Keeping the Centromere under Control: A Promising Role for DNA Methylation


{kind=link}
Abstract
:1. Introduction
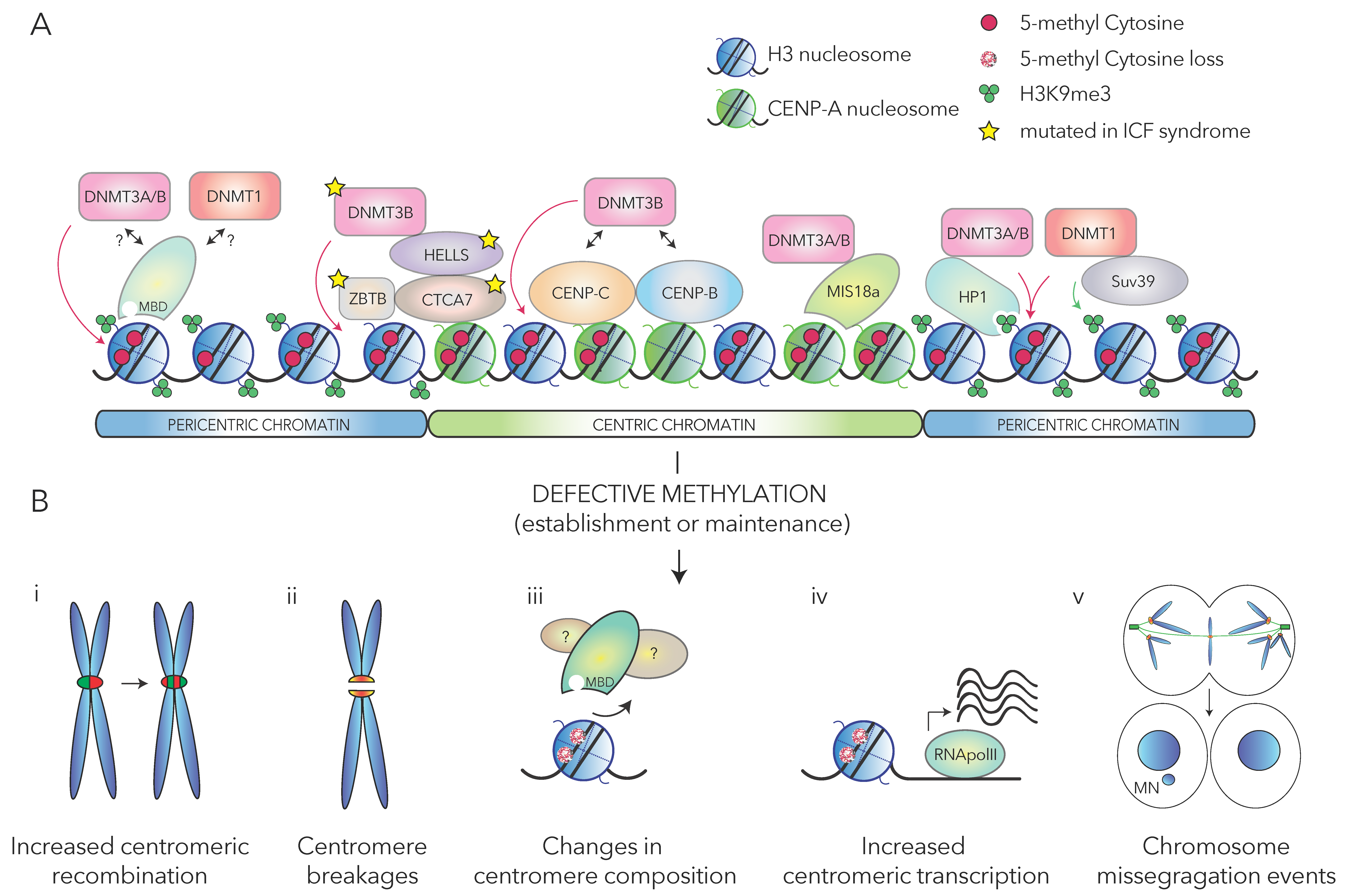
2. DNA Methylation and its Writers
3. Mechanisms Maintaining DNA Methylation at (Peri)Centromeric Regions
4. DNA Methylation and the Maintenance of Centromere Features and Stability
4.1. The Impact of (Peri)Centromeric DNA Methylation on Genomic Stability
4.2. The Role of DNA Methylation in the Regulation of (Peri)Centromeric Features
4.3. DNA Methylation in the Maintaince and Establishiment of Centromeres
5. DNA Methylation and Centromere Stability: Insights from Pathologies
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fukagawa, T.; Earnshaw, W.C. The centromere: Chromatin foundation for the kinetochore machinery. Dev. Cell 2014, 30, 496–508. [Google Scholar] [CrossRef] [PubMed]
- McKinley, K.L.; Cheeseman, I.M. The molecular basis for centromere identity and function. Nat. Rev. Mol. Cell Biol. 2016, 17, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Barra, V.; Fachinetti, D. The dark side of centromeres: Types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Santaguida, S.; Musacchio, A. The life and miracles of kinetochores. EMBO J. 2009, 28, 2511–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fachinetti, D.; Folco, H.D.; Nechemia-Arbely, Y.; Valente, L.P.; Nguyen, K.; Wong, A.J.; Zhu, Q.; Holland, A.J.; Desai, A.; Jansen, L.E.; et al. A two-step mechanism for epigenetic specification of centromere identity and function. Nat. Cell Biol. 2013, 15, 1056–1066. [Google Scholar] [CrossRef] [Green Version]
- Waye, J.S.; Willard, H.F. Structure, organization, and sequence of alpha satellite DNA from human chromosome 17: Evidence for evolution by unequal crossing-over and an ancestral pentamer repeat shared with the human X chromosome. Mol. Cell Biol. 1986, 6, 3156–3165. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Manuelidis, L. Sequence definition and organization of a human repeated DNA. J. Mol. Biol. 1980, 142, 363–386. [Google Scholar] [CrossRef]
- Dumont, M.; Fachinetti, D. DNA Sequences in Centromere Formation and Function. Prog. Mol. Subcell. Biol. 2017, 56, 305–336. [Google Scholar]
- Aldrup-MacDonald, M.E.; Kuo, M.E.; Sullivan, L.L.; Chew, K.; Sullivan, B.A. Genomic variation within alpha satellite DNA influences centromere location on human chromosomes with metastable epialleles. Genome Res. 2016, 26, 1301–1311. [Google Scholar] [CrossRef] [Green Version]
- Muro, Y.; Masumoto, H.; Yoda, K.; Nozaki, N.; Ohashi, M.; Okazaki, T. Centromere protein B assembles human centromeric alpha-satellite DNA at the 17-bp sequence, CENP-B box. J. Cell Biol. 1992, 116, 585–596. [Google Scholar] [CrossRef]
- Klein, S.J.; O’Neill, R.J. Transposable elements: Genome innovation, chromosome diversity, and centromere conflict. Chromosome Res. 2018, 26, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Costanzo, V. Centromere Structure and Function. Prog. Mol. Subcell. Biol. 2017, 56, 515–539. [Google Scholar] [PubMed]
- Knutsen, T.; Gobu, V.; Knaus, R.; Padilla-Nash, H.; Augustus, M.; Strausberg, R.L.; Kirsch, I.R.; Sirotkin, K.; Ried, T. The interactive online SKY/M-FISH & CGH database and the Entrez cancer chromosomes search database: Linkage of chromosomal aberrations with the genome sequence. Genes Chromosom. Cancer 2005, 44, 52–64. [Google Scholar] [PubMed]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Yoder, J.A.; Walsh, C.P.; Bestor, T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13, 335–340. [Google Scholar] [CrossRef]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Aapola, U.; Kawasaki, K.; Scott, H.S.; Ollila, J.; Vihinen, M.; Heino, M.; Shintani, A.; Kawasaki, K.; Minoshima, S.; Krohn, K.; et al. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics 2000, 65, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef]
