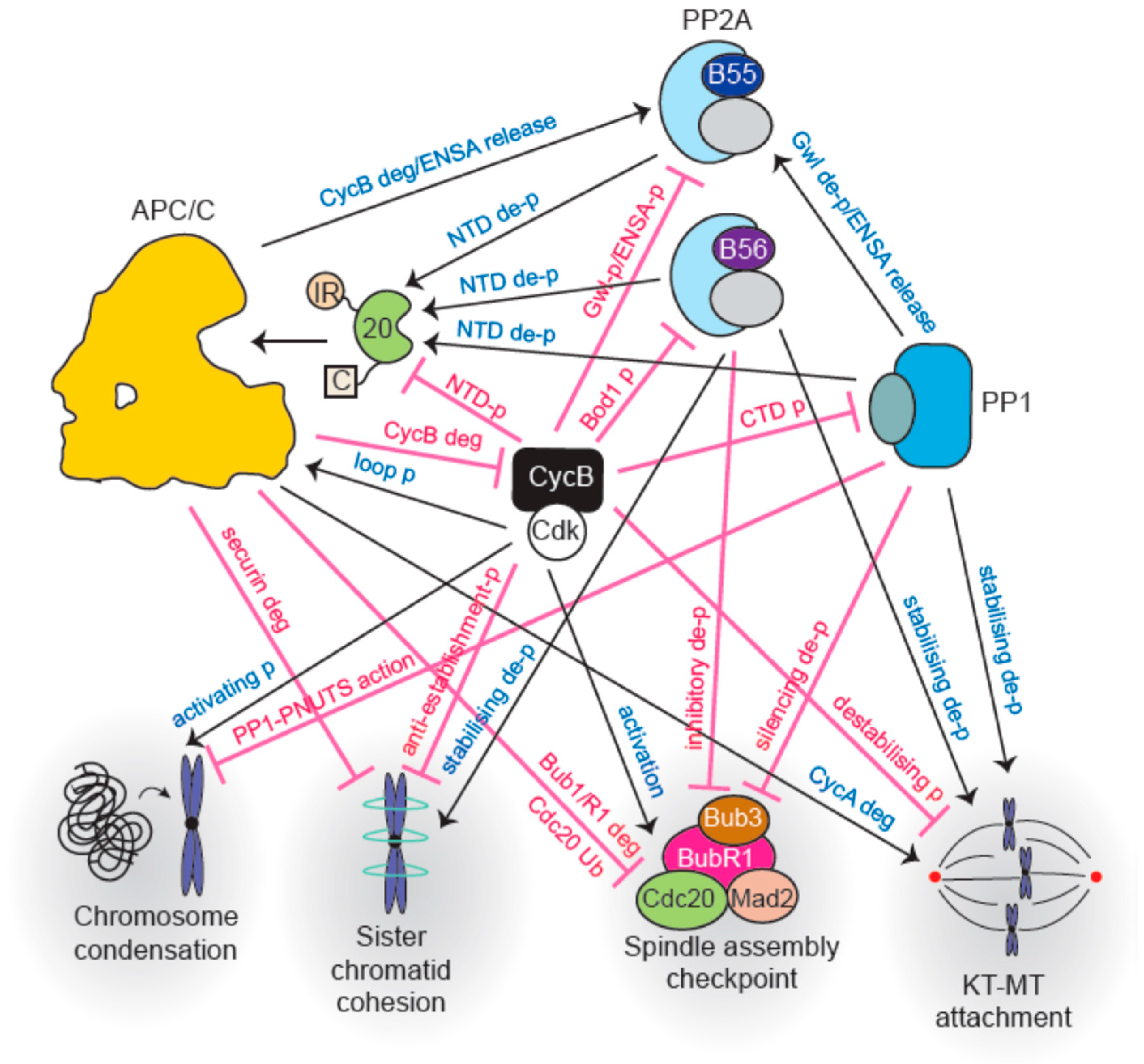
Major interactions between cell cycle phosphatases and the APC/C are outlined in Figure 4.
4. Intricate Interplay between Phosphatases and the APC/C Regulates Metaphase-Anaphase Transition and Mitotic Exit
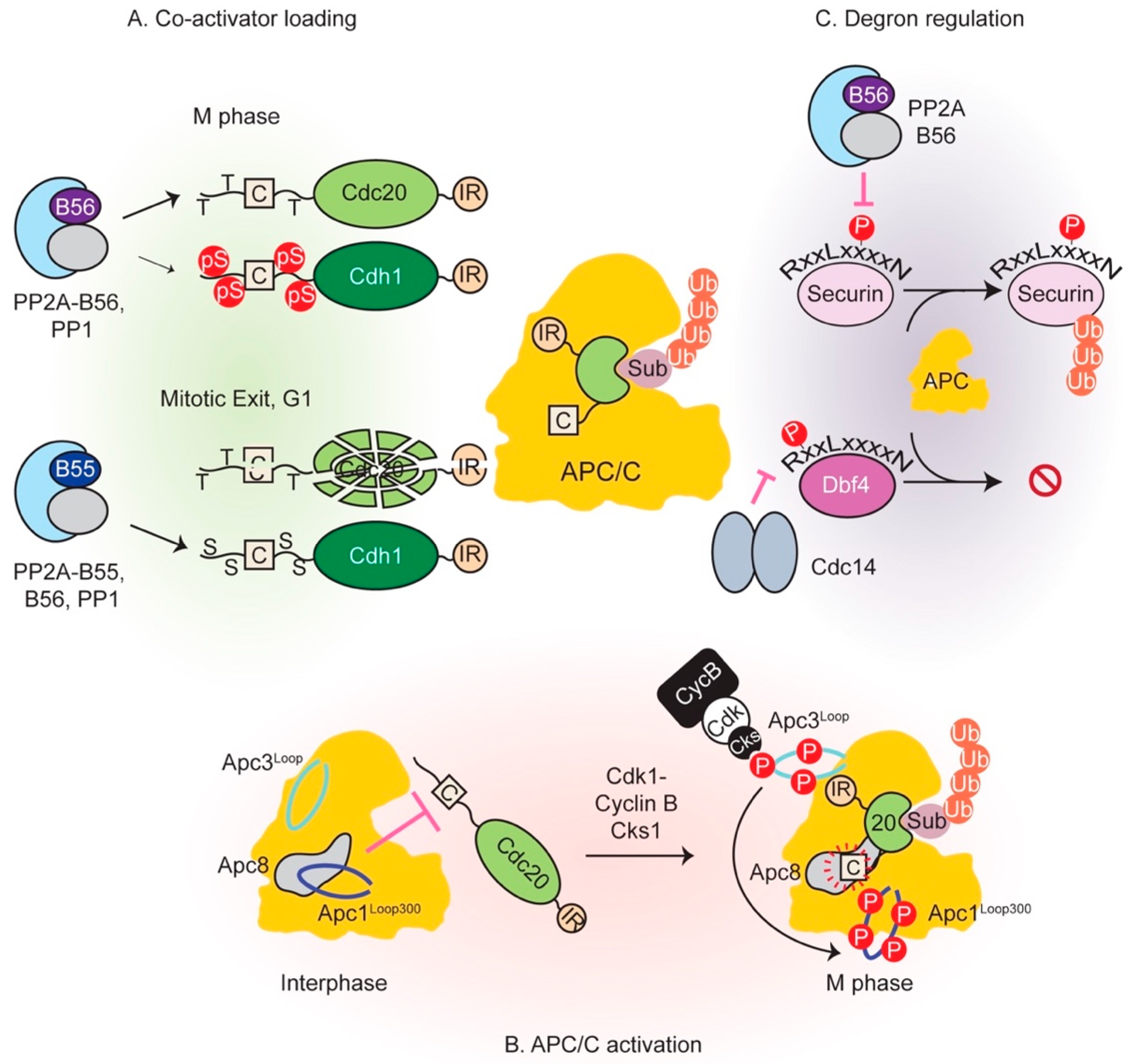
Once all chromosomes are aligned and the SAC is switched off, APC/C-Cdc20 becomes active. In addition to securin destruction, Cyclin B also gets degraded which reduces the overall kinase activity and initiates phosphatase reactivation. Extensive phosphoregulation of both the APC/C and co-activators governs its activity throughout mitosis. Phosphorylation precipitates opposite outcomes in this regard: phosphorylation of the APC/C activates it while co-activator phosphorylation prevents its association with the E3 ligase. Figure 5 summarizes control of APC/C function by phosphatases.
4.1. Phosphorylation of APC/C Enables Cdc20 Binding
Co-activators bind to APC/C at multiple sites, employing evolutionarily conserved regions within their largely intrinsically unstructured N- and C-terminal tails. These tails flank the highly structured WD40 propeller domain, which only mediates substrate recruitment rather than APC/C association. Of chief importance are two short linear binding motifs within co-activators (the C box and IR tail) that cooperatively enable Cdc20 and Cdh1 association with the APC/C by binding to the TPR lobe (the substrate recognition sub-complex). The C box, named so due to its evolutionary conservation within Cdc20 family of proteins [190], typically lies close to the N-terminus of co-activators and docks into one of the copies of the TPR subunit Apc8 [60,124]. The Ile-Arg or IR tail at the very C-terminus of co-activators binds an equivalent site within Apc3 [191]. The equivalence of the C box and IR tail receptor sites is demonstrated by the fact that the IR tail of the Cdc20 molecule within the MCC binds the Apc8 subunit instead [61,64]. Interestingly, the core subunit Apc10, the inhibitor Emi1, the E2 enzyme Ube2S and the prometaphase substrate Nek2A also employ the IR tail or variations thereof to associate with the APC/C [60,61,62,63,64,66].
Although both co-activators possess these binding sites, it had long been known that Cdc20 can only bind APC/C hyperphosphorylated in early mitosis by Cdk1 and Plk kinases [192,193,194,195,196]. Cdh1-mediated activation, however, is not conditional on APC/C phosphorylation status [193] and occurs towards the end of mitosis and G1. At this stage, APC/C phosphorylation is low due to Cdk1 inactivation by kinase degradation and Wee1 activity, and concomitant action of reactivated phosphatases. This dichotomy was resolved by cryo-EM, biochemical and functional analyses and unveiled a sequential phosphorylation relay that activates the APC/C by allowing Cdc20 binding [197,198,199]. Apc1 and Apc3 subunits were found to be hyperphosphorylated within two intrinsically unstructured loop domains: a large ~280 residue segment within Apc3, and a shorter ~100 residue stretch within Apc1 at position 300 from the N-terminus (Apc1Loop300), both of which are conserved in vertebrates. Mutation of Apc3 or Apc1 sites of phosphorylation to alanine considerably reduced APC/C-Cdc20 activity, with simultaneous mutation of both rendering it almost inactive and unable to bind Cdc20 [197]. It was found that Apc3 phosphomutant failed to recruit the phosphoadaptor subunit p9/Suc1/Cks [197], which had been known to interact as a part of cyclin B-Cdk1-Cks complex [200,201]. This led to downstream effects in the form of reduced Apc1 phosphorylation. Thus, initial phosphorylation of Apc3 by Cdk1 and polo-like kinases creates a phosphodocking site for p9/Suc1/Cks, probably through p-Thr residues that conform to its preferred docking motif [202,203], which then leads to intramolecular phosphorylation of Apc1Loop300. These results pointed to an inhibitory function for Apc1Loop300 that is relieved upon its phosphorylation. Cryo-EM studies revealed that this loop contained a stretch similar to the co-activator C box and physically occluded the C box binding site within Apc8 in the non-phosphorylated form of APC/C [198,199]. Phosphorylation of sites within Apc1Loop300 peels it away from this region by disrupting its interactions and allows Cdc20 binding. Consistent with this model, deletion of Apc1Loop300 in the context of the holocomplex permitted Cdc20-dependent activity even in the absence of phosphorylation [198,199]. Recombinant Apc1Loop300 could co-precipitate holo-APC/C while its phosphomimetic counterpart could not, further bolstering this conclusion [197]. Thus, intrinsically unstructured regions of the APC/C act as regulatory hubs to control its activity. It is conceivable that phosphatase recruitment to the APC/C is at least partly mediated through similar docking interactions. Recruited phosphatases could also be involved in dephosphorylating co-activator, thereby activating the complex (see below).
Phosphorylation only affects access of Cdc20 to the APC/C, with Cdh1 evidently outcompeting the autoinhibition of Apc1Loop300 to bind the enzyme. Higher affinity of Cdh1 compared to Cdc20 for the E3 ligase, evidenced by increased interaction sites within its N-terminus with multiple APC/C subunits, and reduced flexibility of the IR tail within Apc3 in cryo-EM structures has been suggested as an explanation for their differential behavior [198,204].
The budding and fission yeast APC/C adopts a very similar architecture to its vertebrate counterpart (barring the absence of Apc7) [205,206,207]. There is compelling evidence for phosphorylation-mediated activation of the yeast enzyme [192,208]. It is likely that this is general mechanism of cell cycle-mediated control of this E3 ligase and is evolutionarily conserved. A loop domain within Apc1, or within a different subunit in both yeasts might be implicated in this regulation.
A majority of APC/C subunits undergo phosphorylation [198,199]. Whether these impinge on the Apc3-Apc1 pathway or regulate other functions of the APC/C (such as MCC, Emi1 or phosphatase binding) remains to be elucidated.
At some point following entry into anaphase, APC/C dephosphorylation occurs and increased electrophoretic mobility of Apc3 is readily observed by SDS-PAGE [124]. Precisely when this occurs in the context of other cell cycle events is unknown, but it likely follows reactivation of PP1 and PP2A-B55 phosphatases. Dephosphorylated Apc3 would no longer support further Apc1 phosphorylation. Thus, it is possible that Apc1Loop300 is protected from, or impervious to dephosphorylation in early anaphase to preserve Cdc20 function. APC/C is not homogenously distributed throughout the cell. For instance, it is localized to mitotic chromosomes by the Ska complex in a dephosphorylated yet active form [209]. How this localization is mediated and how its hypophosphorylated state reconciles with our current view of APC/C function are unknown.
4.2. Co-Activator Dephosphorylation is Essential for APC/C Interaction
As mentioned previously, Cdc20 is an early activator of the APC/C, while Cdh1 takes the reins in late mitosis upon its dephosphorylation. The overall structure and organization of the co-activators is remarkably similar; nonetheless, important differences persist and define their characteristic behaviors. First, Cdc20 can only bind phosphorylated APC/C due to reasons outlined in the previous section. Secondly, both co-activators employ the use of D- and KEN boxes within their substrates; however, only vertebrate Cdc20 appears to bind the ABBA motif which contributes to its inhibition by BubR1 [56]. Budding yeast Cdh1 and Cdc20 also appear to have slight variations within their preferred ABBA motif signatures [58,210]. Finally, in the absence of Cdh1, Cdc20 is able to compensate for most of its functions, possibly due to its stabilization as it is degraded by action of APC/C-Cdh1. Efficient Aurora A and B degradation, however, appears to be dependent on Cdh1 [211].
In contrast to APC/C, Cdk1 phosphorylation of both Cdc20 and Cdh1 is inhibitory and prevents their association with the E3 ligase [124,212,213,214]. In early anaphase, Cdc20 phosphorylation at several sites within its N-terminal tail reduces its affinity for the APC/C in an additive manner. Preserving the function of any one site while mutating the rest does not elicit the same inhibitory effect. Nonetheless, a p-Thr site flanking the Cdc20 C box appeared to be particularly inhibitory and phosphorylation of three threonine residues (Thr64/Thr68/Thr79 in Xenopus Cdc20) largely inhibits the activation role of Cdc20 [124]. Coupled with the observation that the C box flanking phosphosites are disordered within cryo-EM structures [198,199], it appears that negative charges and/or bulky phosphate groups destabilize C box receptor interactions [197].
As Cyclin B is degraded, the previously inhibited phosphatases PP1 and PP2A-B55 get reactivated (discussed later). Cdh1 is dephosphorylated, probably primarily by active PP2A-B55 in vertebrates [21], and replaces Cdc20 as the co-activator. Prior to late anaphase, phosphorylation of Cdh1 at its N-terminus precludes its APC/C binding [193,212]. Analogous to Cdc20 regulation, Cdh1 phosphorylation destabilizes the interaction of its C box and other affinity elements with the APC/C [60].
A recent study addressed how PP2A-B55 creates distinct temporal windows for co-activator dephosphorylation and activation: while Cdc20 is dephosphorylated in early anaphase, Cdh1 is only targeted later. Phosphothreonine residues within Cdc20 are preferred PP2A-B55 substrates compared to phosphoserines, a property likely shared by all members of the PPP family owing to the similarities of their catalytic cores. This allows faster activation of Cdc20 by preferential dephosphorylation of its residues that are mostly Thr. In contrast, p-Ser residues predominate Cdh1 N-terminus and are disfavored PP2A substrates. Their dephosphorylation occurs later in anaphase when other factors such as reduced substrate competition possibly make its dephosphorylation more amenable. Importance of late Cdh1 dephosphorylation is underscored by the aberrant mitotic phenotypes associated with a phosphothreonine variant of the co-activator that is dephosphorylated earlier [21]. How PP2A enzymes select a p-Thr is unknown. It has been suggested that a small hydrophobic cleft close to the metal ion binding sites at the catalytic core could make additional contacts with the extra methyl group that differentiates a threonine from a serine [215]. This could translate into p-Thr residues exhibiting a lower Km, a higher kcat or both, instigating faster turnover. In summary, both Cdc20 and Cdh1 are dephosphorylated at different points in the cell cycle to drive APC/C activation.
Cdc20 dephosphorylation invokes a catch-22 scenario: on the one hand it requires PP2A-B55 activity; on the other, it promotes B55 reactivation by degrading Cyclin B [216]. It begs the question: how does initial Cdc20 activation take place? Could a very small pool of B55 have escaped p-ENSA inhibition at metaphase to promote Cdc20 dephosphorylation? While this possibility cannot be ruled out, given the stoichiometric excess of p-ENSA over PP2A-B55 and subnanomolar Kd [101], it is likely that initial Cdc20 dephosphorylation is commenced by other phosphatases. Kinetochore-localized PP1-Knl1 was shown to opportunistically dephosphorylate Cdc20 recruited by the ABBA motif within Bub1 in C. elegans [122]. However, it is likely that other sources of phosphatase activity also target Cdc20, as abrogation of the PP1 pathway leads to mitotic delay rather than a block. PP2A-B56 represents another possibility as it is active in mitosis. In fact, PP2A-B56 has been shown to associate with the APC/C constitutively [123], although the molecular basis of this interaction remains unknown. It has also been reported that PP2A-B56 promotes the stable association of Ube2S with the APC/C through dephosphorylation of Cdc20 at Ser92, although the detailed mechanism remains elusive [217].
4.3. Multiple Mechanisms Regulate Degron-Mediated Substrate Degradation
Mitotic exit witnesses degradation of at least a 100 protein substrates in a strict temporal order—a prerequisite for successful division. Perturbations in this order upset cell division. Coupled with the fact that protein synthesis exacts a considerable energetic cost, cells have evolved mechanisms to ensure specific, timely and efficient degradation of substrates by the APC/C. Computational modeling of APC/C-Cdc20-mediated substrate degradation in yeast concluded that substrate affinity, catalytic rate of ubiquitin transfer to achieve multi-ubiquitylation and substrate competition can together determine the order of substrate degradation [218]. Co-activator WD40 domains recruit substrates by employing structured groves on their surface that bind degron motifs. Degron requirement can be bypassed by independently recruiting substrates to the E3 ligase by a translational fusion of a substrate-APC/C subunit for instance [52]. Structural and functional analyses have revealed the three major degron-binding pockets: D-box (RxxLxxxxN) binds between a conserved region between WD40 blades 1 and 7, while residues C-terminal to it contact the Apc10 co-receptor; KEN box (composed of residues Lys-Glu-Asn) binding is mediated by the top of the WD40 barrel; and the ABBA motif (Fx[ILV][FY]x[DE]) receptor is situated within a hydrophobic surface formed between blades 2 and 3 [219,220,221].
There is considerable plasticity among what constitutes a ‘degron’ motif and seemingly incompatible residues can be allowed in a particular sequence context. For instance, divergence from consensus can be tolerated if contacts at additional positions stabilize binding, a concept reviewed by Davey and Morgan [58]. The degeneracy of the degron binding pocket permits multiple flavors of degrons to bind likely with a variety of affinities, contributing to their orderly degradation.
Multivalent substrate interactions, either with the WD40 domains by presence of multiple degron motifs, or with other APC/C subunits can elicit higher affinity—a strategy often employed by APC/C inhibitors. Higher order properties can also determine substrate binding and degradation. The example of Nek2A degradation illustrates this concept: the MR tail of Nek2A resembles co-activator and Apc10 IR tails and allows it to be independently bound and degraded by the APC/C during prometaphase, when the SAC is active. Although necessary, the MR tail is not sufficient for degradation; dimerization of Nek2A is also essential [222]. It is likely that dimerization works by increasing affinity of Nek2A for the APC/C, as docking of one molecule by the MR tail would obligatorily bring in a second.
Degron modification is another major mechanism regulating substrate degradation. Acetylation of BubR1 during metaphase blocks its degradation during metaphase [143]. Additionally, phosphorylation within and around degron motifs can occasion substrate degradation or stabilization and is highly context specific. For instance, budding yeast securin phosphorylation on Cdk1 residues situated outside of the D- and KEN box motifs is refractory to degradation. Activation of the Cdk1-antagonizing phosphatase Cdc14 by separase leads to securin dephosphorylation and degradation in a positive feedback loop (as mentioned in Section 3.3) [189]. On the other hand, phosphorylation of human securin Thr66 that lies within the core of the D-box motif enhances its interaction with the co-activator and promotes degradation [186,219]. This forms the molecular basis of separase stabilization by its previously mentioned PP2A-B56 association [186]. Further highlighting the importance of the precise position of phosphoresidues in relation to degron motifs are the following examples of Dbf4 and Cdc6. Phosphorylation of budding yeast DDK kinase subunit Dbf4 one residue after the Arg within its N-terminal D-box delays its degradation in vivo, indicating that phosphorylation at this position is inhibitory [210]. Cyclin E-Cdk2 phosphorylates human Cdc6 at two Ser sites, each N-terminal to a D- and a KEN box and reduces its association with Cdh1 in G1. This blocks Cdc6 degradation to allow timely origin licensing and S phase initiation [223].
5. Phosphatases Escort Cells through Exit from Mitosis
Phosphostate of a protein is always in flux; it is a readout of the equilibrium kinase and phosphatase activities governing it. Any change in one of the parameters alters this equilibrium and is swiftly reflected in the phosphate occupancy of the protein. This is the principle behind rapid phosphatase activation as cells commence exit from mitosis. Cyclin B degradation by APC/C-Cdc20 decreases Cdk1 activity and creates a domino effect. The balance is shifted towards PP1 autoactivation by dephosphorylation of its C-terminal tail and I-1 [108,109,110]. Free PP1 then dephosphorylates and partially inactivates Greatwall [224,225,226], ceasing the supply of phosphorylated ENSA/Arpp19. Any PP2A-B55 dephosphorylated p-ENSA/p-Arpp19 inhibitor molecules are no longer replenished. Williams et al. estimated that these inhibitors are present in a 5-10 fold excess to PP2A-B55, and would be dephosphorylated within a few minutes of Greatwall inactivation to swiftly reactivate the phosphatase [101]. PP2A-B55 completes Greatwall inactivation by full dephosphorylation of the protein. This positive feedback loop, coupled with the rest of the system architecture makes mitotic exit a bistable transition [226]. Once liberated, PP2A-B55 is thought to dephosphorylate hundreds of substrates in conjunction with PP1 and PP2A-B56. In fission yeast, a similar principle of phosphatase activation governs, although the molecular underpinnings are unclear. PP1 liberated upon Cyclin B degradation activates PP2A-B55 by directly binding it. Active PP2A-B55 then dephosphorylates a PP1 docking site within PP2A-B56, thereby allowing PP1 to bind and activate it [227]. PP1 could act by reversing inhibitory B55 phosphorylation mentioned previously [105,106]. How it effects B55- and B56-bound PP2A enzymes is a question that remains to be answered.
This leads us to an important discussion point: there likely is not one ‘mitotic exit phosphatase’ in higher organisms. In contrast, inactivation of Cdc14 in budding yeast elicits a complete block and arrests cells in telophase with elongated spindles due to its role in downregulating kinase activity [213,228,229]. Mitotic exit in human cells and other vertebrates such as Xenopus has only been prevented upon pan-phosphatase inhibition by toxins such as okadaic acid (OA) (following Cdk1 inhibition, for instance) [19,230,231,232], which inhibits multiple phosphatases to varying degrees. Nonetheless, that phosphatases collaborate to bring about robust mitotic exit in all organisms is incontrovertible.
Assembly of a central spindle, nuclear envelope reformation, chromosome decondensation and finally abscission into two daughter cells along the cytokinetic furrow are some of the events associated with mitotic exit. PP2A-B55 directly dephosphorylates p-Thr481 within Prc1, allowing its association with the central spindle where it bundles microtubules in an antiparallel array and defines the position of the cytokinetic furrow. PP2A-B55 also controls dephosphorylation of Aurora A regulator Tpx2 and its central spindle localization [20]. PP2A-B56 recruited by Kif4a also influences central spindle growth by opposing Aurora B phosphorylation of Mklp2 and Kif4a [21,233]. PP2A-B56 is also recruited by the centralspindlin protein Racgap1 and regulates its phosphorylation for accurate execution of cytokinesis. As mentioned previously, APC/C-Cdh1 activity later degrades Aurora B and halts further signaling.
PP1 also plays a major role in regulating mitotic exit and cytokinesis [234,235]. For instance, PP1 in conjunction with its regulatory subunit Repo-Man dephosphorylates H3 p-Thr3 to evict Aurora B from centromeres to the central spindle. Repo-Man association with histones is held in check by inhibitory phosphorylation by Cdk1. Cyclin B degradation changes the balance of kinase/phosphatase activities and allows PP1 and PP2A-B56-mediated (via a LxxIxE motif) dephosphorylation of Repo-Man, efficient PP1 binding and centromere targeting. PP1-Repo-Man has also been implicated in reestablishment of the nuclear envelope [145,146,236] and dephosphorylation of importin β to initiate nuclear envelope reassembly [236]. PNUTS (PP1 Nuclear Targeting Subunit), another PP1 interacting protein binds chromatin, and has been shown to enhance in vitro chromosome decondensation [237].
This is far from an exhaustive list of phosphatase regulation of mitosis; one can nonetheless appreciate that it is essential that these events occur in a specific order to avoid mitotic failure. For instance, premature recruitment of Prc1 to the central spindle upon Greatwall depletion induces early cytokinesis and causes a ‘cut’ phenotype in a majority of cells [232]. Therefore, not only must phosphatases be activated at the right time, they must also dephosphorylate hundreds of substrates in the right order. In Xenopus egg extracts and human cells, high levels of non-degradable Cyclin B block cells in metaphase, intermediate levels allow progression into a pseudo-anaphase state with separated sister chromatids, while persistent low levels arrest cells in telophase [238,239]. These observations suggest that ordering mechanisms exit in higher vertebrates. Differences in PP2A-B55 activation as a function of Cyclin B levels can partly explain some of these observations [232]. However, once liberated PP2A-B55 must still contend with a multitude of substrates in the backdrop of opposing activities of mitotic kinases such as Plk1. In budding yeast, the relative catalytic efficiencies of Cdk1 kinase and Cdc14 phosphatase towards a particular substrate was shown to set its dephosphorylation time and order mitotic exit [240]. It is likely that similar mechanisms exist in higher organisms, albeit with different molecular players.
How are hundreds of proteins discriminated by mitotic phosphatases? Part of the answer lies in intrinsic preference for phosphothreonines exhibited by PP2A phosphatases. Accordingly, survey of proteins dephosphorylated very early during an induced mitotic exit in human cells revealed that p-Thr-Pro (p-TP) sites were significantly more dephosphorylated than p-Ser-Pro (p-SP) sites by OA sensitive phosphatases [19]. A small non-polar residue at the +2 position potentiated, while a +2 Pro made p-TP refractory to dephosphorylation [19]. In another study of human cells undergoing mitotic exit as a result of phosphatase activation upon mitotic extract dilution, 45% of candidate PP2A-B55 dependent substrates were p-Thr, compared to 25% p-Thr detected in the total phosphoproteome [20], corroborating previous observations. Patches of basic residues both N- and C-terminal to the phosphosite enhanced dephosphorylation and depended on an acidic surface within B55. In fact, a correlation between motif basophilicity and PP2A-B55 was uncovered [20]. Highly basophilic motifs would be disfavored by Plk1, perhaps further contributing to their enhanced dephosphorylation [241]. A phosphoproteomic study uncovered that PP6, a phosphatase highly similar to PP2A, instead prefers acidophilic motifs within substrate proteins and opposes CK2 phosphorylation [242].
A majority of cellular phosphorylation events are catalyzed on serine residues (98.2%) [243]. This could be due to their greater overall numbers in the proteome and higher surface accessibility compared to the slightly more hydrophobic nature of threonines. How is their dephosphorylation controlled? Presence of bulky aromatics upstream to the p-Ser enhances their dephosphorylation by PP2A-B55 and constitutes one mechanism, although its molecular basis is unknown [20]. It is also possible that, on an average, p-Ser are targeted later in mitosis when p-Thr residues no longer outcompete them, as substrate competition has been shown to be an ordering mechanism [240]. Finally, it is also likely that a serine-specific phosphatase collaborates with PP2A enzymes. Cdc14 is a p-SPxK/R-directed phosphatase [244,245] and could aid dephosphorylation of some substrates, particularly in early anaphase. Indeed, human Cdc14B has been shown to catalyze phosphoserine-rich Cdh1 dephosphorylation upon DNA damage in G2 to induce Plk1 degradation and halt the cell cycle [246]. Whether a supportive role for Cdc14 in normal mitotic dephosphorylation also exists remains a question for the future.
Finally, most proteins contain multiple phosphosites that could be potential phosphatase targets upon binding to substrate docking motifs such as LxxIxE. How the presence of these substrate affinity elements colludes with phosphosite preferences in the context of the whole protein is unknown.
Our discussion thus far has been simplified by referring to each phosphatase as a homogeneous entity. This view, however, does the complexity of the system a disservice: each phosphatase has multiple isoforms. Do they exist to provide redundancy and buffer the system against inactivating mutations? Or do they perform distinct functions within the cell? A recent report examined the importance of multiple B56 isoforms, all of which contain an LxxIxE-binding pocket but exhibit distinct localizations within cells: while B56α binds Sgo2 at centromeres, B56γ prefers BubR1 binding via its LxxIxE motif. A small loop within B56α is thought to prevent docking interactions with BubR1 by repressing its binding to LxxIxE motifs, and enabling Sgo2 interactions instead [247]. Specific binding of PP1γ to its regulatory proteins Repo-Man and Ki-67 follows a similar theme: presence of an N-terminal Arg residue generates a regulator binding pocket unique to PP1γ [248]. Therefore, there has been considerable subfunctionalization between different phosphatase isoforms, providing finer brushstrokes on the canvas of mitosis. Which substrates are targeted by different phosphatase pools remains unknown. Coupled with structural basis for differential substrate targeting, this knowledge can aid development of drugs specific to a particular flavor of phosphatase.
6. Roles in Diseases and Avenues for Therapies
As the APC/C and phosphatases have pivotal roles in the cell cycle and beyond, they have been implicated in various diseases such as cancer. In case of the APC/C, heterozygous mutations within core subunits Apc3, Apc8 and Apc6 are found in human colon cancer cells [249]. Involvement of the APC/C co-activators has also been found in tumorigenesis and disease progression. Overexpression of Cdc20 in particular has been correlated to poor prognosis in various cancers such as non-small cell lung cancer [250], colorectal cancer [251], multiple myeloma [252], etc. Studies in cancer cell lines showed that depletion of Cdc20 caused cell cycle arrest and a reduction in cell viability, underlining active participation of Cdc20 in disease state in addition to its value as a prognostic marker. Further bolstering the role of Cdc20 as a potential oncogene are observations in mouse skin cancer models, where inducible depletion of Cdc20 led to regression of tumors due to cell cycle arrest followed by apoptosis [253]. Cdh1 is thought of as a tumor suppressor, perhaps due to its role in restraining the entry of cells into the cell cycle and maintaining genome stability. Accordingly, several cancers exhibit a reduction in Cdh1 levels or function [254,255,256], due to hyperphosphorylation [257], for instance. Cdh1 heterozygous deletion mice are more susceptible to developing tumors later in life [258]. Furthermore, Cdh1 also regulates myriad aspects of development and functioning of the nervous system [259], and its downregulation has been implicated in Alzheimer’s disease [259,260,261].
APC/C has been proposed as a potential target in cancer therapy, as its inhibition leads to an arrest in mitosis followed by cell death [262]. Two small molecule inhibitors of the APC/C, TAME (tosyl-l-arginine methyl ester) and apicin were isolated from screens in Xenopus extracts [263]. TAME blocks the structurally equivalent C-box and IR binding sites within the APC/C and prevents efficient co-activator loading [198]. Apicin occludes the D-box receptor within Cdc20 and competes with APC/C substrates. Simultaneous inhibition of the APC/C with these two inhibitors has a synergistic effect in blocking mitotic exit [264]—a strategy in clinical use with SAC inhibitors. Indeed, SAC inhibition alone appears to be inefficient in blocking mitotic exit, as cells escape the arrest due to leaky Cyclin B degradation [265]. Combination therapy with APC/C inhibition is likely to constitute a more effective anti-cancer strategy. Additionally, due to the tumor suppressive activity of APC/C-Cdh1, it is desirable to specifically target APC/C-Cdc20 complexes. It is thus important to understand the differences between loading of these co-activators.