- Barau, J.; Teissandier, A.; Zamudio, N.; Roy, S.; Nalesso, V.; Herault, Y.; Guillou, F.; Bourc’his, D. The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science 2016, 354, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Walton, E.L.; Francastel, C.; Velasco, G. Maintenance of DNA methylation: Dnmt3b joins the dance. Epigenetics 2011, 6, 1373–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Jeong, S.; Escobar, S.G.; Cortez, C.C.; Li, T.W.; Saito, Y.; Yoo, C.B.; Jones, P.A.; Liang, G. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc. Natl. Acad. Sci. USA 2006, 103, 14080–14085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatemi, M.; Hermann, A.; Gowher, H.; Jeltsch, A. Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur. J. Biochem. 2002, 269, 4981–4984. [Google Scholar] [CrossRef] [PubMed]
- Jair, K.W.; Bachman, K.E.; Suzuki, H.; Ting, A.H.; Rhee, I.; Yen, R.W.; Baylin, S.B.; Schuebel, K.E. De novo CpG island methylation in human cancer cells. Cancer Res. 2006, 66, 682–692. [Google Scholar] [CrossRef]
- Gowher, H.; Liebert, K.; Hermann, A.; Xu, G.; Jeltsch, A. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Biol. Chem. 2005, 280, 13341–13348. [Google Scholar] [CrossRef]
- Chedin, F.; Lieber, M.R.; Hsieh, C.L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl. Acad. Sci. USA 2002, 99, 16916–16921. [Google Scholar] [CrossRef]
- Zhang, Y.; Jurkowska, R.; Soeroes, S.; Rajavelu, A.; Dhayalan, A.; Bock, I.; Rathert, P.; Brandt, O.; Reinhardt, R.; Fischle, W.; et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acid. Res. 2010, 38, 4246–4253. [Google Scholar] [CrossRef] [Green Version]
- Otani, J.; Nankumo, T.; Arita, K.; Inamoto, S.; Ariyoshi, M.; Shirakawa, M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009, 10, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cedar, H.; Bergman, Y. Programming of DNA methylation patterns. Annu. Rev. Biochem. 2012, 81, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schubeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef]
- Wong, N.C.; Wong, L.H.; Quach, J.M.; Canham, P.; Craig, J.M.; Song, J.Z.; Clark, S.J.; Choo, K.H. Permissive transcriptional activity at the centromere through pockets of DNA hypomethylation. PLoS Genet. 2006, 2. [Google Scholar] [CrossRef]
- Chen, T.; Tsujimoto, N.; Li, E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell Biol. 2004, 24, 9048–9058. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.P.; Schermelleh, L.; Leonhardt, H.; Cardoso, M.C. Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep. 2004, 5, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef] [PubMed]
- Fachinetti, D.; Han, J.S.; McMahon, M.A.; Ly, P.; Abdullah, A.; Wong, A.J.; Cleveland, D.W. DNA Sequence-Specific Binding of CENP-B Enhances the Fidelity of Human Centromere Function. Dev. Cell 2015, 33, 314–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Kurumizaka, H.; Yokoyama, S. CpG methylation of the CENP-B box reduces human CENP-B binding. FEBS J. 2005, 272, 282–289. [Google Scholar] [CrossRef]
- Mitchell, A.R.; Jeppesen, P.; Nicol, L.; Morrison, H.; Kipling, D. Epigenetic control of mammalian centromere protein binding: Does DNA methylation have a role? J. Cell Sci. 1996, 109, 2199–2206. [Google Scholar] [PubMed]