PP2A has been heavily implicated in several diseases such as cancer, Alzheimer’s disease, diabetes, etc. [266,267]. Traditionally, PP2A has been thought of as a tumor suppressor, with its downregulation promoting cancer development. For instance, PP2A subunits have been frequently found to be mutated in cancer. Cancer-associated mutations within the scaffolding [268,269], catalytic [270] and regulatory subunits [271] typically tend to affect assembly of the holoenzyme. Endogenous inhibitors of PP2A such as SET have also been found to be upregulated in cancer cells [272,273], thereby leading to a reduction in PP2A activity. Following these observations, activation of PP2A has been suggested as a potential cancer therapy, by inhibiting SET [274], or in combination therapy to prevent PP2A inhibition-mediated drug resistance [275]. Conversely, PP2A inhibition has been employed to overcome resistance of some cancers to radiation therapy [276]. To this end, a small molecule PP2A inhibitor called LB-100 has entered clinical trials for treatment of solid cancers among others [277]. However, LB-100 also exhibits inhibition of PP5 [278], owing to the previously mentioned similarities in the catalytic sites of PPP family of phosphatases. Therefore, it is vital to further understand the differences between phosphatases relevant to diseases to develop specific inhibitors. Targeting PP1 has also been proposed as an anti-cancer therapy as its inhibition in mouse xenograft models impaired tumor growth [279]. As PP1 binds to hundreds of regulatory proteins, its blanket inhibition is likely to be highly toxic. Recent drug screening approaches have sought to target specific PP1-regulator holocomplexes in treatment of Huntington’s disease [280], for instance.
Cell cycle phosphatases have been implicated in numerous signaling pathways, regulating myriad functions. A systems-level analysis of the consequences of phosphatase perturbation in various contexts would be a beneficial complement to various studies.
7. Outlook
Mitosis is a tour de force achieved by phosphatases and the APC/C working in concert with mitotic kinases. They integrate a variety of information to ensure timely and accurate execution of cell division. Inhibitors, regulatory modifications and opposing activities modulate their activities to fine-tune passage through cell cycle transitions. The past few years have unfurled layers of regulation by critical molecular pathways that control their activities.
A larger theme emerging in phosphatase and APC/C function is the ubiquity of regulation by intrinsically unstructured loop domains. These act as regulatory hubs to occasion myriad effects: they underlie efficient catalysis by increasing substrate residence time or local concentrations of regulators with respect to the enzyme. The example of PP1 shows that one enzyme can possess multiple pockets that bind their own short linear motifs. Thus, it is likely that many other such interactions within other phosphatase and the APC/C await discovery.
Equilibrium protein modification relies on many factors. Some of them are intrinsic to the substrate, such as the nature of the phosphosite presented to phosphatases, or indeed the accessibility of the substrate lysines by the APC/C. These can be further potentiated by the strength of degrons, or presence of docking motif interactions. However, many factors extrinsic to the substrates are likely to exert equally important effects on protein modifications. Higher local concentrations of enzymes, achieved by tethering them to cellular structures for instance, would aid the formation of substrate-enzyme complex and catalysis. Competition provided by more efficient substrates could also delay enzyme action on less preferred ones, as would the presence of opposing enzyme activities. Hence, unraveling how these factors work together would help us better understand how two cells are successfully produced from one.
Funding
This work was supported by the Wellcome Trust (205150/Z/16/Z).
Acknowledgments
We are grateful to F Uhlmann, SA Touati and members of the Yamano laboratory for helpful discussions and critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cori, G.T.; Green, A.A. Crystalline muscle phosphorylase: II. Prosthetic group. J. Biol. Chem. 1943, 151, 31–38. [Google Scholar]
- Fischer, E.H.; Krebs, E.G. Conversion of phosphorylase b to phosphorylase a in muscle extracts. J. Biol. Chem. 1955, 216, 121–132. [Google Scholar]
- Krebs, E.G.; Fischer, E.H. The phosphorylase b to a converting enzyme of rabbit skeletal muscle. Biochim. Biophys. Acta 1956, 20, 150–157. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833. [Google Scholar] [CrossRef]
- Shi, Y. Serine/Threonine Phosphatases: Mechanism through Structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.J.; Dixon, J.E.; Manning, G. Genomics and evolution of protein phosphatases. Sci. Signal. 2017, 10, eaag1796. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishito, Y.; Usui, H.; Shinzawa-Itoh, K.; Inoue, R.; Tanabe, O.; Nagase, T.; Murakami, T.; Takeda, M. Direct metal analyses of Mn2+-dependent and -independent protein phosphatase 2A from human erythrocytes detect zinc and iron only in the Mn2+-independent one. FEBS Lett. 1999, 447, 29–33. [Google Scholar] [CrossRef]
- Heroes, E.; Rip, J.; Beullens, M.; Van Meervelt, L.; De Gendt, S.; Bollen, M. Metals in the active site of native protein phosphatase-1. J. Inorg. Biochem. 2015, 149, 1–5. [Google Scholar] [CrossRef]
- Barford, D.; Das, A.K.; Egloff, M.-P. The structure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 133–164. [Google Scholar] [CrossRef]
- Heroes, E.; Lesage, B.; Görnemann, J.; Beullens, M.; Van Meervelt, L.; Bollen, M. The PP1 binding code: A molecular-lego strategy that governs specificity. FEBS J. 2013, 280, 584–595. [Google Scholar] [CrossRef]
- Peti, W.; Nairn, A.C.; Page, R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 2013, 280, 596–611. [Google Scholar] [CrossRef]
- Weith, M.; Seiler, J.; van den Boom, J.; Kracht, M.; Hülsmann, J.; Primorac, I.; del Pino Garcia, J.; Kaschani, F.; Kaiser, M.; Musacchio, A.; et al. Ubiquitin-Independent Disassembly by a p97 AAA-ATPase Complex Drives PP1 Holoenzyme Formation. Mol. Cell 2018, 72, 766–777.e6. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Yamano, H.; Kinoshita, N.; Yanagida, M. Mitotic regulation of protein phosphatases by the fission yeast sds22 protein. Curr. Biol. 1993, 3, 13–26. [Google Scholar] [CrossRef]
- Stern, B.; Nurse, P. A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trends Genet. 1996, 12, 345–350. [Google Scholar] [CrossRef]
- Coudreuse, D.; Nurse, P. Driving the cell cycle with a minimal CDK control network. Nature 2010, 468, 1074–1079. [Google Scholar] [CrossRef]
- Agostinis, P.; Goris, J.; Pinna, L.A.; Marchiori, F.; Perich, J.W.; Meyer, H.E.; Merlevede, W. Synthetic peptides as model substrates for the study of the specificity of the polycation-stimulated protein phosphatases. Eur. J. Biochem. 1990, 189, 235–241. [Google Scholar] [CrossRef]
- Godfrey, M.; Touati, S.A.; Kataria, M.; Jones, A.; Snijders, A.P.; Uhlmann, F. PP2ACdc55 Phosphatase Imposes Ordered Cell-Cycle Phosphorylation by Opposing Threonine Phosphorylation. Mol. Cell 2017, 65, 393–402.e3. [Google Scholar] [CrossRef]
- McCloy, R.A.; Parker, B.L.; Rogers, S.; Chaudhuri, R.; Gayevskiy, V.; Hoffman, N.J.; Ali, N.; Watkins, D.N.; Daly, R.J.; James, D.E.; et al. Global Phosphoproteomic Mapping of Early Mitotic Exit in Human Cells Identifies Novel Substrate Dephosphorylation Motifs. Mol. Cell. Proteom.: Mcp 2015, 14, 2194–2212. [Google Scholar] [CrossRef] [Green Version]
- Cundell, M.J.; Hutter, L.H.; Nunes Bastos, R.; Poser, E.; Holder, J.; Mohammed, S.; Novak, B.; Barr, F.A. A PP2A-B55 recognition signal controls substrate dephosphorylation kinetics during mitotic exit. J. Cell Biol. 2016, 214, 539–554. [Google Scholar] [CrossRef] [Green Version]
- Hein, J.B.; Hertz, E.P.T.; Garvanska, D.H.; Kruse, T.; Nilsson, J. Distinct kinetics of serine and threonine dephosphorylation are essential for mitosis. Nat. Cell Biol. 2017. [Google Scholar] [CrossRef]
- Kataria, M.; Mouilleron, S.; Seo, M.-H.; Corbi-Verge, C.; Kim, P.M.; Uhlmann, F. A PxL motif promotes timely cell cycle substrate dephosphorylation by the Cdc14 phosphatase. Nat. Struct. Mol. Biol. 2018, 25, 1093–1102. [Google Scholar] [CrossRef]
- Pinna, L.A.; Donella, A.; Clari, G.; Moret, V. Preferential dephosphorylation of protein bound phosphorylthreonine and phosphorylserine residues by cytosol and mitochondrial “casein phosphatases”. Biochem. Biophys. Res. Commun. 1976, 70, 1308–1315. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; Yu, T.; Yang, H.; Virshup, D.M.; Kops, G.J.; Lee, S.H.; Zhou, W.; Li, X.; Xu, W.; et al. Crystal structure of a PP2A B56-BubR1 complex and its implications for PP2A substrate recruitment and localization. Protein Cell 2016, 7, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Bajaj, R.; Bollen, M.; Peti, W.; Page, R. Expanding the PP2A Interactome by Defining a B56-Specific SLiM. Structure 2016, 24, 2174–2181. [Google Scholar] [CrossRef] [Green Version]
- Kruse, T.; Zhang, G.; Larsen, M.S.; Lischetti, T.; Streicher, W.; Kragh Nielsen, T.; Bjorn, S.P.; Nilsson, J. Direct binding between BubR1 and B56-PP2A phosphatase complexes regulate mitotic progression. J. Cell Sci. 2013, 126, 1086–1092. [Google Scholar] [CrossRef]
- Hertz, E.P.T.; Kruse, T.; Davey, N.E.; Lopez-Mendez, B.; Sigurethsson, J.O.; Montoya, G.; Olsen, J.V.; Nilsson, J. A Conserved Motif Provides Binding Specificity to the PP2A-B56 Phosphatase. Mol. Cell 2016, 63, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wisniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef]
- Touati, S.A.; Kataria, M.; Jones, A.W.; Snijders, A.P.; Uhlmann, F. Phosphoproteome dynamics during mitotic exit in budding yeast. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Ciehanover, A.; Hod, Y.; Hershko, A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 1978, 81, 1100–1105. [Google Scholar] [CrossRef]
- Ciechanover, A.; Finley, D.; Varshavsky, A. Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85. Cell 1984, 37, 57–66. [Google Scholar] [CrossRef]
- Glotzer, M.; Murray, A.W.; Kirschner, M.W. Cyclin is degraded by the ubiquitin pathway. Nature 1991, 349, 132–138. [Google Scholar] [CrossRef]
- Holloway, S.L.; Glotzer, M.; King, R.W.; Murray, A.W. Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell 1993, 73, 1393–1402. [Google Scholar] [CrossRef]
- Funabiki, H.; Yamano, H.; Kumada, K.; Nagao, K.; Hunt, T.; Yanagida, M. Cut2 proteolysis required for sister-chromatid seperation in fission yeast. Nature 1996, 381, 438–441. [Google Scholar] [CrossRef]
- Cohen-Fix, O.; Peters, J.M.; Kirschner, M.W.; Koshland, D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996, 10, 3081–3093. [Google Scholar] [CrossRef]
- Uhlmann, F.; Lottspeich, F.; Nasmyth, K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 1999, 400, 37–42. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Vlasschaert, C.; Cook, D.; Xia, X.; Gray, D.A. The evolution and functional diversification of the deubiquitinating enzyme superfamily. Genome Biol. Evol. 2017, 9, 558–573. [Google Scholar] [CrossRef]
- Sivakumar, S.; Gorbsky, G.J. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 2015, 16, 82–94. [Google Scholar] [CrossRef] [Green Version]
- Sudakin, V.; Ganoth, D.; Dahan, A.; Heller, H.; Hershko, J.; Luca, F.C.; Ruderman, J.V.; Hershko, A. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell 1995, 6, 185–197. [Google Scholar] [CrossRef]
- King, R.W.; Peters, J.-M.; Tugendreich, S.; Rolfe, M.; Hieter, P.; Kirschner, M.W. A 20s complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 1995, 81, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Irniger, S.; Piatti, S.; Michaelis, C.; Nasmyth, K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell 1995, 81, 269–278. [Google Scholar] [CrossRef]
- Schwab, M.; Lutum, A.S.; Seufert, W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 1997, 90, 683–693. [Google Scholar] [CrossRef]
- Visintin, R.; Prinz, S.; Amon, A. CDC20 and CDH1: A Family of Substrate-Specific Activators of APC-Dependent Proteolysis. Science 1997, 278, 460–463. [Google Scholar] [CrossRef]
- Kraft, C.; Vodermaier, H.C.; Maurer-Stroh, S.; Eisenhaber, F.; Peters, J.M. The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol. Cell 2005, 18, 543–553. [Google Scholar] [CrossRef]
- Cooper, K.F.; Mallory, M.J.; Egeland, D.B.; Jarnik, M.; Strich, R. Ama1p is a meiosis-specific regulator of the anaphase promoting complex/cyclosome in yeast. Proc. Natl. Acad. Sci. USA 2000, 97, 14548–14553. [Google Scholar] [CrossRef] [Green Version]
- Blanco, M.A.; Pelloquin, L.; Moreno, S. Fission yeast mfr1 activates APC and coordinates meiotic nuclear division with sporulation. J. Cell Sci. 2001, 114, 2135–2143. [Google Scholar]
- Kimata, Y.; Kitamura, K.; Fenner, N.; Yamano, H. Mes1 controls the meiosis I to meiosis II transition by distinctly regulating the anaphase-promoting complex/cyclosome coactivators Fzr1/Mfr1 and Slp1 in fission yeast. Mol. Biol. Cell 2011, 22, 1486–1494. [Google Scholar] [CrossRef]
- Page, A.W.; Orr-Weaver, T.L. The Drosophila genes grauzone and cortex are necessary for proper female meiosis. J. Cell Sci. 1996, 109 Pt 7, 1707–1715. [Google Scholar]
- Van Voorhis, V.A.; Morgan, D.O. Activation of the APC/C ubiquitin ligase by enhanced E2 efficiency. Curr. Biol. 2014, 24, 1556–1562. [Google Scholar] [CrossRef]
- Kimata, Y.; Baxter, J.E.; Fry, A.M.; Yamano, H. A role for the Fizzy/Cdc20 family of proteins in activation of the APC/C distinct from substrate recruitment. Mol. Cell 2008, 32, 576–583. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Culotti, J.; Reid, B. Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc. Natl. Acad. Sci. USA 1970, 66, 352–359. [Google Scholar] [CrossRef]
- Pfleger, C.M.; Kirschner, M.W. The KEN box: An APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 2000, 14, 655–665. [Google Scholar]
- Di Fiore, B.; Davey, N.E.; Hagting, A.; Izawa, D.; Mansfeld, J.; Gibson, T.J.; Pines, J. The ABBA motif binds APC/C activators and is shared by APC/C substrates and regulators. Dev. Cell 2015, 32, 358–372. [Google Scholar] [CrossRef]
- Diaz-Martinez, L.A.; Tian, W.; Li, B.; Warrington, R.; Jia, L.; Brautigam, C.A.; Luo, X.; Yu, H. The Cdc20-binding Phe box of the spindle checkpoint protein BubR1 maintains the mitotic checkpoint complex during mitosis. J. Biol. Chem. 2015, 290, 2431–2443. [Google Scholar] [CrossRef]
- Davey, N.E.; Morgan, D.O. Building a Regulatory Network with Short Linear Sequence Motifs: Lessons from the Degrons of the Anaphase-Promoting Complex. Mol. Cell 2016, 64, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.F.; Zhang, Z.; Yang, J.; McLaughlin, S.H.; Barford, D. Molecular architecture and mechanism of the anaphase-promoting complex. Nature 2014, 513, 388–393. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Zhang, Z.; Yang, J.; McLaughlin, S.H.; Barford, D. Atomic structure of the APC/C and its mechanism of protein ubiquitination. Nature 2015, 522, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, M.; VanderLinden, R.; Weissmann, F.; Qiao, R.; Dube, P.; Brown, N.G.; Haselbach, D.; Zhang, W.; Sidhu, S.S.; Peters, J.M.; et al. Cryo EM of Mitotic Checkpoint Complex-bound APC/C reveals reciprocal and conformational regulation of ubiquitin ligation. Mol. Cell 2016, 63, 593–607. [Google Scholar] [CrossRef]
- Frye, J.J.; Brown, N.G.; Petzold, G.; Watson, E.R.; Grace, C.R.; Nourse, A.; Jarvis, M.A.; Kriwacki, R.W.; Peters, J.M.; Stark, H.; et al. Electron microscopy structure of human APC/C(CDH1)-EMI1 reveals multimodal mechanism of E3 ligase shutdown. Nat. Struct. Mol. Biol. 2013, 20, 827–835. [Google Scholar] [CrossRef]
- Brown, N.G.; VanderLinden, R.; Watson, E.R.; Weissmann, F.; Ordureau, A.; Wu, K.P.; Zhang, W.; Yu, S.; Mercredi, P.Y.; Harrison, J.S.; et al. Dual RING E3 Architectures Regulate Multiubiquitination and Ubiquitin Chain Elongation by APC/C. Cell 2016, 165, 1440–1453. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, C.; Chang, L.; Zhang, Z.; Yang, J.; Maslen, S.; Skehel, M.; Barford, D. Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 2016, 536, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Min, M.; Mayor, U.; Dittmar, G.; Lindon, C. Using in vivo biotinylated ubiquitin to describe a mitotic exit ubiquitome from human cells. Mol. Cell. Proteom.: Mcp 2014, 13, 2411–2425. [Google Scholar] [CrossRef]
- Hayes, M.J.; Kimata, Y.; Wattam, S.L.; Lindon, C.; Mao, G.; Yamano, H.; Fry, A.M. Early mitotic degradation of Nek2A depends on Cdc20-independent interaction with the APC/C. Nat. Cell Biol. 2006, 8, 607–614. [Google Scholar] [CrossRef]
- Di Fiore, B.; Pines, J. How cyclin A destruction escapes the spindle assembly checkpoint. J. Cell Biol. 2010, 190, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.; Morgan, D.O. Finishing mitosis, one step at a time. Nat. Rev. Mol. Cell Biol. 2007, 8, 894–903. [Google Scholar] [CrossRef]
- Peters, J.M. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006, 7, 644–656. [Google Scholar] [CrossRef]
- Primorac, I.; Musacchio, A. Panta rhei: The APC/C at steady state. J. Cell Biol. 2013, 201, 177–189. [Google Scholar] [CrossRef]
- Pines, J. Cubism and the cell cycle: The many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 2011, 12, 427–438. [Google Scholar] [CrossRef]
- López-Avilés, S.; Kapuy, O.; Novák, B.; Uhlmann, F. Irreversibility of mitotic exit is the consequence of systems-level feedback. Nature 2009, 459, 592–595. [Google Scholar] [CrossRef] [Green Version]
- Potapova, T.A.; Sivakumar, S.; Flynn, J.N.; Li, R.; Gorbsky, G.J. Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited. Mol. Biol. Cell 2011, 22, 1191–1206. [Google Scholar] [CrossRef]
- Saurin, A.T. Kinase and Phosphatase Cross-Talk at the Kinetochore. Front. Cell Dev. Biol. 2018, 6, 62. [Google Scholar] [CrossRef] [Green Version]
- Manic, G.; Corradi, F.; Sistigu, A.; Siteni, S.; Vitale, I. Molecular Regulation of the Spindle Assembly Checkpoint by Kinases and Phosphatases. Int. Rev. Cell Mol. Biol. 2017, 328, 105–161. [Google Scholar] [CrossRef]
- Joglekar, A.P.; Kukreja, A.A. How Kinetochore Architecture Shapes the Mechanisms of Its Function. Curr. Biol. 2017, 27, R816–R824. [Google Scholar] [CrossRef]
- Gelens, L.; Qian, J.; Bollen, M.; Saurin, A.T. The Importance of Kinase-Phosphatase Integration: Lessons from Mitosis. Trends Cell Biol. 2018, 28, 6–21. [Google Scholar] [CrossRef]
- Musacchio, A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. 2015, 25, R1002–R1018. [Google Scholar] [CrossRef] [Green Version]
- Atherton-Fessler, S.; Parker, L.L.; Geahlen, R.L.; Piwnica-Worms, H. Mechanisms of p34cdc2 regulation. Mol. Cell. Biol. 1993, 13, 1675–1685. [Google Scholar] [CrossRef]
- Solomon, M.J.; Glotzer, M.; Lee, T.H.; Philippe, M.; Kirschner, M.W. Cyclin activation of p34cdc2. Cell 1990, 63, 1013–1024. [Google Scholar] [CrossRef]
- Kinoshita, N.; Yamano, H.; Niwa, H.; Yoshida, T.; Yanagida, M. Negative regulation of mitosis by the fission yeast protein phosphatase ppa2. Genes Dev. 1993, 7, 1059–1071. [Google Scholar] [CrossRef]
- Felix, M.A.; Cohen, P.; Karsenti, E. Cdc2 H1 kinase is negatively regulated by a type 2A phosphatase in the Xenopus early embryonic cell cycle: Evidence from the effects of okadaic acid. EMBO J. 1990, 9, 675–683. [Google Scholar] [CrossRef]
- Clarke, P.R.; Hoffmann, I.; Draetta, G.; Karsenti, E. Dephosphorylation of cdc25-C by a type-2A protein phosphatase: Specific regulation during the cell cycle in Xenopus egg extracts. Mol. Biol. Cell 1993, 4, 397–411. [Google Scholar] [CrossRef]
- Mochida, S.; Rata, S.; Hino, H.; Nagai, T.; Novák, B. Two Bistable Switches Govern M Phase Entry. Curr. Biol. 2016, 26, 3361–3367. [Google Scholar] [CrossRef] [Green Version]
- Rata, S.; Suarez Peredo Rodriguez, M.F.; Joseph, S.; Peter, N.; Echegaray Iturra, F.; Yang, F.; Madzvamuse, A.; Ruppert, J.G.; Samejima, K.; Platani, M.; et al. Two Interlinked Bistable Switches Govern Mitotic Control in Mammalian Cells. Curr. Biol. 2018. [Google Scholar] [CrossRef]
- Vigneron, S.; Sundermann, L.; Labbe, J.C.; Pintard, L.; Radulescu, O.; Castro, A.; Lorca, T. Cyclin A-cdk1-Dependent Phosphorylation of Bora Is the Triggering Factor Promoting Mitotic Entry. Dev. Cell 2018, 45, 637–650.e7. [Google Scholar] [CrossRef]
- Gavet, O.; Pines, J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef]
- Santos, S.D.M.; Wollman, R.; Meyer, T.; Ferrell, J.E., Jr. Spatial positive feedback at the onset of mitosis. Cell 2012, 149, 1500–1513. [Google Scholar] [CrossRef]
- Hagting, A.; Jackman, M.; Simpson, K.; Pines, J. Translocation of cyclin B1 to the nucleus at prophase requires a phosphorylation-dependent nuclear import signal. Curr. Biol. 1999, 9, 680–689. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Meyer, A.N.; Donoghue, D.J. Nuclear localization of cyclin B1 mediates its biological activity and is regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 1997, 94, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 checkpoint control: Regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef]
- Kumagai, A.; Yakowec, P.S.; Dunphy, W.G. 14-3-3 proteins act as negative regulators of the mitotic inducer Cdc25 in Xenopus egg extracts. Mol. Biol. Cell 1998, 9, 345–354. [Google Scholar] [CrossRef]
- Margolis, S.S.; Walsh, S.; Weiser, D.C.; Yoshida, M.; Shenolikar, S.; Kornbluth, S. PP1 control of M phase entry exerted through 14-3-3-regulated Cdc25 dephosphorylation. EMBO J. 2003, 22, 5734–5745. [Google Scholar] [CrossRef] [Green Version]
- Margolis, S.S.; Perry, J.A.; Weitzel, D.H.; Freel, C.D.; Yoshida, M.; Haystead, T.A.; Kornbluth, S. A role for PP1 in the Cdc2/Cyclin B-mediated positive feedback activation of Cdc25. Mol. Biol. Cell 2006, 17, 1779–1789. [Google Scholar] [CrossRef]
- Donzelli, M.; Squatrito, M.; Ganoth, D.; Hershko, A.; Pagano, M.; Draetta, G.F. Dual mode of degradation of Cdc25 A phosphatase. EMBO J. 2002, 21, 4875–4884. [Google Scholar] [CrossRef] [Green Version]
- Vigneron, S.; Gharbi-Ayachi, A.; Raymond, A.-A.; Burgess, A.; Labbé, J.-C.; Labesse, G.; Monsarrat, B.; Lorca, T.; Castro, A. Characterization of the Mechanisms Controlling Greatwall Activity. Mol. Cell. Biol. 2011, 31, 2262. [Google Scholar] [CrossRef]
- Blake-Hodek, K.A.; Williams, B.C.; Zhao, Y.; Castilho, P.V.; Chen, W.; Mao, Y.; Yamamoto, T.M.; Goldberg, M.L. Determinants for Activation of the Atypical AGC Kinase Greatwall during M Phase Entry. Mol. Cell. Biol. 2012, 32, 1337–1353. [Google Scholar] [CrossRef] [Green Version]
- Mochida, S.; Maslen, S.L.; Skehel, M.; Hunt, T. Greatwall Phosphorylates an Inhibitor of Protein Phosphatase 2Α That Is Essential for Mitosis. Science 2010, 330, 1670. [Google Scholar] [CrossRef]
- Gharbi-Ayachi, A.; Labbé, J.-C.; Burgess, A.; Vigneron, S.; Strub, J.-M.; Brioudes, E.; Van-Dorsselaer, A.; Castro, A.; Lorca, T. The Substrate of Greatwall Kinase, Arpp19, Controls Mitosis by Inhibiting Protein Phosphatase 2A. Science 2010, 330, 1673. [Google Scholar] [CrossRef]
- Mochida, S. Regulation of α–endosulfine, an inhibitor of protein phosphatase 2A, by multisite phosphorylation. FEBS J. 2014, 281, 1159–1169. [Google Scholar] [CrossRef]
- Williams, B.C.; Filter, J.J.; Blake-Hodek, K.A.; Wadzinski, B.E.; Fuda, N.J.; Shalloway, D.; Goldberg, M.L. Greatwall-phosphorylated Endosulfine is both an inhibitor and a substrate of PP2A-B55 heterotrimers. eLife 2014, 3, e01695. [Google Scholar] [CrossRef]
- Schleicher, K.; Porter, M.; Ten Have, S.; Sundaramoorthy, R.; Porter, I.M.; Swedlow, J.R. The Ndc80 complex targets Bod1 to human mitotic kinetochores. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Porter, I.M.; Schleicher, K.; Porter, M.; Swedlow, J.R. Bod1 regulates protein phosphatase 2A at mitotic kinetochores. Nat. Commun. 2013, 4, 2677. [Google Scholar] [CrossRef] [Green Version]
- Longin, S.; Zwaenepoel, K.; Louis, J.V.; Dilworth, S.; Goris, J.; Janssens, V. Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic Subunit. J. Biol. Chem. 2007, 282, 26971–26980. [Google Scholar] [CrossRef]
- Jativa, S.; Calabria, I.; Moyano-Rodriguez, Y.; Garcia, P.; Queralt, E. Cdc14 activation requires coordinated Cdk1-dependent phosphorylation of Net1 and PP2A-Cdc55 at anaphase onset. Cell Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Schmitz, M.H.A.; Held, M.; Janssens, V.; Hutchins, J.R.A.; Hudecz, O.; Ivanova, E.; Goris, J.; Trinkle-Mulcahy, L.; Lamond, A.I.; Poser, I.; et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat. Cell Biol. 2010, 12, 886–893. [Google Scholar] [CrossRef]
- Kwon, Y.G.; Lee, S.Y.; Choi, Y.; Greengard, P.; Nairn, A.C. Cell cycle-dependent phosphorylation of mammalian protein phosphatase 1 by cdc2 kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 2168–2173. [Google Scholar] [CrossRef] [Green Version]
- Yamano, H.; Ishii, K.; Yanagida, M. Phosphorylation of dis2 protein phosphatase at the C-terminal cdc2 consensus and its potential role in cell cycle regulation. EMBO J. 1994, 13, 5310–5318. [Google Scholar] [CrossRef]
- Dohadwala, M.; da Cruz e Silva, E.F.; Hall, F.L.; Williams, R.T.; Carbonaro-Hall, D.A.; Nairn, A.C.; Greengard, P.; Berndt, N. Phosphorylation and inactivation of protein phosphatase 1 by cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA 1994, 91, 6408–6412. [Google Scholar] [CrossRef]
- Wu, J.Q.; Guo, J.Y.; Tang, W.; Yang, C.-S.; Freel, C.D.; Chen, C.; Nairn, A.C.; Kornbluth, S. PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat. Cell Biol. 2009, 11, 644–651. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P. The discovery of protein phosphatases: From chaos and confusion to an understanding of their role in cell regulation and human disease. Bioessays 1994, 16, 583–588. [Google Scholar] [CrossRef]
- Miller, M.L.; Jensen, L.J.; Diella, F.; Jørgensen, C.; Tinti, M.; Li, L.; Hsiung, M.; Parker, S.A.; Bordeaux, J.; Sicheritz-Ponten, T.; et al. Linear Motif Atlas for Phosphorylation-Dependent Signaling. Sci. Signal. 2008, 1, ra2. [Google Scholar] [CrossRef]
- Nasa, I.; Rusin, S.F.; Kettenbach, A.N.; Moorhead, G.B. Aurora B opposes PP1 function in mitosis by phosphorylating the conserved PP1-binding RVxF motif in PP1 regulatory proteins. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Zeng, K.; Bastos, R.N.; Barr, F.A.; Gruneberg, U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J. Cell Biol. 2010, 191, 1315–1332. [Google Scholar] [CrossRef] [Green Version]