- Okada, T.; Ohzeki, J.; Nakano, M.; Yoda, K.; Brinkley, W.R.; Larionov, V.; Masumoto, H. CENP-B controls centromere formation depending on the chromatin context. Cell 2007, 131, 1287–1300. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Sullivan, B.A.; Trazzi, S.; Della Valle, G.; Robertson, K.D. DNMT3B interacts with constitutive centromere protein CENP-C to modulate DNA methylation and the histone code at centromeric regions. Hum. Mol. Genet. 2009, 18, 3178–3193. [Google Scholar] [CrossRef] [Green Version]
- Fujita, Y.; Hayashi, T.; Kiyomitsu, T.; Toyoda, Y.; Kokubu, A.; Obuse, C.; Yanagida, M. Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev. Cell 2007, 12, 17–30. [Google Scholar] [CrossRef]
- Kim, I.S.; Lee, M.; Park, K.C.; Jeon, Y.; Park, J.H.; Hwang, E.J.; Jeon, T.I.; Ko, S.; Lee, H.; Baek, S.H.; et al. Roles of Mis18alpha in epigenetic regulation of centromeric chromatin and CENP-A loading. Mol. Cell 2012, 46, 260–273. [Google Scholar] [CrossRef]
- Saksouk, N.; Barth, T.K.; Ziegler-Birling, C.; Olova, N.; Nowak, A.; Rey, E.; Mateos-Langerak, J.; Urbach, S.; Reik, W.; Torres-Padilla, M.E.; et al. Redundant mechanisms to form silent chromatin at pericentromeric regions rely on BEND3 and DNA methylation. Mol. Cell 2014, 56, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; Li, E.; Jenuwein, T.; Peters, A.H. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003, 13, 1192–1200. [Google Scholar] [CrossRef]
- Guetg, C.; Lienemann, P.; Sirri, V.; Grummt, I.; Hernandez-Verdun, D.; Hottiger, M.O.; Fussenegger, M.; Santoro, R. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J. 2010, 29, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Hurd, P.J.; Deplus, R.; Kouzarides, T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acid. Res. 2003, 31, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Luciani, J.J.; Depetris, D.; Missirian, C.; Mignon-Ravix, C.; Metzler-Guillemain, C.; Megarbane, A.; Moncla, A.; Mattei, M.G. Subcellular distribution of HP1 proteins is altered in ICF syndrome. Eur. J. Hum. Genet. 2005, 13, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, R.J.; McDowell, T.L.; Raman, S.; O’Rourke, D.M.; Garrick, D.; Ayyub, H.; Higgs, D.R. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat. Genet. 2000, 24, 368–371. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Tate, P.; Li, E.; Bird, A. DNA methylation specifies chromosomal localization of MeCP2. Mol. Cell Biol. 1996, 16, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Hou, J.; Maclean, A.; Nasir, J.; Lafuente, M.J.; Shu, X.; Kriaucionis, S.; Bird, A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. USA 2007, 104, 2709–2714. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.; Gartler, S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 14412–14417. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Karpf, A.R.; Matsui, S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005, 65, 8635–8639. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.E.; Okano, M.; Dick, F.; Tsujimoto, N.; Chen, T.; Wang, S.; Ueda, Y.; Dyson, N.; Li, E. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 2005, 280, 17986–17991. [Google Scholar] [CrossRef] [PubMed]
- Tsumura, A.; Hayakawa, T.; Kumaki, Y.; Takebayashi, S.; Sakaue, M.; Matsuoka, C.; Shimotohno, K.; Ishikawa, F.; Li, E.; Ueda, H.R.; et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes. Cells 2006, 11, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Teitz, L.S.; Kim, D.H.; Shoshani, O.; Skaletsky, H.; Fachinetti, D.; Page, D.C.; Cleveland, D.W. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol. 2017, 19, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Fujii, M.; Ayusawa, D. Demethylation of classical satellite 2 and 3 DNA with chromosomal instability in senescent human fibroblasts. Exp. Gerontol. 2002, 37, 1005–1014. [Google Scholar] [CrossRef]