- Kettenbach, A.N.; Schlosser, K.A.; Lyons, S.P.; Nasa, I.; Gui, J.; Adamo, M.E.; Gerber, S.A. Global assessment of its network dynamics reveals that the kinase Plk1 inhibits the phosphatase PP6 to promote Aurora A activity. Sci. Signal. 2018, 11, eaaq1441. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.T.; Philp, A.; Vazquez-Martin, C. Protein phosphatase 4--from obscurity to vital functions. FEBS Lett. 2005, 579, 3278–3286. [Google Scholar] [CrossRef]
- Lee, D.-H.; Acharya, S.S.; Kwon, M.; Drane, P.; Guan, Y.; Adelmant, G.; Kalev, P.; Shah, J.; Pellman, D.; Marto, J.A.; et al. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol. Cell 2014, 54, 512–525. [Google Scholar] [CrossRef]
- Lipinszki, Z.; Lefevre, S.; Savoian, M.S.; Singleton, M.R.; Glover, D.M.; Przewloka, M.R. Centromeric binding and activity of Protein Phosphatase 4. Nat. Commun. 2015, 6, 5894. [Google Scholar] [CrossRef]
- Helps, N.R.; Brewis, N.D.; Lineruth, K.; Davis, T.; Kaiser, K.; Cohen, P.T. Protein phosphatase 4 is an essential enzyme required for organisation of microtubules at centrosomes in Drosophila embryos. J. Cell Sci. 1998, 111 Pt 10, 1331–1340. [Google Scholar]
- Sumiyoshi, E.; Sugimoto, A.; Yamamoto, M. Protein phosphatase 4 is required for centrosome maturation in mitosis and sperm meiosis in C. elegans. J. Cell Sci. 2002, 115, 1403–1410. [Google Scholar]
- Voss, M.; Campbell, K.; Saranzewa, N.; Campbell, D.G.; Hastie, C.J.; Peggie, M.W.; Martin-Granados, C.; Prescott, A.R.; Cohen, P.T. Protein phosphatase 4 is phosphorylated and inactivated by Cdk in response to spindle toxins and interacts with gamma-tubulin. Cell Cycle 2013, 12, 2876–2887. [Google Scholar] [CrossRef]
- Kim, T.; Lara-Gonzalez, P.; Prevo, B.; Meitinger, F.; Cheerambathur, D.K.; Oegema, K.; Desai, A. Kinetochores accelerate or delay APC/C activation by directing Cdc20 to opposing fates. Genes Dev. 2017, 31, 1089–1094. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Rodriguez-Bravo, V.; Kim, H.; Datta, S.; Foley, E.A. The PP2AB56 phosphatase promotes the association of Cdc20 with APC/C in mitosis. J. Cell Sci. 2017, 130, 1760. [Google Scholar] [CrossRef]
- Labit, H.; Fujimitsu, K.; Bayin, N.S.; Takaki, T.; Gannon, J.; Yamano, H. Dephosphorylation of Cdc20 is required for its C-box-dependent activation of the APC/C. EMBO J. 2012, 31, 3351–3362. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Craney, A.; Rape, M. Microtubule-dependent regulation of mitotic protein degradation. Mol. Cell 2014, 53, 179–192. [Google Scholar] [CrossRef]
- Magidson, V.; O’Connell, C.B.; Lončarek, J.; Paul, R.; Mogilner, A.; Khodjakov, A. The spatial arrangement of chromosomes during prometaphase facilitates spindle assembly. Cell 2011, 146, 555–567. [Google Scholar] [CrossRef]
- Tanaka, K.; Mukae, N.; Dewar, H.; van Breugel, M.; James, E.K.; Prescott, A.R.; Antony, C.; Tanaka, T.U. Molecular mechanisms of kinetochore capture by spindle microtubules. Nature 2005, 434, 987–994. [Google Scholar] [CrossRef]
- Foley, E.A.; Kapoor, T.M. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat. Rev. Mol. Cell Biol. 2013, 14, 25–37. [Google Scholar] [CrossRef]
- Kelly, A.E.; Ghenoiu, C.; Xue, J.Z.; Zierhut, C.; Kimura, H.; Funabiki, H. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science 2010, 330, 235–239. [Google Scholar] [CrossRef]
- Kawashima, S.A.; Yamagishi, Y.; Honda, T.; Ishiguro, K.-i.; Watanabe, Y. Phosphorylation of H2A by Bub1 Prevents Chromosomal Instability Through Localizing Shugoshin. Science 2010, 327, 172. [Google Scholar] [CrossRef]
- Hindriksen, S.; Lens, S.M.A.; Hadders, M.A. The Ins and Outs of Aurora B Inner Centromere Localization. Front. Cell Dev. Biol. 2017, 5, 112. [Google Scholar] [CrossRef] [Green Version]
- DeLuca, J.G.; Gall, W.E.; Ciferri, C.; Cimini, D.; Musacchio, A.; Salmon, E.D. Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 2006, 127, 969–982. [Google Scholar] [CrossRef]
- Schmidt, J.C.; Arthanari, H.; Boeszoermenyi, A.; Dashkevich, N.M.; Wilson-Kubalek, E.M.; Monnier, N.; Markus, M.; Oberer, M.; Milligan, R.A.; Bathe, M.; et al. The kinetochore-bound Ska1 complex tracks depolymerizing microtubules and binds to curved protofilaments. Dev. Cell 2012, 23, 968–980. [Google Scholar] [CrossRef]
- Chan, Y.W.; Jeyaprakash, A.A.; Nigg, E.A.; Santamaria, A. Aurora B controls kinetochore-microtubule attachments by inhibiting Ska complex-KMN network interaction. J. Cell Biol. 2012, 196, 563–571. [Google Scholar] [CrossRef]
- Foley, E.A.; Maldonado, M.; Kapoor, T.M. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat. Cell Biol. 2011, 13, 1265–1271. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Vleugel, M.; Backer, C.B.; Hori, T.; Fukagawa, T.; Cheeseman, I.M.; Lampson, M.A. Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J. Cell Biol. 2010, 188, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Holland, A.J.; Lan, W.; Cleveland, D.W. Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell 2010, 142, 444–455. [Google Scholar] [CrossRef]
- Sivakumar, S.; Janczyk, P.L.; Qu, Q.; Brautigam, C.A.; Stukenberg, P.T.; Yu, H.; Gorbsky, G.J. The human SKA complex drives the metaphase-anaphase cell cycle transition by recruiting protein phosphatase 1 to kinetochores. eLife 2016, 5. [Google Scholar] [CrossRef]
- Kabeche, L.; Compton, D.A. Cyclin A regulates kinetochore microtubules to promote faithful chromosome segregation. Nature 2013, 502, 110–113. [Google Scholar] [CrossRef] [Green Version]
- Wolthuis, R.; Clay-Farrace, L.; van Zon, W.; Yekezare, M.; Koop, L.; Ogink, J.; Medema, R.; Pines, J. Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol. Cell 2008, 30, 290–302. [Google Scholar] [CrossRef]
- Wan, X.; O’Quinn, R.P.; Pierce, H.L.; Joglekar, A.P.; Gall, W.E.; DeLuca, J.G.; Carroll, C.W.; Liu, S.-T.; Yen, T.J.; McEwen, B.F.; et al. Protein Architecture of the Human Kinetochore Microtubule Attachment Site. Cell 2009, 137, 672–684. [Google Scholar] [CrossRef] [Green Version]
- Musacchio, A.; Desai, A. A Molecular View of Kinetochore Assembly and Function. Biol. (Basel) 2017, 6, 5. [Google Scholar] [CrossRef]
- Choi, E.; Choe, H.; Min, J.; Choi, J.Y.; Kim, J.; Lee, H. BubR1 acetylation at prometaphase is required for modulating APC/C activity and timing of mitosis. EMBO J. 2009, 28, 2077–2089. [Google Scholar] [CrossRef] [Green Version]
- King, E.M.; van der Sar, S.J.; Hardwick, K.G. Mad3 KEN boxes mediate both Cdc20 and Mad3 turnover, and are critical for the spindle checkpoint. PLoS ONE 2007, 2, e342. [Google Scholar] [CrossRef]
- Vagnarelli, P.; Ribeiro, S.; Sennels, L.; Sanchez-Pulido, L.; de Lima Alves, F.; Verheyen, T.; Kelly, D.A.; Ponting, C.P.; Rappsilber, J.; Earnshaw, W.C. Repo-Man coordinates chromosomal reorganization with nuclear envelope reassembly during mitotic exit. Dev. Cell 2011, 21, 328–342. [Google Scholar] [CrossRef]
- Qian, J.; Lesage, B.; Beullens, M.; Van Eynde, A.; Bollen, M. PP1/Repo-man dephosphorylates mitotic histone H3 at T3 and regulates chromosomal aurora B targeting. Curr. Biol. 2011, 21, 766–773. [Google Scholar] [CrossRef]
- Mirchenko, L.; Uhlmann, F. Sli15(INCENP) dephosphorylation prevents mitotic checkpoint reengagement due to loss of tension at anaphase onset. Curr. Biol. 2010, 20, 1396–1401. [Google Scholar] [CrossRef]
- Luo, X.; Tang, Z.; Rizo, J.; Yu, H. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol. Cell 2002, 9, 59–71. [Google Scholar] [CrossRef]
- Faesen, A.C.; Thanasoula, M.; Maffini, S.; Breit, C.; Müller, F.; van Gerwen, S.; Bange, T.; Musacchio, A. Basis of catalytic assembly of the mitotic checkpoint complex. Nature 2017, 542, 498–502. [Google Scholar] [CrossRef] [Green Version]
- De Antoni, A.; Pearson, C.G.; Cimini, D.; Canman, J.C.; Sala, V.; Nezi, L.; Mapelli, M.; Sironi, L.; Faretta, M.; Salmon, E.D.; et al. The Mad1/Mad2 Complex as a Template for Mad2 Activation in the Spindle Assembly Checkpoint. Curr. Biol. 2005, 15, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Izawa, D.; Pines, J. The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature 2015, 517, 631–634. [Google Scholar] [CrossRef]
- Morin, V.; Prieto, S.; Melines, S.; Hem, S.; Rossignol, M.; Lorca, T.; Espeut, J.; Morin, N.; Abrieu, A. CDK-dependent potentiation of MPS1 kinase activity is essential to the mitotic checkpoint. Curr. Biol. 2012, 22, 289–295. [Google Scholar] [CrossRef]
- von Schubert, C.; Cubizolles, F.; Bracher, J.M.; Sliedrecht, T.; Kops, G.; Nigg, E.A. Plk1 and Mps1 Cooperatively Regulate the Spindle Assembly Checkpoint in Human Cells. Cell Rep. 2015, 12, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, J.; Yekezare, M.; Minshull, J.; Pines, J. The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat. Cell Biol. 2008, 10, 1411–1420. [Google Scholar] [CrossRef]
- Reddy, S.K.; Rape, M.; Margansky, W.A.; Kirschner, M.W. Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature 2007, 446, 921–925. [Google Scholar] [CrossRef]
- Stegmeier, F.; Rape, M.; Draviam, V.M.; Nalepa, G.; Sowa, M.E.; Ang, X.L.; McDonald, E.R., 3rd; Li, M.Z.; Hannon, G.J.; Sorger, P.K.; et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature 2007, 446, 876–881. [Google Scholar] [CrossRef]
- Yang, M.; Li, B.; Tomchick, D.R.; Machius, M.; Rizo, J.; Yu, H.; Luo, X. p31comet blocks Mad2 activation through structural mimicry. Cell 2007, 131, 744–755. [Google Scholar] [CrossRef]
- Habu, T.; Kim, S.H.; Weinstein, J.; Matsumoto, T. Identification of a MAD2-binding protein, CMT2, and its role in mitosis. EMBO J. 2002, 21, 6419–6428. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, C.; Chang, L.; Barford, D. Mechanism for remodelling of the cell cycle checkpoint protein MAD2 by the ATPase TRIP13. Nature 2018, 559, 274–278. [Google Scholar] [CrossRef]
- Mo, M.; Arnaoutov, A.; Dasso, M. Phosphorylation of Xenopus p31comet potentiates mitotic checkpoint exit. Cell Cycle 2015, 14, 3978–3985. [Google Scholar] [CrossRef]
- Date, D.A.; Burrows, A.C.; Summers, M.K. Phosphorylation regulates the p31Comet-mitotic arrest-deficient 2 (Mad2) interaction to promote spindle assembly checkpoint (SAC) activity. J. Biol. Chem. 2014, 289, 11367–11373. [Google Scholar] [CrossRef]
- Kaisari, S.; Shomer, P.; Ziv, T.; Sitry-Shevah, D.; Miniowitz-Shemtov, S.; Teichner, A.; Hershko, A. Role of Polo-like kinase 1 in the regulation of the action of p31(comet) in the disassembly of mitotic checkpoint complexes. Proc. Natl. Acad. Sci. USA 2019, 116, 11725–11730. [Google Scholar] [CrossRef]
- Hiruma, Y.; Sacristan, C.; Pachis, S.T.; Adamopoulos, A.; Kuijt, T.; Ubbink, M.; von Castelmur, E.; Perrakis, A.; Kops, G.J. CELL DIVISION CYCLE. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science 2015, 348, 1264–1267. [Google Scholar] [CrossRef]
- Qian, J.; Garcia-Gimeno, M.A.; Beullens, M.; Manzione, M.G.; Van der Hoeven, G.; Igual, J.C.; Heredia, M.; Sanz, P.; Gelens, L.; Bollen, M. An Attachment-Independent Biochemical Timer of the Spindle Assembly Checkpoint. Mol. Cell 2017, 68, 715–730.e5. [Google Scholar] [CrossRef]
- Espert, A.; Uluocak, P.; Bastos, R.N.; Mangat, D.; Graab, P.; Gruneberg, U. PP2A-B56 opposes Mps1 phosphorylation of Knl1 and thereby promotes spindle assembly checkpoint silencing. J. Cell Biol. 2014, 206, 833–842. [Google Scholar] [CrossRef]
- Nijenhuis, W.; Vallardi, G.; Teixeira, A.; Kops, G.J.; Saurin, A.T. Negative feedback at kinetochores underlies a responsive spindle checkpoint signal. Nat. Cell Biol. 2014, 16, 1257–1264. [Google Scholar] [CrossRef] [Green Version]
- Clute, P.; Pines, J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol. 1999, 1, 82–87. [Google Scholar] [CrossRef]
- Uhlmann, F. SMC complexes: From DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 2016, 17, 399–412. [Google Scholar] [CrossRef]
- Hirano, T. Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell 2016, 164, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Shintomi, K.; Takahashi, T.S.; Hirano, T. Reconstitution of mitotic chromatids with a minimum set of purified factors. Nat. Cell Biol. 2015, 17, 1014–1023. [Google Scholar] [CrossRef]
- St-Pierre, J.; Douziech, M.; Bazile, F.; Pascariu, M.; Bonneil, E.; Sauve, V.; Ratsima, H.; D’Amours, D. Polo kinase regulates mitotic chromosome condensation by hyperactivation of condensin DNA supercoiling activity. Mol. Cell 2009, 34, 416–426. [Google Scholar] [CrossRef]
- Mora-Bermudez, F.; Gerlich, D.; Ellenberg, J. Maximal chromosome compaction occurs by axial shortening in anaphase and depends on Aurora kinase. Nat. Cell Biol. 2007, 9, 822–831. [Google Scholar] [CrossRef]
- Lipp, J.J.; Hirota, T.; Poser, I.; Peters, J.-M. Aurora B controls the association of condensin I but not condensin II with mitotic chromosomes. J. Cell Sci. 2007, 120, 1245. [Google Scholar] [CrossRef]
- Abe, S.; Nagasaka, K.; Hirayama, Y.; Kozuka-Hata, H.; Oyama, M.; Aoyagi, Y.; Obuse, C.; Hirota, T. The initial phase of chromosome condensation requires Cdk1-mediated phosphorylation of the CAP-D3 subunit of condensin II. Genes Dev. 2011, 25, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Hirano, M.; Kobayashi, R.; Hirano, T. Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science 1998, 282, 487–490. [Google Scholar] [CrossRef]
- Sutani, T.; Yuasa, T.; Tomonaga, T.; Dohmae, N.; Takio, K.; Yanagida, M. Fission yeast condensin complex: Essential roles of non-SMC subunits for condensation and Cdc2 phosphorylation of Cut3/SMC4. Genes Dev. 1999, 13, 2271–2283. [Google Scholar] [CrossRef]