- Guttenbach, M.; Schmid, M. Exclusion of specific human chromosomes into micronuclei by 5-azacytidine treatment of lymphocyte cultures. Exp. Cell Res. 1994, 211, 127–132. [Google Scholar] [CrossRef]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.; Smith, G.R. Repression of harmful meiotic recombination in centromeric regions. Semin. Cell Dev. Biol. 2016, 54, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Koehler, K.E.; Boulton, C.L.; Collins, H.E.; French, R.L.; Herman, K.C.; Lacefield, S.M.; Madden, L.D.; Schuetz, C.D.; Hawley, R.S. Spontaneous X chromosome MI and MII nondisjunction events in Drosophila melanogaster oocytes have different recombinational histories. Nat. Genet. 1996, 14, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Lamb, N.E.; Sherman, S.L.; Hassold, T.J. Effect of meiotic recombination on the production of aneuploid gametes in humans. Cytogenet. Genome Res. 2005, 111, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.K.; Doyle, G.G.; Brigham, B.; Carter, J.; Hooker, K.D.; Lai, A.; Rice, M.; Stack, S.M. High-resolution crossover maps for each bivalent of Zea mays using recombination nodules. Genetics 2003, 165, 849–865. [Google Scholar] [PubMed]
- Lambie, E.J.; Roeder, G.S. Repression of meiotic crossing over by a centromere (CEN3) in Saccharomyces cerevisiae. Genetics 1986, 114, 769–789. [Google Scholar]
- Talbert, P.B.; Henikoff, S. Centromeres convert but don’t cross. PLoS Biol. 2010, 8. [Google Scholar] [CrossRef]
- Yelina, N.E.; Lambing, C.; Hardcastle, T.J.; Zhao, X.; Santos, B.; Henderson, I.R. DNA methylation epigenetically silences crossover hot spots and controls chromosomal domains of meiotic recombination in Arabidopsis. Genes Dev. 2015, 29, 2183–2202. [Google Scholar] [CrossRef]
- Melamed-Bessudo, C.; Levy, A.A. Deficiency in DNA methylation increases meiotic crossover rates in euchromatic but not in heterochromatic regions in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, E981–E988. [Google Scholar] [CrossRef] [Green Version]
- Mirouze, M.; Lieberman-Lazarovich, M.; Aversano, R.; Bucher, E.; Nicolet, J.; Reinders, J.; Paszkowski, J. Loss of DNA methylation affects the recombination landscape in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 5880–5885. [Google Scholar] [CrossRef]
- Jaco, I.; Canela, A.; Vera, E.; Blasco, M.A. Centromere mitotic recombination in mammalian cells. J. Cell Biol. 2008, 181, 885–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichikawa, K.; Tomioka, S.; Suzuki, Y.; Nakamura, R.; Doi, K.; Yoshimura, J.; Kumagai, M.; Inoue, Y.; Uchida, Y.; Irie, N.; et al. Centromere evolution and CpG methylation during vertebrate speciation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Belmont, A.S. Mitotic chromosome structure and condensation. Curr. Opin. Cell Biol. 2006, 18, 632–638. [Google Scholar] [CrossRef]
- Hirota, T.; Gerlich, D.; Koch, B.; Ellenberg, J.; Peters, J.M. Distinct functions of condensin I and II in mitotic chromosome assembly. J. Cell Sci. 2004, 117, 6435–6445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T.; Kobayashi, R.; Hirano, M. Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell 1997, 89, 511–521. [Google Scholar] [CrossRef]
- Kokalj-Vokac, N.; Almeida, A.; Viegas-Pequignot, E.; Jeanpierre, M.; Malfoy, B.; Dutrillaux, B. Specific induction of uncoiling and recombination by azacytidine in classical satellite-containing constitutive heterochromatin. Cytogenet. Cell Genet. 1993, 63, 11–15. [Google Scholar] [CrossRef]
- Fazzio, T.G.; Panning, B. Condensin complexes regulate mitotic progression and interphase chromatin structure in embryonic stem cells. J. Cell Biol. 2010, 188, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Geiman, T.M.; Sankpal, U.T.; Robertson, A.K.; Chen, Y.; Mazumdar, M.; Heale, J.T.; Schmiesing, J.A.; Kim, W.; Yokomori, K.; Zhao, Y.; et al. Isolation and characterization of a novel DNA methyltransferase complex linking DNMT3B with components of the mitotic chromosome condensation machinery. Nucleic Acid. Res. 2004, 32, 2716–2729. [Google Scholar] [CrossRef] [Green Version]
- Flagiello, D.; Bernardino-Sgherri, J.; Dutrillaux, B. Complex relationships between 5-aza-dC induced DNA demethylation and chromosome compaction at mitosis. Chromosoma 2002, 111, 37–44. [Google Scholar]
- Collings, C.K.; Waddell, P.J.; Anderson, J.N. Effects of DNA methylation on nucleosome stability. Nucleic Acid. Res. 2013, 41, 2918–2931. [Google Scholar] [CrossRef] [Green Version]
- Choy, J.S.; Wei, S.; Lee, J.Y.; Tan, S.; Chu, S.; Lee, T.H. DNA methylation increases nucleosome compaction and rigidity. J. Am. Chem. Soc. 2010, 132, 1782–1783. [Google Scholar] [CrossRef] [PubMed]
- Keshet, I.; Lieman-Hurwitz, J.; Cedar, H. DNA methylation affects the formation of active chromatin. Cell 1986, 44, 535–543. [Google Scholar] [CrossRef]
- Fernandez, J.L.; Goyanes, V.; Pereira, S.; Lopez-Fernandez, C.; Gosalvez, J. 5-azacytidine produces differential undercondensation of alpha, beta and classical human satellite DNAs. Chromosom. Res. 1994, 2, 29–35. [Google Scholar] [CrossRef]
- Osakabe, A.; Adachi, F.; Arimura, Y.; Maehara, K.; Ohkawa, Y.; Kurumizaka, H. Influence of DNA methylation on positioning and DNA flexibility of nucleosomes with pericentric satellite DNA. Open Biol. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, B.A.; Karpen, G.H. Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat. Struct. Mol. Biol. 2004, 11, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, J.H.; Rodriguez, M.G.; Martins, N.M.; Kimura, H.; Kelly, D.A.; Masumoto, H.; Larionov, V.; Jansen, L.E.; Earnshaw, W.C. Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. EMBO J. 2011, 30, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Schalch, T.; Steiner, F.A. Structure of centromere chromatin: From nucleosome to chromosomal architecture. Chromosoma 2017, 126, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Heit, R.; Rattner, J.B.; Chan, G.K.; Hendzel, M.J. G2 histone methylation is required for the proper segregation of chromosomes. J. Cell Sci. 2009, 122, 2957–2968. [Google Scholar] [CrossRef] [Green Version]
- Ekwall, K.; Olsson, T.; Turner, B.M.; Cranston, G.; Allshire, R.C. Transient inhibition of histone deacetylation alters the structural and functional imprint at fission yeast centromeres. Cell 1997, 91, 1021–1032. [Google Scholar] [CrossRef]
- Xin, H.; Yoon, H.G.; Singh, P.B.; Wong, J.; Qin, J. Components of a pathway maintaining histone modification and heterochromatin protein 1 binding at the pericentric heterochromatin in Mammalian cells. J. Biol. Chem. 2004, 279, 9539–9546. [Google Scholar] [CrossRef]
- Monier, K.; Mouradian, S.; Sullivan, K.F. DNA methylation promotes Aurora-B-driven phosphorylation of histone H3 in chromosomal subdomains. J. Cell Sci. 2007, 120, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, K.B.; Salmon, E.D. Chromosome dynamics: New light on Aurora B kinase function. Curr. Biol. 2002, 12, R458–R460. [Google Scholar] [CrossRef]
- Hsu, J.Y.; Sun, Z.W.; Li, X.; Reuben, M.; Tatchell, K.; Bishop, D.K.; Grushcow, J.M.; Brame, C.J.; Caldwell, J.A.; Hunt, D.F.; et al. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell 2000, 102, 279–291. [Google Scholar] [CrossRef]