- Kao, L.; Wang, Y.-T.; Chen, Y.-C.; Tseng, S.-F.; Jhang, J.-C.; Chen, Y.-J.; Teng, S.-C. Global analysis of cdc14 dephosphorylation sites reveals essential regulatory role in mitosis and cytokinesis. Mol. Cell. Proteom.: Mcp 2014, 13, 594–605. [Google Scholar] [CrossRef]
- Xing, H.; Vanderford, N.L.; Sarge, K.D. The TBP-PP2A mitotic complex bookmarks genes by preventing condensin action. Nat. Cell Biol. 2008, 10, 1318–1323. [Google Scholar] [CrossRef]
- Takemoto, A.; Kimura, K.; Yanagisawa, J.; Yokoyama, S.; Hanaoka, F. Negative regulation of condensin I by CK2-mediated phosphorylation. EMBO J. 2006, 25, 5339–5348. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, T.; Sykora, M.M.; Huis in ‘t Veld, P.J.; Mechtler, K.; Peters, J.-M. Aurora B and Cdk1 mediate Wapl activation and release of acetylated cohesin from chromosomes by phosphorylating Sororin. Proc. Natl. Acad. Sci. USA 2013, 110, 13404–13409. [Google Scholar] [CrossRef] [Green Version]
- Sumara, I.; Vorlaufer, E.; Stukenberg, P.T.; Kelm, O.; Redemann, N.; Nigg, E.A.; Peters, J.M. The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol. Cell 2002, 9, 515–525. [Google Scholar] [CrossRef]
- Liu, H.; Rankin, S.; Yu, H. Phosphorylation-enabled binding of SGO1-PP2A to cohesin protects sororin and centromeric cohesion during mitosis. Nat. Cell Biol. 2013, 15, 40–49. [Google Scholar] [CrossRef]
- Kitajima, T.S.; Kawashima, S.A.; Watanabe, Y. The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature 2004, 427, 510–517. [Google Scholar] [CrossRef]
- Xu, Z.; Cetin, B.; Anger, M.; Cho, U.S.; Helmhart, W.; Nasmyth, K.; Xu, W. Structure and function of the PP2A-shugoshin interaction. Mol. Cell 2009, 35, 426–441. [Google Scholar] [CrossRef]
- Qi, W.; Yu, H. KEN-box-dependent degradation of the Bub1 spindle checkpoint kinase by the anaphase-promoting complex/cyclosome. J. Biol. Chem. 2007, 282, 3672–3679. [Google Scholar] [CrossRef]
- Hellmuth, S.; Böttger, F.; Pan, C.; Mann, M.; Stemmann, O. PP2A delays APC/C-dependent degradation of separase-associated but not free securin. EMBO J. 2014, 33, 1134–1147. [Google Scholar] [CrossRef]
- Holland, A.J.; Böttger, F.; Stemmann, O.; Taylor, S.S. Protein Phosphatase 2A and Separase Form a Complex Regulated by Separase Autocleavage. J. Biol. Chem. 2007, 282, 24623–24632. [Google Scholar] [CrossRef] [Green Version]
- Shindo, N.; Kumada, K.; Hirota, T. Separase sensor reveals dual roles for separase coordinating cohesin cleavage and cdk1 inhibition. Dev. Cell 2012, 23, 112–123. [Google Scholar] [CrossRef]
- Holt, L.J.; Krutchinsky, A.N.; Morgan, D.O. Positive feedback sharpens the anaphase switch. Nature 2008, 454, 353–357. [Google Scholar] [CrossRef] [Green Version]
- Schwab, M.; Neutzner, M.; Mocker, D.; Seufert, W. Yeast Hct1 recognizes the mitotic cyclin Clb2 and other substrates of the ubiquitin ligase APC. EMBO J. 2001, 20, 5165–5175. [Google Scholar] [CrossRef] [Green Version]
- Vodermaier, H.C.; Gieffers, C.; Maurer-Stroh, S.; Eisenhaber, F.; Peters, J.M. TPR subunits of the anaphase-promoting complex mediate binding to the activator protein CDH1. Curr. Biol. 2003, 13, 1459–1468. [Google Scholar] [CrossRef]
- Rudner, A.D.; Murray, A.W. Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol. 2000, 149, 1377–1390. [Google Scholar] [CrossRef]
- Kramer, E.R.; Scheuringer, N.; Podtelejnikov, A.V.; Mann, M.; Peters, J.-M. Mitotic Regulation of the APC Activator Proteins CDC20 and CDH1. Mol. Biol. Cell 2000, 11, 1555–1569. [Google Scholar] [CrossRef] [Green Version]
- Shteinberg, M.; Protopopov, Y.; Listovsky, T.; Brandeis, M.; Hershko, A. Phosphorylation of the cyclosome is required for its stimulation by Fizzy/cdc20. Biochem. Biophys. Res. Commun. 1999, 260, 193–198. [Google Scholar] [CrossRef]
- Kraft, C.; Herzog, F.; Gieffers, C.; Mechtler, K.; Hagting, A.; Pines, J.; Peters, J.-M. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 2003, 22, 6598–6609. [Google Scholar] [CrossRef] [Green Version]
- Golan, A.; Yudkovsky, Y.; Hershko, A. The cyclin-ubiquitin ligase activity of cyclosome/APC is jointly activated by protein kinases Cdk1-cyclin B and Plk. J. Biol. Chem. 2002, 277, 15552–15557. [Google Scholar] [CrossRef]
- Fujimitsu, K.; Grimaldi, M.; Yamano, H. Cyclin-dependent kinase 1-dependent activation of APC/C ubiquitin ligase. Science 2016, 352, 1121–1124. [Google Scholar] [CrossRef]
- Zhang, S.; Chang, L.; Alfieri, C.; Zhang, Z.; Yang, J.; Maslen, S.; Skehel, M.; Barford, D. Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature 2016, 533, 260–264. [Google Scholar] [CrossRef] [Green Version]
- Qiao, R.; Weissmann, F.; Yamaguchi, M.; Brown, N.G.; VanderLinden, R.; Imre, R.; Jarvis, M.A.; Brunner, M.R.; Davidson, I.F.; Litos, G.; et al. Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2016, 113, E2570–E2578. [Google Scholar] [CrossRef]
- Shteinberg, M.; Hershko, A. Role of Suc1 in the activation of the cyclosome by protein kinase Cdk1/cyclin B. Biochem. Biophys. Res. Commun. 1999, 257, 12–18. [Google Scholar] [CrossRef]
- Patra, D.; Dunphy, W.G. Xe-p9, a Xenopus Suc1/Cks protein, is essential for the Cdc2-dependent phosphorylation of the anaphase- promoting complex at mitosis. Genes Dev. 1998, 12, 2549–2559. [Google Scholar] [CrossRef]
- McGrath, D.A.; Balog, E.R.M.; Kõivomägi, M.; Lucena, R.; Mai, M.V.; Hirschi, A.; Kellogg, D.R.; Loog, M.; Rubin, S.M. Cks confers specificity to phosphorylation-dependent CDK signaling pathways. Nat. Struct. Mol. Biol. 2013, 20, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Kõivomägi, M.; Ord, M.; Iofik, A.; Valk, E.; Venta, R.; Faustova, I.; Kivi, R.; Balog, E.R.M.; Rubin, S.M.; Loog, M. Multisite phosphorylation networks as signal processors for Cdk1. Nat. Struct. Mol. Biol. 2013, 20, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, C.; Zhang, S.; Barford, D. Visualizing the complex functions and mechanisms of the anaphase promoting complex/cyclosome (APC/C). Open Biol. 2017, 7. [Google Scholar] [CrossRef]
- Passmore, L.A.; Booth, C.R.; Venien-Bryan, C.; Ludtke, S.J.; Fioretto, C.; Johnson, L.N.; Chiu, W.; Barford, D. Structural analysis of the anaphase-promoting complex reveals multiple active sites and insights into polyubiquitylation. Mol. Cell 2005, 20, 855–866. [Google Scholar] [CrossRef]
- Ohi, M.D.; Feoktistova, A.; Ren, L.; Yip, C.; Cheng, Y.; Chen, J.S.; Yoon, H.J.; Wall, J.S.; Huang, Z.; Penczek, P.A.; et al. Structural organization of the anaphase-promoting complex bound to the mitotic activator Slp1. Mol. Cell 2007, 28, 871–885. [Google Scholar] [CrossRef]
- Schreiber, A.; Stengel, F.; Zhang, Z.; Enchev, R.I.; Kong, E.H.; Morris, E.P.; Robinson, C.V.; da Fonseca, P.C.; Barford, D. Structural basis for the subunit assembly of the anaphase-promoting complex. Nature 2011, 470, 227–232. [Google Scholar] [CrossRef]
- Yamada, H.; Kumada, K.; Yanagida, M. Distinct subunit functions and cell cycle regulated phosphorylation of 20S APC/cyclosome required for anaphase in fission yeast. J. Cell Sci. 1997, 110 Pt 15, 1793–1804. [Google Scholar]
- Sivakumar, S.; Daum, J.R.; Tipton, A.R.; Rankin, S.; Gorbsky, G.J. The spindle and kinetochore-associated (Ska) complex enhances binding of the anaphase-promoting complex/cyclosome (APC/C) to chromosomes and promotes mitotic exit. Mol. Biol. Cell 2014, 25, 594–605. [Google Scholar] [CrossRef]
- Lu, D.; Hsiao, J.Y.; Davey, N.E.; Van Voorhis, V.A.; Foster, S.A.; Tang, C.; Morgan, D.O. Multiple mechanisms determine the order of APC/C substrate degradation in mitosis. J. Cell Biol. 2014, 207, 23–39. [Google Scholar] [CrossRef] [Green Version]
- Floyd, S.; Pines, J.; Lindon, C. APC/C Cdh1 targets aurora kinase to control reorganization of the mitotic spindle at anaphase. Curr. Biol. 2008, 18, 1649–1658. [Google Scholar] [CrossRef]
- Zachariae, W.; Schwab, M.; Nasmyth, K.; Seufert, W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science 1998, 282, 1721–1724. [Google Scholar] [CrossRef]
- Visintin, R.; Craig, K.; Hwang, E.S.; Prinz, S.; Tyers, M.; Amon, A. The Phosphatase Cdc14 Triggers Mitotic Exit by Reversal of Cdk-Dependent Phosphorylation. Mol. Cell 1998, 2, 709–718. [Google Scholar] [CrossRef]
- Yudkovsky, Y.; Shteinberg, M.; Listovsky, T.; Brandeis, M.; Hershko, A. Phosphorylation of Cdc20/fizzy negatively regulates the mammalian cyclosome/APC in the mitotic checkpoint. Biochem. Biophys. Res. Commun. 2000, 271, 299–304. [Google Scholar] [CrossRef]
- Rogers, S.; McCloy, R.; Watkins, D.N.; Burgess, A. Mechanisms regulating phosphatase specificity and the removal of individual phosphorylation sites during mitotic exit. BioEssays 2016, 38, S24–S32. [Google Scholar] [CrossRef]
- Nilsson, J. Protein phosphatases in the regulation of mitosis. J. Cell Biol. 2019, 218, 395. [Google Scholar] [CrossRef]
- Craney, A.; Kelly, A.; Jia, L.; Fedrigo, I.; Yu, H.; Rape, M. Control of APC/C-dependent ubiquitin chain elongation by reversible phosphorylation. Proc. Natl. Acad. Sci. 2016, 113, 1540. [Google Scholar] [CrossRef]
- Lu, D.; Girard, J.R.; Li, W.; Mizrak, A.; Morgan, D.O. Quantitative framework for ordered degradation of APC/C substrates. BMC Biol. 2015, 13, 96. [Google Scholar] [CrossRef]
- He, J.; Chao, W.C.H.; Zhang, Z.; Yang, J.; Cronin, N.; Barford, D. Insights into degron recognition by APC/C coactivators from the structure of an Acm1-Cdh1 complex. Mol. Cell 2013, 50, 649–660. [Google Scholar] [CrossRef]
- Tian, W.; Li, B.; Warrington, R.; Tomchick, D.R.; Yu, H.; Luo, X. Structural analysis of human Cdc20 supports multisite degron recognition by APC/C. Proc. Natl. Acad. Sci. USA 2012, 109, 18419–18424. [Google Scholar] [CrossRef] [Green Version]
- Chao, W.C.; Kulkarni, K.; Zhang, Z.; Kong, E.H.; Barford, D. Structure of the mitotic checkpoint complex. Nature 2012, 484, 208–213. [Google Scholar] [CrossRef]
- Sedgwick, G.G.; Hayward, D.G.; Di Fiore, B.; Pardo, M.; Yu, L.; Pines, J.; Nilsson, J. Mechanisms controlling the temporal degradation of Nek2A and Kif18A by the APC/C-Cdc20 complex. EMBO J. 2013, 32, 303–314. [Google Scholar] [CrossRef]
- Mailand, N.; Diffley, J.F. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 2005, 122, 915–926. [Google Scholar] [CrossRef]
- Ma, S.; Vigneron, S.; Robert, P.; Strub, J.M.; Cianferani, S.; Castro, A.; Lorca, T. Greatwall dephosphorylation and inactivation upon mitotic exit is triggered by PP1. J. Cell Sci. 2016, 129, 1329–1339. [Google Scholar] [CrossRef]
- Heim, A.; Konietzny, A.; Mayer, T.U. Protein phosphatase 1 is essential for Greatwall inactivation at mitotic exit. EMBO Rep. 2015, 16, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Rogers, S.; Fey, D.; McCloy, R.A.; Parker, B.L.; Mitchell, N.J.; Payne, R.J.; Daly, R.J.; James, D.E.; Caldon, C.E.; Watkins, D.N.; et al. PP1 initiates the dephosphorylation of MASTL, triggering mitotic exit and bistability in human cells. J. Cell Sci. 2016, 129, 1340–1354. [Google Scholar] [CrossRef] [Green Version]
- Grallert, A.; Boke, E.; Hagting, A.; Hodgson, B.; Connolly, Y.; Griffiths, J.R.; Smith, D.L.; Pines, J.; Hagan, I.M. A PP1-PP2A phosphatase relay controls mitotic progression. Nature 2015, 517, 94–98. [Google Scholar] [CrossRef]
- De Wulf, P.; Montani, F.; Visintin, R. Protein phosphatases take the mitotic stage. Curr. Opin. Cell Biol. 2009, 21, 806–815. [Google Scholar] [CrossRef]
- Queralt, E.; Uhlmann, F. Cdk-counteracting phosphatases unlock mitotic exit. Curr. Opin. Cell Biol. 2008, 20, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Skoufias, D.A.; Indorato, R.-L.; Lacroix, F.; Panopoulos, A.; Margolis, R.L. Mitosis persists in the absence of Cdk1 activity when proteolysis or protein phosphatase activity is suppressed. J. Cell Biol. 2007, 179, 671–685. [Google Scholar] [CrossRef] [Green Version]
- Mochida, S.; Ikeo, S.; Gannon, J.; Hunt, T. Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 2009, 28, 2777–2785. [Google Scholar] [CrossRef]
- Cundell, M.J.; Bastos, R.N.; Zhang, T.; Holder, J.; Gruneberg, U.; Novak, B.; Barr, F.A. The BEG (PP2A-B55/ENSA/Greatwall) pathway ensures cytokinesis follows chromosome separation. Mol. Cell 2013, 52, 393–405. [Google Scholar] [CrossRef]
- Bastos, R.N.; Cundell, M.J.; Barr, F.A. KIF4A and PP2A–B56 form a spatially restricted feedback loop opposing Aurora B at the anaphase central spindle. J. Cell Biol. 2014, 207, 683. [Google Scholar] [CrossRef]
- Gil, R.S.; Vagnarelli, P. Protein phosphatases in chromatin structure and function. Biochim. Et Biophys. Acta. Mol. Cell Res. 2019, 1866, 90–101. [Google Scholar] [CrossRef]
- Bhowmick, R.; Thakur, R.S.; Venegas, A.B.; Liu, Y.; Nilsson, J.; Barisic, M.; Hickson, I.D. The RIF1-PP1 Axis Controls Abscission Timing in Human Cells. Curr. Biol. 2019, 29, 1232–1242.e5. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Beullens, M.; Huang, J.; De Munter, S.; Lesage, B.; Bollen, M. Cdk1 orders mitotic events through coordination of a chromosome-associated phosphatase switch. Nat. Commun. 2015, 6, 10215. [Google Scholar] [CrossRef]
- Landsverk, H.B.; Kirkhus, M.; Bollen, M.; Küntziger, T.; Collas, P. PNUTS enhances in vitro chromosome decondensation in a PP1-dependent manner. Biochem. J. 2005, 390, 709–717. [Google Scholar] [CrossRef]
- Wolf, F.; Wandke, C.; Isenberg, N.; Geley, S. Dose-dependent effects of stable cyclin B1 on progression through mitosis in human cells. EMBO J. 2006, 25, 2802–2813. [Google Scholar] [CrossRef]