- Sugimura, K.; Fukushima, Y.; Ishida, M.; Ito, S.; Nakamura, M.; Mori, Y.; Okumura, K. Cell cycle-dependent accumulation of histone H3.3 and euchromatic histone modifications in pericentromeric heterochromatin in response to a decrease in DNA methylation levels. Exp. Cell Res. 2010, 316, 2731–2746. [Google Scholar] [CrossRef] [PubMed]
- Bouzinba-Segard, H.; Guais, A.; Francastel, C. Accumulation of small murine minor satellite transcripts leads to impaired centromeric architecture and function. Proc. Natl. Acad. Sci. USA 2006, 103, 8709–8714. [Google Scholar] [CrossRef] [Green Version]
- Ichida, K.; Suzuki, K.; Fukui, T.; Takayama, Y.; Kakizawa, N.; Watanabe, F.; Ishikawa, H.; Muto, Y.; Kato, T.; Saito, M.; et al. Overexpression of satellite alpha transcripts leads to chromosomal instability via segregation errors at specific chromosomes. Int. J. Oncol. 2018, 52, 1685–1693. [Google Scholar] [CrossRef]
- Scott, K.C.; Sullivan, B.A. Neocentromeres: A place for everything and everything in its place. Trends Genet. 2014, 30, 66–74. [Google Scholar] [CrossRef]
- Amor, D.J.; Choo, K.H. Neocentromeres: Role in human disease, evolution, and centromere study. Am. J. Hum. Genet. 2002, 71, 695–714. [Google Scholar] [CrossRef]
- Santos, F.; Dean, W. Epigenetic reprogramming during early development in mammals. Reproduction 2004, 127, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, K.; Yamazaki, T.; Miki, H.; Ogonuki, N.; Inoue, K.; Ogura, A.; Baba, T. Centromeric DNA hypomethylation as an epigenetic signature discriminates between germ and somatic cell lineages. Dev. Biol. 2007, 312, 419–426. [Google Scholar] [CrossRef]
- Gisselsson, D.; Shao, C.; Tuck-Muller, C.M.; Sogorovic, S.; Palsson, E.; Smeets, D.; Ehrlich, M. Interphase chromosomal abnormalities and mitotic missegregation of hypomethylated sequences in ICF syndrome cells. Chromosoma 2005, 114, 118–126. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. DNA methylation and genetic instability in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 1997, 94, 2545–2550. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Galindo, R.; Fischer, S.; Saha, A.K.; Lundy, J.D.; Cervantes, P.W.; Mourad, M.; Wang, C.; Qian, B.; Dai, M.; Meng, F.; et al. Rapid molecular assays to study human centromere genomics. Genome Res. 2017, 27, 2040–2049. [Google Scholar] [CrossRef] [Green Version]
- Drpic, D.; Almeida, A.C.; Aguiar, P.; Renda, F.; Damas, J.; Lewin, H.A.; Larkin, D.M.; Khodjakov, A.; Maiato, H. Chromosome Segregation Is Biased by Kinetochore Size. Curr. Biol. 2018, 28, 1344–1356. [Google Scholar] [CrossRef]
- Irvine, D.V.; Amor, D.J.; Perry, J.; Sirvent, N.; Pedeutour, F.; Choo, K.H.; Saffery, R. Chromosome size and origin as determinants of the level of CENP-A incorporation into human centromeres. Chromosom. Res. 2004, 12, 805–815. [Google Scholar] [CrossRef]
- Sullivan, L.L.; Boivin, C.D.; Mravinac, B.; Song, I.Y.; Sullivan, B.A. Genomic size of CENP-A domain is proportional to total alpha satellite array size at human centromeres and expands in cancer cells. Chromosom. Res. 2011, 19, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.; Lam, W.C.; Lai, P.B.; Pang, E.; Lau, W.Y.; Johnson, P.J. Hypomethylation of chromosome 1 heterochromatin DNA correlates with q-arm copy gain in human hepatocellular carcinoma. Am. J. Pathol. 2001, 159, 465–471. [Google Scholar] [CrossRef]
- Ehrlich, M.; Jiang, G.; Fiala, E.; Dome, J.S.; Yu, M.C.; Long, T.I.; Youn, B.; Sohn, O.S.; Widschwendter, M.; Tomlinson, G.E.; et al. Hypomethylation and hypermethylation of DNA in Wilms tumors. Oncogene 2002, 21, 6694–6702. [Google Scholar] [CrossRef] [Green Version]
- Vilain, A.; Bernardino, J.; Gerbault-Seureau, M.; Vogt, N.; Niveleau, A.; Lefrancois, D.; Malfoy, B.; Dutrillaux, B. DNA methylation and chromosome instability in lymphoblastoid cell lines. Cytogenet. Cell Genet. 2000, 90, 93–101. [Google Scholar] [CrossRef]
- Miniou, P.; Jeanpierre, M.; Bourc’his, D.; Coutinho Barbosa, A.C.; Blanquet, V.; Viegas-Pequignot, E. alpha-satellite DNA methylation in normal individuals and in ICF patients: Heterogeneous methylation of constitutive heterochromatin in adult and fetal tissues. Hum. Genet. 1997, 99, 738–745. [Google Scholar] [CrossRef]
- Ehrlich, M.; Jackson, K.; Weemaes, C. Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF). Orphanet J. Rare Dis. 2006, 1. [Google Scholar] [CrossRef]
- Jefferson, A.; Colella, S.; Moralli, D.; Wilson, N.; Yusuf, M.; Gimelli, G.; Ragoussis, J.; Volpi, E.V. Altered intra-nuclear organisation of heterochromatin and genes in ICF syndrome. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Matarazzo, M.R.; Boyle, S.; D’Esposito, M.; Bickmore, W.A. Chromosome territory reorganization in a human disease with altered DNA methylation. Proc. Natl. Acad. Sci. USA 2007, 104, 16546–16551. [Google Scholar] [CrossRef] [Green Version]
- Weemaes, C.M.; van Tol, M.J.; Wang, J.; van Ostaijen-ten Dam, M.M.; van Eggermond, M.C.; Thijssen, P.E.; Aytekin, C.; Brunetti-Pierri, N.; van der Burg, M.; Graham Davies, E.; et al. Heterogeneous clinical presentation in ICF syndrome: Correlation with underlying gene defects. Eur. J. Hum. Genet. 2013, 21, 1219–1225. [Google Scholar] [CrossRef]
- Ji, W.; Hernandez, R.; Zhang, X.Y.; Qu, G.Z.; Frady, A.; Varela, M.; Ehrlich, M. DNA demethylation and pericentromeric rearrangements of chromosome 1. Mutat. Res. 1997, 379, 33–41. [Google Scholar] [CrossRef]
- Hernandez, R.; Frady, A.; Zhang, X.Y.; Varela, M.; Ehrlich, M. Preferential induction of chromosome 1 multibranched figures and whole-arm deletions in a human pro-B cell line treated with 5-azacytidine or 5-azadeoxycytidine. Cytogenet. Cell Genet. 1997, 76, 196–201. [Google Scholar] [CrossRef]
- Jeanpierre, M.; Turleau, C.; Aurias, A.; Prieur, M.; Ledeist, F.; Fischer, A.; Viegas-Pequignot, E. An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum. Mol. Genet. 1993, 2, 731–735. [Google Scholar] [CrossRef]
- Ge, Y.Z.; Pu, M.T.; Gowher, H.; Wu, H.P.; Ding, J.P.; Jeltsch, A.; Xu, G.L. Chromatin targeting of de novo DNA methyltransferases by the PWWP domain. J. Biol. Chem. 2004, 279, 25447–25454. [Google Scholar] [CrossRef]
- de Greef, J.C.; Wang, J.; Balog, J.; den Dunnen, J.T.; Frants, R.R.; Straasheijm, K.R.; Aytekin, C.; van der Burg, M.; Duprez, L.; Ferster, A.; et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am. J. Hum. Genet. 2011, 88, 796–804. [Google Scholar] [CrossRef]
- Thijssen, P.E.; Ito, Y.; Grillo, G.; Wang, J.; Velasco, G.; Nitta, H.; Unoki, M.; Yoshihara, M.; Suyama, M.; Sun, Y.; et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Velasco, G.; Grillo, G.; Touleimat, N.; Ferry, L.; Ivkovic, I.; Ribierre, F.; Deleuze, J.F.; Chantalat, S.; Picard, C.; Francastel, C. Comparative methylome analysis of ICF patients identifies heterochromatin loci that require ZBTB24, CDCA7 and HELLS for their methylated state. Hum. Mol. Genet. 2018, 27, 2409–2424. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.L.; Rigolet, M.; Bourc’his, D.; Nigon, F.; Bokesoy, I.; Fryns, J.P.; Hulten, M.; Jonveaux, P.; Maraschio, P.; Megarbane, A.; et al. DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum. Mutat. 2005, 25, 56–63. [Google Scholar] [CrossRef]