- Stemmann, O.; Zou, H.; Gerber, S.A.; Gygi, S.P.; Kirschner, M.W. Dual inhibition of sister chromatid separation at metaphase. Cell 2001, 107, 715–726. [Google Scholar] [CrossRef]
- Bouchoux, C.; Uhlmann, F. A quantitative model for ordered Cdk substrate dephosphorylation during mitotic exit. Cell 2011, 147, 803–814. [Google Scholar] [CrossRef]
- Nakajima, H.; Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J. Biol. Chem. 2003, 278, 25277–25280. [Google Scholar] [CrossRef]
- Rusin, S.F.; Adamo, M.E.; Kettenbach, A.N. Identification of Candidate Casein Kinase 2 Substrates in Mitosis by Quantitative Phosphoproteomics. Front. Cell Dev. Biol. 2017, 5, 97. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef]
- Bremmer, S.C.; Hall, H.; Martinez, J.S.; Eissler, C.L.; Hinrichsen, T.H.; Rossie, S.; Parker, L.L.; Hall, M.C.; Charbonneau, H. Cdc14 phosphatases preferentially dephosphorylate a subset of cyclin-dependent kinase (Cdk) sites containing phosphoserine. J. Biol. Chem. 2012, 287, 1662–1669. [Google Scholar] [CrossRef]
- Eissler, C.L.; Mazon, G.; Powers, B.L.; Savinov, S.N.; Symington, L.S.; Hall, M.C. The Cdk/Cdc14 module controls activation of the Yen1 holliday junction resolvase to promote genome stability. Mol. Cell 2014, 54, 80–93. [Google Scholar] [CrossRef]
- Bassermann, F.; Frescas, D.; Guardavaccaro, D.; Busino, L.; Peschiaroli, A.; Pagano, M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 2008, 134, 256–267. [Google Scholar] [CrossRef]
- Vallardi, G.; Allan, L.A.; Crozier, L.; Saurin, A.T. Division of labour between PP2A-B56 isoforms at the centromere and kinetochore. eLife 2019, 8. [Google Scholar] [CrossRef]
- Kumar, G.S.; Gokhan, E.; De Munter, S.; Bollen, M.; Vagnarelli, P.; Peti, W.; Page, R. The Ki-67 and RepoMan mitotic phosphatases assemble via an identical, yet novel mechanism. eLife 2016, 5. [Google Scholar] [CrossRef]
- Wang, Q.; Moyret-Lalle, C.; Couzon, F.; Surbiguet-Clippe, C.; Saurin, J.-C.; Lorca, T.; Navarro, C.; Puisieux, A. Alterations of anaphase-promoting complex genes in human colon cancer cells. Oncogene 2003, 22, 1486–1490. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Daigo, Y.; Aragaki, M.; Ishikawa, K.; Sato, M.; Kaji, M. Overexpression of CDC20 predicts poor prognosis in primary non-small cell lung cancer patients. J. Surg. Oncol. 2012, 106, 423–430. [Google Scholar] [CrossRef]
- Wu, W.J.; Hu, K.S.; Wang, D.S.; Zeng, Z.L.; Zhang, D.S.; Chen, D.L.; Bai, L.; Xu, R.H. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J. Transl. Med. 2013, 11, 142. [Google Scholar] [CrossRef]
- Lub, S.; Maes, A.; Maes, K.; De Veirman, K.; De Bruyne, E.; Menu, E.; Fostier, K.; Kassambara, A.; Moreaux, J.; Hose, D.; et al. Inhibiting the anaphase promoting complex/cyclosome induces a metaphase arrest and cell death in multiple myeloma cells. Oncotarget 2016, 7, 4062–4076. [Google Scholar] [CrossRef]
- Manchado, E.; Guillamot, M.; de Carcer, G.; Eguren, M.; Trickey, M.; Garcia-Higuera, I.; Moreno, S.; Yamano, H.; Canamero, M.; Malumbres, M. Targeting mitotic exit leads to tumor regression in vivo: Modulation by Cdk1, Mastl, and the PP2A/B55alpha,delta phosphatase. Cancer Cell 2010, 18, 641–654. [Google Scholar] [CrossRef]
- Fujita, T.; Liu, W.; Doihara, H.; Date, H.; Wan, Y. Dissection of the APCCdh1-Skp2 cascade in breast cancer. Clin. Cancer Res. 2008, 14, 1966–1975. [Google Scholar] [CrossRef]
- Fujita, T.; Liu, W.; Doihara, H.; Wan, Y. Regulation of Skp2-p27 axis by the Cdh1/anaphase-promoting complex pathway in colorectal tumorigenesis. Am. J. Pathol. 2008, 173, 217–228. [Google Scholar] [CrossRef]
- Engelbert, D.; Schnerch, D.; Baumgarten, A.; Wasch, R. The ubiquitin ligase APC(Cdh1) is required to maintain genome integrity in primary human cells. Oncogene 2008, 27, 907–917. [Google Scholar] [CrossRef]
- De, K.; Grubb, T.M.; Zalenski, A.A.; Pfaff, K.E.; Pal, D.; Majumder, S.; Summers, M.K.; Venere, M. Hyperphosphorylation of CDH1 in Glioblastoma Cancer Stem Cells Attenuates APC/C(CDH1) Activity and Pharmacologic Inhibition of APC/C(CDH1/CDC20) Compromises Viability. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Garcia-Higuera, I.; Manchado, E.; Dubus, P.; Canamero, M.; Mendez, J.; Moreno, S.; Malumbres, M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 2008, 10, 802–811. [Google Scholar] [CrossRef]
- Fuchsberger, T.; Lloret, A.; Vina, J. New Functions of APC/C Ubiquitin Ligase in the Nervous System and Its Role in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1057. [Google Scholar] [CrossRef]
- Silva, P.N.; Gigek, C.O.; Leal, M.F.; Bertolucci, P.H.; de Labio, R.W.; Payao, S.L.; Smith Mde, A. Promoter methylation analysis of SIRT3, SMARCA5, HTERT and CDH1 genes in aging and Alzheimer’s disease. J. Alzheimers Dis. 2008, 13, 173–176. [Google Scholar] [CrossRef]
- Vina, J.; Fuchsberger, T.; Giraldo, E.; Lloret, A. APC/Cdh E3 ubiquitin ligase in the pathophysiology of Alzheimers disease. Free Radic. Biol. Med. 2014, 75 (Suppl. 1), S4. [Google Scholar] [CrossRef]
- Zeng, X.; Sigoillot, F.; Gaur, S.; Choi, S.; Pfaff, K.L.; Oh, D.C.; Hathaway, N.; Dimova, N.; Cuny, G.D.; King, R.W. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 2010, 18, 382–395. [Google Scholar] [CrossRef]
- Verma, R.; Peters, N.R.; D’Onofrio, M.; Tochtrop, G.P.; Sakamoto, K.M.; Varadan, R.; Zhang, M.; Coffino, P.; Fushman, D.; Deshaies, R.J.; et al. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 2004, 306, 117–120. [Google Scholar] [CrossRef]
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.B.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649. [Google Scholar] [CrossRef] [Green Version]
- Brito, D.A.; Rieder, C.L. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef]
- Sangodkar, J.; Farrington, C.C.; McClinch, K.; Galsky, M.D.; Kastrinsky, D.B.; Narla, G. All roads lead to PP2A: Exploiting the therapeutic potential of this phosphatase. FEBS J. 2016, 283, 1004–1024. [Google Scholar] [CrossRef]
- Baskaran, R.; Velmurugan, B.K. Protein phosphatase 2A as therapeutic targets in various disease models. Life Sci. 2018, 210, 40–46. [Google Scholar] [CrossRef]
- Chen, W.; Arroyo, J.D.; Timmons, J.C.; Possemato, R.; Hahn, W.C. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 2005, 65, 8183–8192. [Google Scholar] [CrossRef]
- Ruediger, R.; Ruiz, J.; Walter, G. Human cancer-associated mutations in the Aα subunit of protein phosphatase 2A increase lung cancer incidence in Aα knock-in and knockout mice. Mol. Cell. Biol. 2011, 31, 3832–3844. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Singh, S.; Srivastava, S.K.; Honkanen, R.E.; Reed, E.; Singh, A.P. Modulation of protein phosphatase 2A activity alters androgen-independent growth of prostate cancer cells: Therapeutic implications. Mol. Cancer 2011, 10, 720–731. [Google Scholar] [CrossRef]
- Nobumori, Y.; Shouse, G.P.; Wu, Y.; Lee, K.J.; Shen, B.; Liu, X. B56γ tumor-associated mutations provide new mechanisms for B56γ-PP2A tumor suppressor activity. Mol. Cancer Res. Mcr. 2013, 11, 995–1003. [Google Scholar] [CrossRef]
- Li, M.; Makkinje, A.; Damuni, Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J. Biol. Chem. 1996, 271, 11059–11062. [Google Scholar] [CrossRef]
- Liu, H.; Gu, Y.; Wang, H.; Yin, J.; Zheng, G.; Zhang, Z.; Lu, M.; Wang, C.; He, Z. Overexpression of PP2A inhibitor SET oncoprotein is associated with tumor progression and poor prognosis in human non-small cell lung cancer. Oncotarget 2015, 6, 14913–14925. [Google Scholar] [CrossRef]
- Szymiczek, A.; Pastorino, S.; Larson, D.; Tanji, M.; Pellegrini, L.; Xue, J.; Li, S.; Giorgi, C.; Pinton, P.; Takinishi, Y.; et al. FTY720 inhibits mesothelioma growth in vitro and in a syngeneic mouse model. J. Transl. Med. 2017, 15, 58. [Google Scholar] [CrossRef]
- Kauko, O.; O’Connor, C.M.; Kulesskiy, E.; Sangodkar, J.; Aakula, A.; Izadmehr, S.; Yetukuri, L.; Yadav, B.; Padzik, A.; Laajala, T.D.; et al. PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Wei, D.; Parsels, L.A.; Karnak, D.; Davis, M.A.; Parsels, J.D.; Marsh, A.C.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Sun, Y.; et al. Inhibition of protein phosphatase 2A radiosensitizes pancreatic cancers by modulating CDC25C/CDK1 and homologous recombination repair. Clin. Cancer Res. 2013, 19, 4422–4432. [Google Scholar] [CrossRef]
- Chung, V.; Mansfield, A.S.; Braiteh, F.; Richards, D.; Durivage, H.; Ungerleider, R.S.; Johnson, F.; Kovach, J.S. Safety, Tolerability, and Preliminary Activity of LB-100, an Inhibitor of Protein Phosphatase 2A, in Patients with Relapsed Solid Tumors: An Open-Label, Dose Escalation, First-in-Human, Phase I Trial. Clin. Cancer Res. 2017, 23, 3277–3284. [Google Scholar] [CrossRef]
- D’Arcy, B.M.; Swingle, M.R.; Papke, C.M.; Abney, K.A.; Bouska, E.S.; Prakash, A.; Honkanen, R.E. The Antitumor Drug LB-100 Is a Catalytic Inhibitor of Protein Phosphatase 2A (PPP2CA) and 5 (PPP5C) Coordinating with the Active-Site Catalytic Metals in PPP5C. Mol. Cancer 2019, 18, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Winkler, C.; De Munter, S.; Van Dessel, N.; Lesage, B.; Heroes, E.; Boens, S.; Beullens, M.; Van Eynde, A.; Bollen, M. The selective inhibition of protein phosphatase-1 results in mitotic catastrophe and impaired tumor growth. J. Cell Sci. 2015, 128, 4526–4537. [Google Scholar] [CrossRef] [Green Version]
- Krzyzosiak, A.; Sigurdardottir, A.; Luh, L.; Carrara, M.; Das, I.; Schneider, K.; Bertolotti, A. Target-Based Discovery of an Inhibitor of the Regulatory Phosphatase PPP1R15B. Cell 2018, 174, 1216–1228.e19. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Protein phosphatases involved in mitosis. Protein phosphatases can be divided into two superfamilies: Protein Tyrosine Phosphatases (PTPs) and Protein Ser/Thr Phosphatases (PSPs). Although these families contain a vast array of phosphatases, those pertinent to mitosis and discussed herein are shown.
Figure 1.
Protein phosphatases involved in mitosis. Protein phosphatases can be divided into two superfamilies: Protein Tyrosine Phosphatases (PTPs) and Protein Ser/Thr Phosphatases (PSPs). Although these families contain a vast array of phosphatases, those pertinent to mitosis and discussed herein are shown.
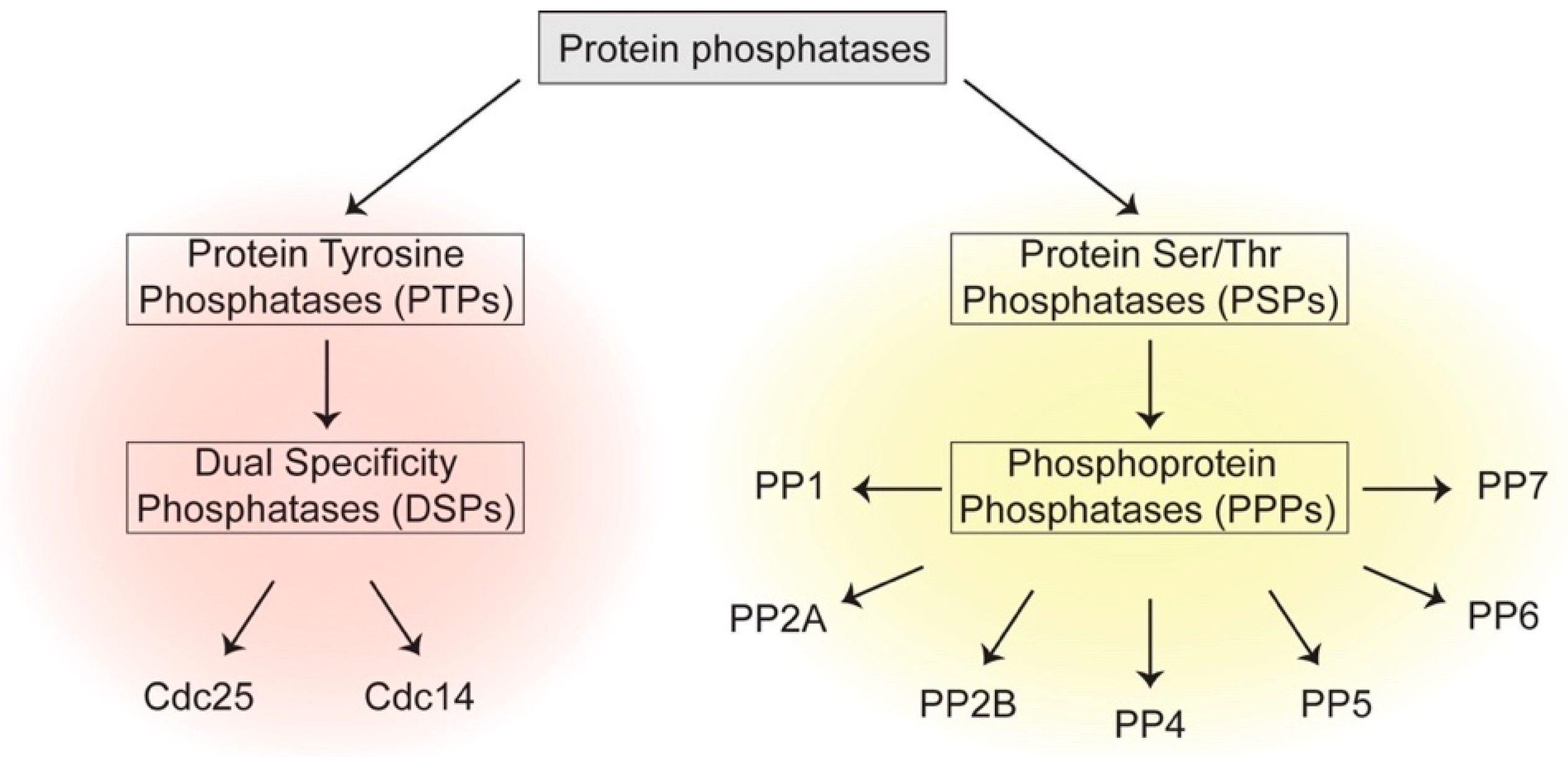
Figure 2.
Catalytic mechanisms of protein tyrosine phosphatase (PTP), protein serine/threonine phosphatase (PSP) and anaphase-promoting complex/cyclosome (APC/C). (A) PTP; the sulphur atom of the catalytic cysteine, part of the signature PTP loop sequence, initiates a nucleophilic attack on the phosphoryl group of the substrate and forms a phosphoryl-enzyme intermediate, while the dephosphorylated substrate receives a proton from the aspartate residue that forms part of the WPD loop. A water molecule leads to hydrolysis of the phosphoryl group. (B) PSP; enzyme-bound two metal ions (e.g., Fe2+, Zn2+ or Mn2+) coordinate a water molecule that mounts a nucleophilic attack on the substrate phosphoryl group, leading to substrate dephosphorylation. (C) APC/C E3 ubiquitin ligase; ubiquitylation requires the action of three enzymes (E1, E2 and E3). First, the E1 ubiquitin-activating enzyme uses ATP and catalyzes ubiquitin C-terminal adenylation. Then, an active site cysteine on the E1 enzyme attacks the C-terminus of ubiquitin and forms a thioester-linked E1~ubiquitin intermediate. The activated ubiquitin is then transferred to an active site cysteine of an E2 ubiquitin-conjugating enzyme. Finally, co-activator bound-APC/C (E3 ubiquitin ligase) simultaneously binds the ubiquitin-charged E2 and a substrate, leading to transfer of the ubiquitin molecule to a primary amine within the substrate. This process repeats several times, yielding polyubiquitylated substrate. Abbreviations: Sub, substrate.
Figure 2.
Catalytic mechanisms of protein tyrosine phosphatase (PTP), protein serine/threonine phosphatase (PSP) and anaphase-promoting complex/cyclosome (APC/C). (A) PTP; the sulphur atom of the catalytic cysteine, part of the signature PTP loop sequence, initiates a nucleophilic attack on the phosphoryl group of the substrate and forms a phosphoryl-enzyme intermediate, while the dephosphorylated substrate receives a proton from the aspartate residue that forms part of the WPD loop. A water molecule leads to hydrolysis of the phosphoryl group. (B) PSP; enzyme-bound two metal ions (e.g., Fe2+, Zn2+ or Mn2+) coordinate a water molecule that mounts a nucleophilic attack on the substrate phosphoryl group, leading to substrate dephosphorylation. (C) APC/C E3 ubiquitin ligase; ubiquitylation requires the action of three enzymes (E1, E2 and E3). First, the E1 ubiquitin-activating enzyme uses ATP and catalyzes ubiquitin C-terminal adenylation. Then, an active site cysteine on the E1 enzyme attacks the C-terminus of ubiquitin and forms a thioester-linked E1~ubiquitin intermediate. The activated ubiquitin is then transferred to an active site cysteine of an E2 ubiquitin-conjugating enzyme. Finally, co-activator bound-APC/C (E3 ubiquitin ligase) simultaneously binds the ubiquitin-charged E2 and a substrate, leading to transfer of the ubiquitin molecule to a primary amine within the substrate. This process repeats several times, yielding polyubiquitylated substrate. Abbreviations: Sub, substrate.
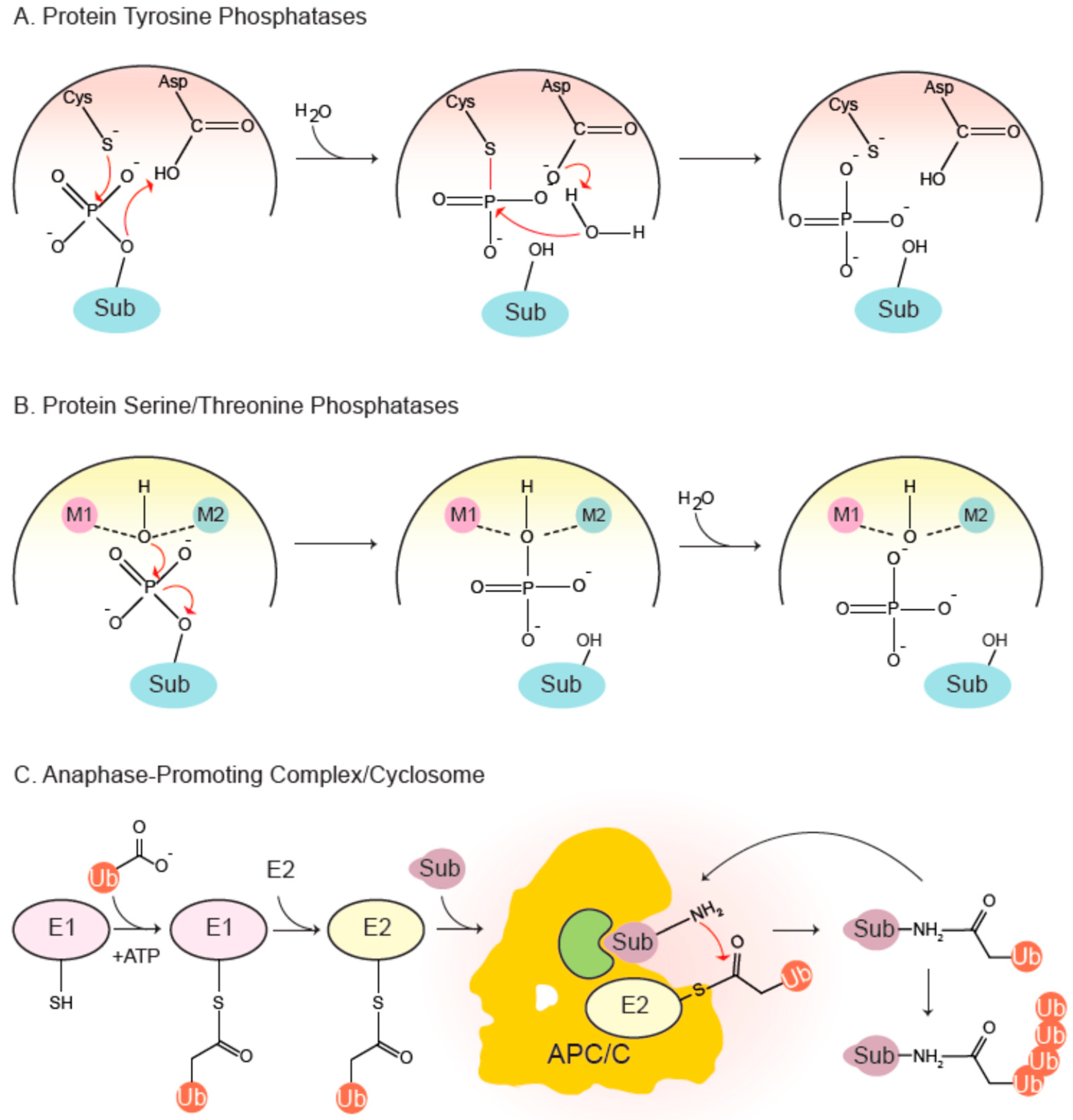
Figure 3.
Activity dynamics of cell cycle phosphatases and the APC/C throughout mitosis. Only major serine/threonine phosphatases are depicted. Precise activity dynamics of PP2A-B56, PP4 and PP6 are not well understood in the context of other cell cycle events. During early mitosis, the APC/C-Cdc20 activity (dotted light green line) is held in check by the spindle assembly checkpoint (SAC; yellow bar) until correct bipolar attachment of chromosomes to the mitotic spindle is achieved. Only substrates that can directly bind the APC/C (such as Cyclin A and Nek2A) are degraded at this stage. Once the SAC is silenced, the APC/C-Cdc20 becomes fully active (solid light green line) and ubiquitylates substrates such as securin and Cyclin B for degradation. Most cell cycle phosphatases are under regulatory control of Cdk1, albeit with different mechanisms, in order to avoid futile phosphorylation events in mitosis. See text for details. Abbreviations: KT-MT, kinetochore-microtubule; NEBD: nuclear envelope breakdown.
Figure 3.
Activity dynamics of cell cycle phosphatases and the APC/C throughout mitosis. Only major serine/threonine phosphatases are depicted. Precise activity dynamics of PP2A-B56, PP4 and PP6 are not well understood in the context of other cell cycle events. During early mitosis, the APC/C-Cdc20 activity (dotted light green line) is held in check by the spindle assembly checkpoint (SAC; yellow bar) until correct bipolar attachment of chromosomes to the mitotic spindle is achieved. Only substrates that can directly bind the APC/C (such as Cyclin A and Nek2A) are degraded at this stage. Once the SAC is silenced, the APC/C-Cdc20 becomes fully active (solid light green line) and ubiquitylates substrates such as securin and Cyclin B for degradation. Most cell cycle phosphatases are under regulatory control of Cdk1, albeit with different mechanisms, in order to avoid futile phosphorylation events in mitosis. See text for details. Abbreviations: KT-MT, kinetochore-microtubule; NEBD: nuclear envelope breakdown.
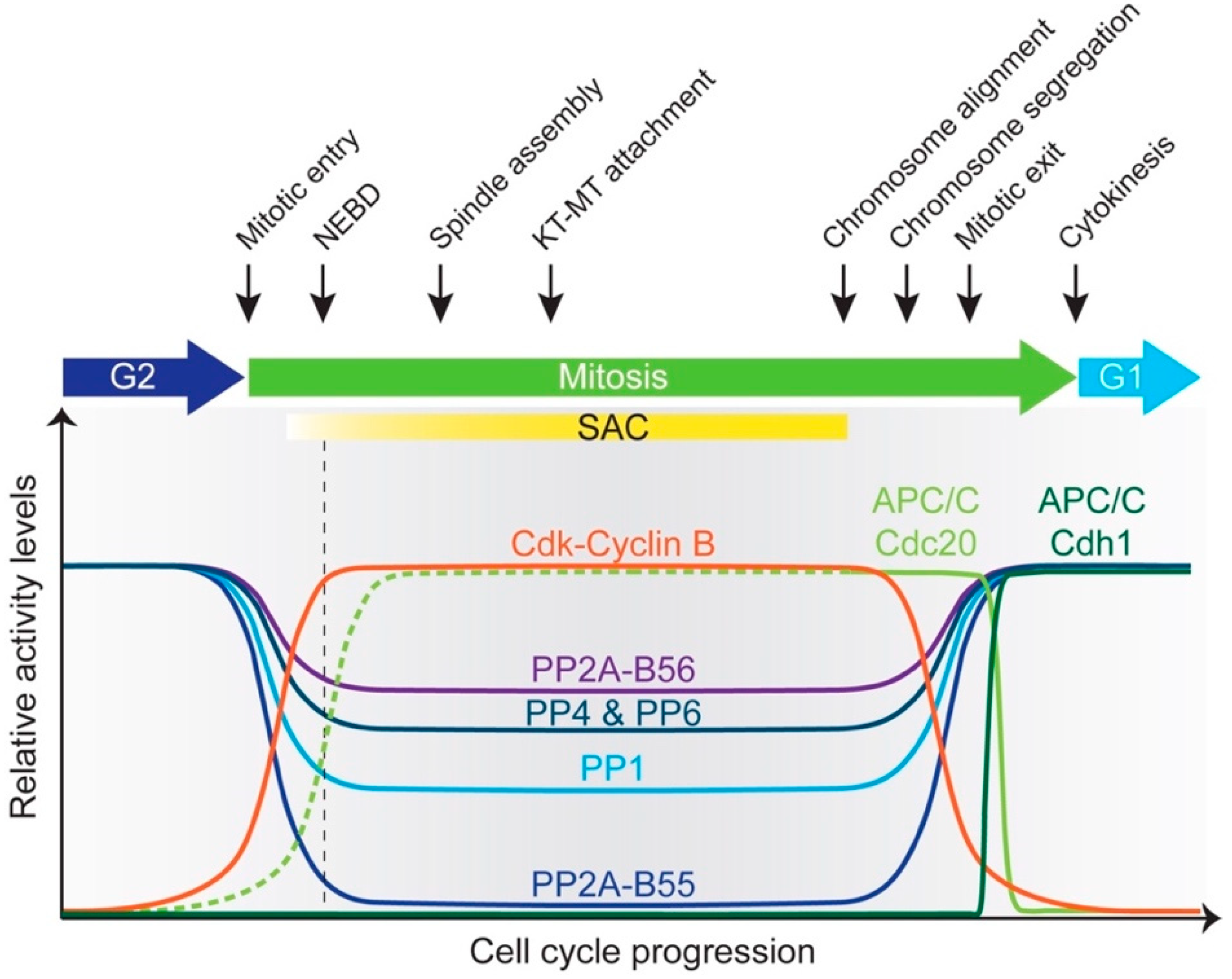
Figure 4.
The APC/C and cell cycle phosphatases display complex interactions, influencing not only each other but also numerous events in mitosis and transition into anaphase. Black arrows indicate stimulation; red T-bars indicate inhibition. See text for details. Abbreviations: CycB, Cyclin B; de-p, dephosphorylation; deg, degradation; KT-MT, kinetochore-microtubule; p, phosphorylation; Ub, ubiquitylation.
Figure 4.
The APC/C and cell cycle phosphatases display complex interactions, influencing not only each other but also numerous events in mitosis and transition into anaphase. Black arrows indicate stimulation; red T-bars indicate inhibition. See text for details. Abbreviations: CycB, Cyclin B; de-p, dephosphorylation; deg, degradation; KT-MT, kinetochore-microtubule; p, phosphorylation; Ub, ubiquitylation.
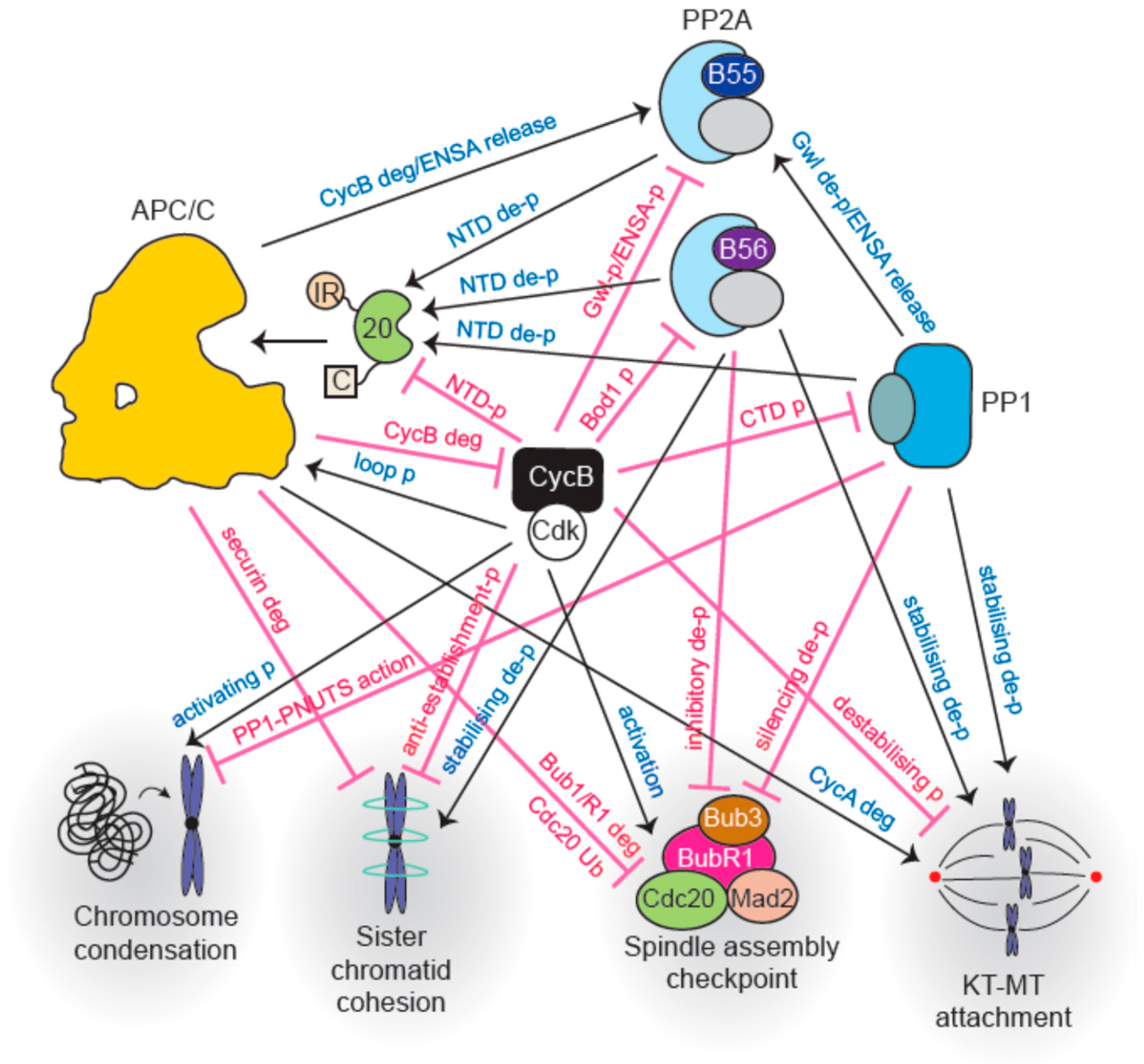
Figure 5.
Phosphatases regulate multiple aspects of APC/C function. (A) In mitosis, PP2A-B56 and PP1 dephosphorylate phosphothreonine-rich Cdc20 N-terminus to promote its interaction with the APC/C. During mitotic exit and in G1, PP2A-B55 (and other mitotic exit phosphatases) dephosphorylate serine-rich Cdh1 N-terminus. In conjunction with other unknown mechanisms, this promotes co-activator exchange and Cdc20 degradation. (B) Apc1Loop300 occludes the C box binding site within Apc8 and prevents Cdc20 loading. Recruitment of mitotic kinases by Apc3Loop leads to Apc1Loop300 phosphorylation (likely opposed by as-yet-unknown phosphatases) and Cdc20 loading. (C) Phosphoregulation of degrons influences substrate degradation by the APC/C. Securin phosphorylation at position 6 within the D-box stimulates its degradation. PP2A-B56 opposes this process. Budding yeast Dbf4 phosphorylation at position 2, removed by Cdc14, is refractory to its degradation. See text for details. Abbreviations: CycB, Cyclin B; Sub, substrate.
Figure 5.
Phosphatases regulate multiple aspects of APC/C function. (A) In mitosis, PP2A-B56 and PP1 dephosphorylate phosphothreonine-rich Cdc20 N-terminus to promote its interaction with the APC/C. During mitotic exit and in G1, PP2A-B55 (and other mitotic exit phosphatases) dephosphorylate serine-rich Cdh1 N-terminus. In conjunction with other unknown mechanisms, this promotes co-activator exchange and Cdc20 degradation. (B) Apc1Loop300 occludes the C box binding site within Apc8 and prevents Cdc20 loading. Recruitment of mitotic kinases by Apc3Loop leads to Apc1Loop300 phosphorylation (likely opposed by as-yet-unknown phosphatases) and Cdc20 loading. (C) Phosphoregulation of degrons influences substrate degradation by the APC/C. Securin phosphorylation at position 6 within the D-box stimulates its degradation. PP2A-B56 opposes this process. Budding yeast Dbf4 phosphorylation at position 2, removed by Cdc14, is refractory to its degradation. See text for details. Abbreviations: CycB, Cyclin B; Sub, substrate.
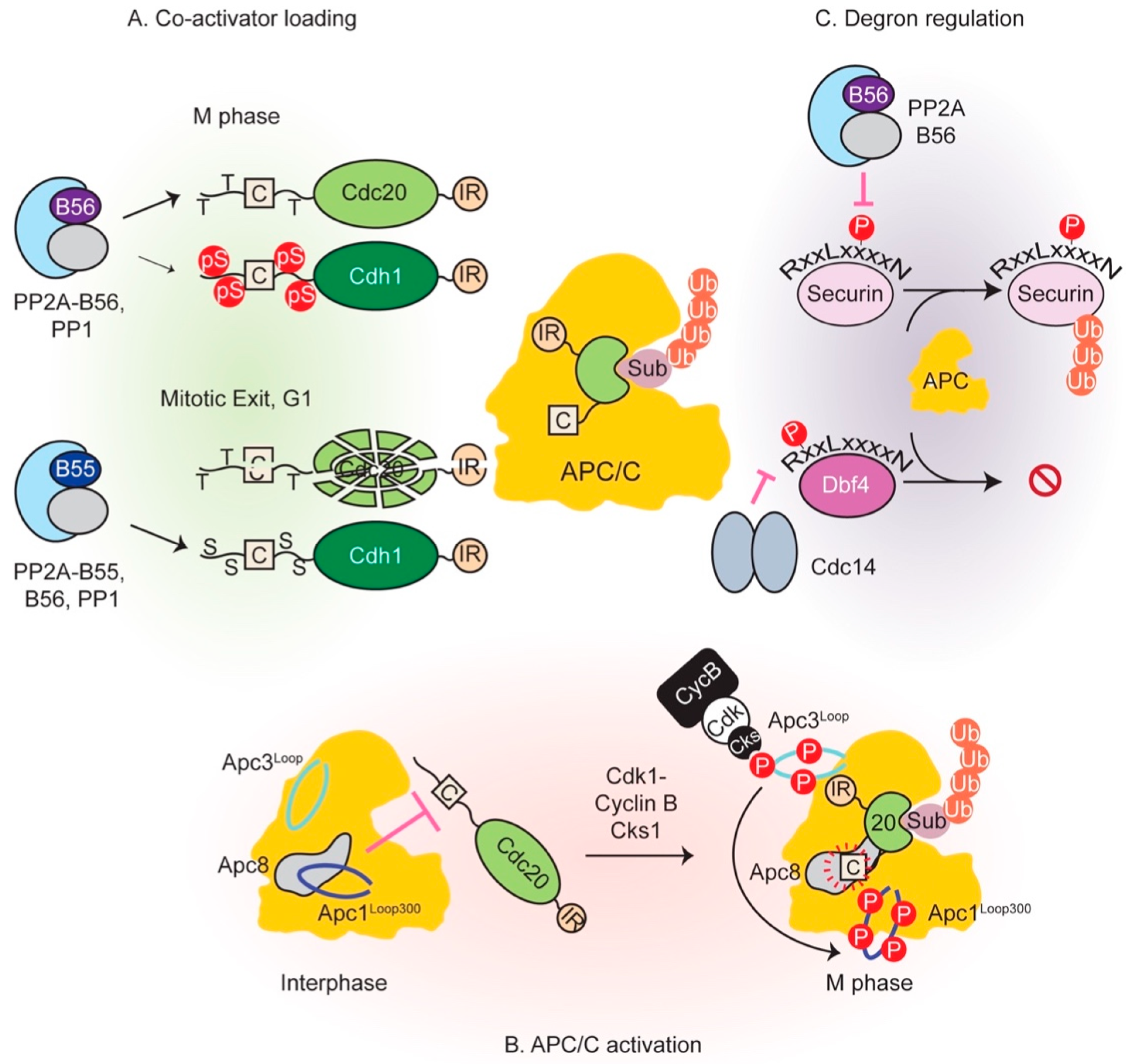
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kataria, M.; Yamano, H. Interplay between Phosphatases and the Anaphase-Promoting Complex/Cyclosome in Mitosis. Cells 2019, 8, 814. https://doi.org/10.3390/cells8080814
AMA Style
Kataria M, Yamano H. Interplay between Phosphatases and the Anaphase-Promoting Complex/Cyclosome in Mitosis. Cells. 2019; 8(8):814. https://doi.org/10.3390/cells8080814
Chicago/Turabian StyleKataria, Meghna, and Hiroyuki Yamano. 2019. "Interplay between Phosphatases and the Anaphase-Promoting Complex/Cyclosome in Mitosis" Cells 8, no. 8: 814. https://doi.org/10.3390/cells8080814
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.