- Myant, K.; Stancheva, I. LSH cooperates with DNA methyltransferases to repress transcription. Mol. Cell Biol. 2008, 28, 215–226. [Google Scholar] [CrossRef]
- Rajshekar, S.; Yao, J.; Arnold, P.K.; Payne, S.G.; Zhang, Y.; Bowman, T.V.; Schmitz, R.J.; Edwards, J.R.; Goll, M. Pericentromeric hypomethylation elicits an interferon response in an animal model of ICF syndrome. Elife 2018, 7. [Google Scholar] [CrossRef]
- Jenness, C.; Giunta, S.; Muller, M.M.; Kimura, H.; Muir, T.W.; Funabiki, H. HELLS and CDCA7 comprise a bipartite nucleosome remodeling complex defective in ICF syndrome. Proc. Natl. Acad. Sci. USA 2018, 115, E876–E885. [Google Scholar] [CrossRef]
- Muegge, K. Lsh, a guardian of heterochromatin at repeat elements. Biochem. Cell Biol. 2005, 83, 548–554. [Google Scholar] [CrossRef]
- Huang, J.; Fan, T.; Yan, Q.; Zhu, H.; Fox, S.; Issaq, H.J.; Best, L.; Gangi, L.; Munroe, D.; Muegge, K. Lsh, an epigenetic guardian of repetitive elements. Nucleic Acid. Res. 2004, 32, 5019–5028. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Thijssen, P.E.; de Klerk, E.; Vonk, K.K.; Wang, J.; den Hamer, B.; Aytekin, C.; van der Maarel, S.M.; Daxinger, L. Converging disease genes in ICF syndrome: ZBTB24 controls expression of CDCA7 in mammals. Hum. Mol. Genet. 2016, 25, 4041–4051. [Google Scholar] [CrossRef]
- Ren, R.; Hardikar, S.; Horton, J.R.; Lu, Y.; Zeng, Y.; Singh, A.K.; Lin, K.; Coletta, L.D.; Shen, J.; Lin Kong, C.S.; et al. Structural basis of specific DNA binding by the transcription factor ZBTB24. Nucleic Acid. Res. 2019. [Google Scholar] [CrossRef]
- Burrage, J.; Termanis, A.; Geissner, A.; Myant, K.; Gordon, K.; Stancheva, I. The SNF2 family ATPase LSH promotes phosphorylation of H2AX and efficient repair of DNA double-strand breaks in mammalian cells. J. Cell Sci. 2012, 125, 5524–5534. [Google Scholar] [CrossRef]
- Unoki, M.; Funabiki, H.; Velasco, G.; Francastel, C.; Sasaki, H. CDCA7 and HELLS mutations undermine nonhomologous end joining in centromeric instability syndrome. J. Clin. Invest. 2019, 129, 78–92. [Google Scholar] [CrossRef]
- Kabeche, L.; Nguyen, H.D.; Buisson, R.; Zou, L. A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science 2018, 359, 108–114. [Google Scholar] [CrossRef]
- Nakama, M.; Kawakami, K.; Kajitani, T.; Urano, T.; Murakami, Y. DNA-RNA hybrid formation mediates RNAi-directed heterochromatin formation. Genes. Cells 2012, 17, 218–233. [Google Scholar]
- Aze, A.; Sannino, V.; Soffientini, P.; Bachi, A.; Costanzo, V. Centromeric DNA replication reconstitution reveals DNA loops and ATR checkpoint suppression. Nat. Cell Biol. 2016, 18, 684–691. [Google Scholar] [CrossRef] [Green Version]
- Dabin, J.; Fortuny, A.; Polo, S.E. Epigenome Maintenance in Response to DNA Damage. Mol. Cell 2016, 62, 712–727. [Google Scholar] [CrossRef] [Green Version]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scelfo, A.; Fachinetti, D. Keeping the Centromere under Control: A Promising Role for DNA Methylation. Cells 2019, 8, 912. https://doi.org/10.3390/cells8080912
Scelfo A, Fachinetti D. Keeping the Centromere under Control: A Promising Role for DNA Methylation. Cells. 2019; 8(8):912. https://doi.org/10.3390/cells8080912
Chicago/Turabian StyleScelfo, Andrea, and Daniele Fachinetti. 2019. "Keeping the Centromere under Control: A Promising Role for DNA Methylation" Cells 8, no. 8: 912. https://doi.org/10.3390/cells8080912
APA StyleScelfo, A., & Fachinetti, D. (2019). Keeping the Centromere under Control: A Promising Role for DNA Methylation. Cells, 8(8), 912. https://doi.org/10.3390/cells8080912