Knockout of the HvCKX1 or HvCKX3 Gene in Barley (Hordeum vulgare L.) by RNA-Guided Cas9 Nuclease Affects the Regulation of Cytokinin Metabolism and Root Morphology


Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Target Sequences and sgRNA Design
2.2. Plant Transformation and Selection of Mutant Lines
2.3. CKX Activity Assays
2.4. Quantitative RT-PCR
2.5. Transcriptome Analysis by RNA-seq
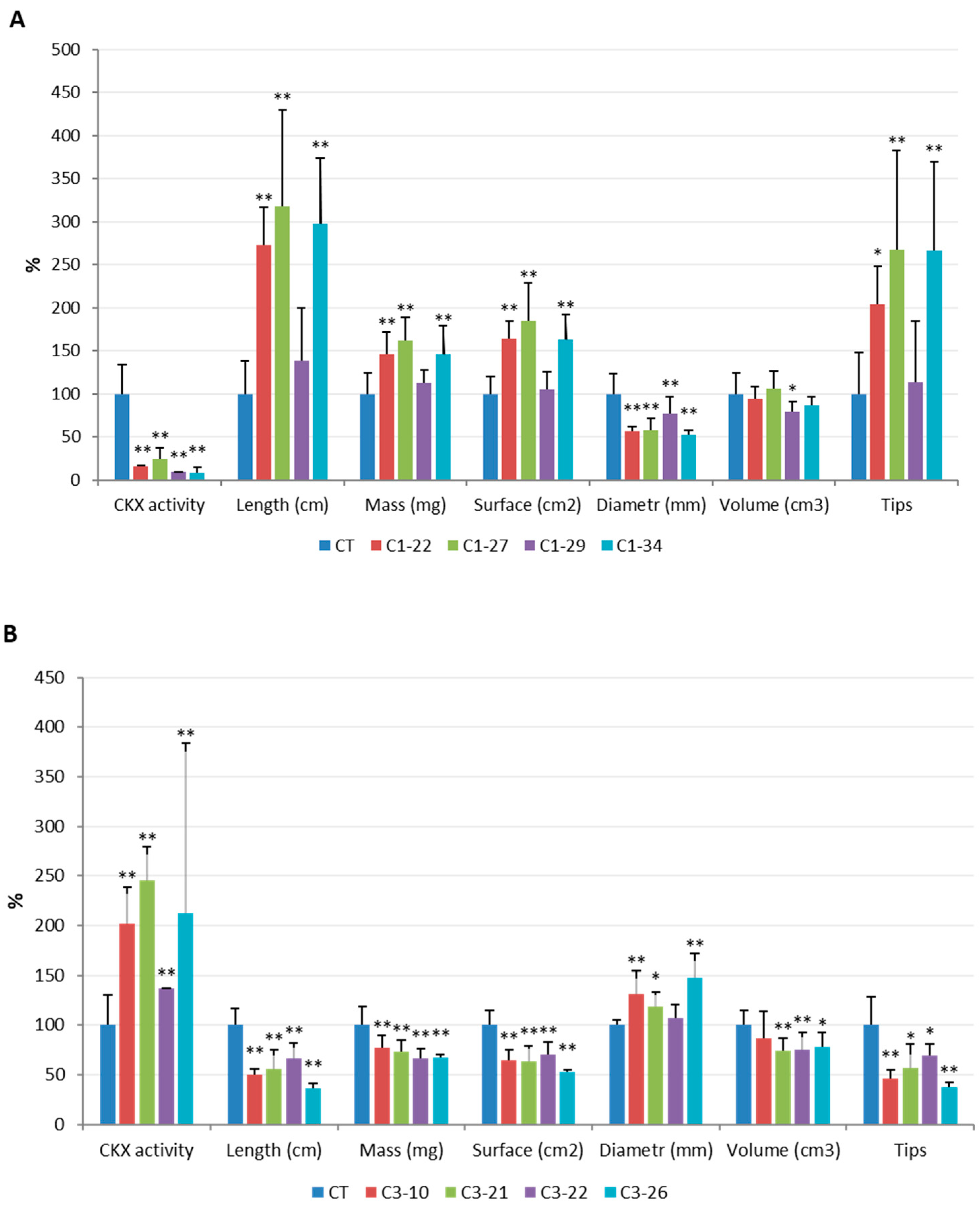
2.6. Analysis of Phenotypic traits
2.7. Statistical Analysis
3. Results
3.1. Generation of Targeted Mutations in the HvCKX1 and HvCKX3 Genes in Barley
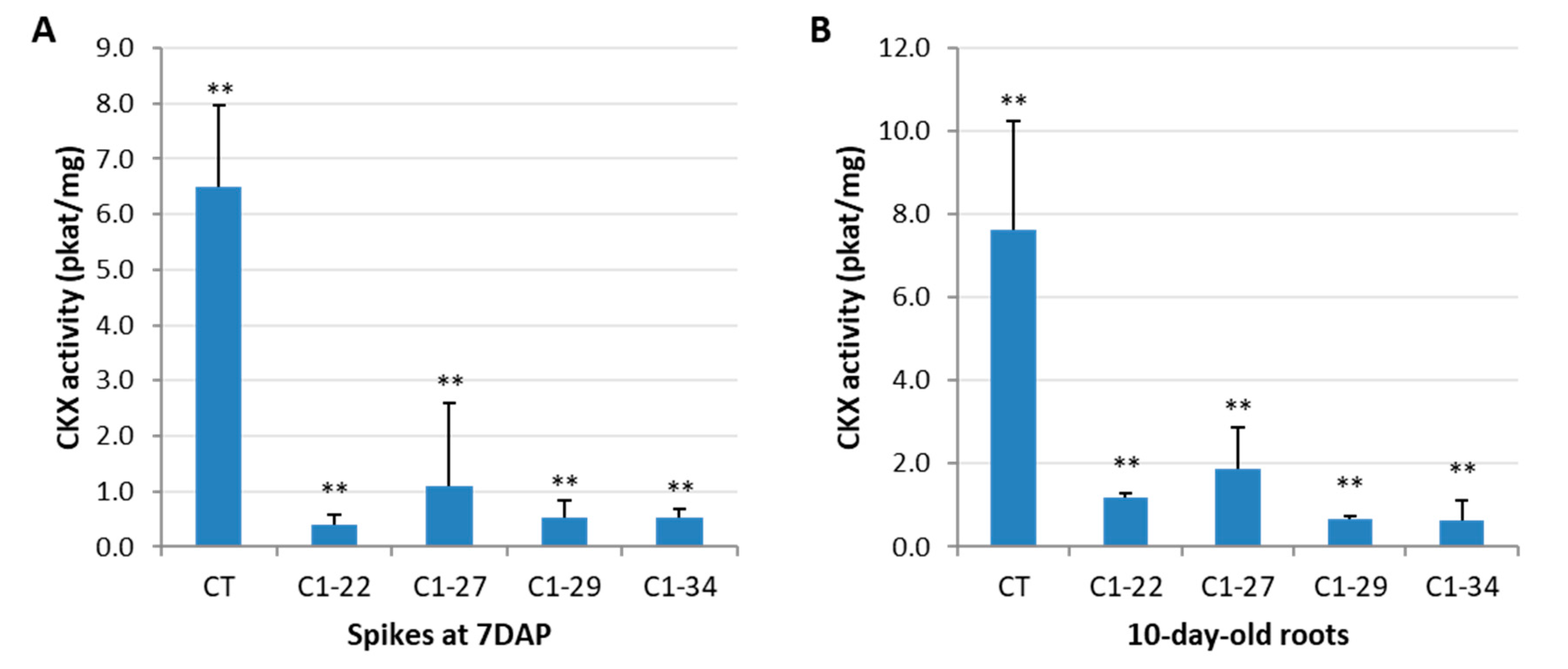
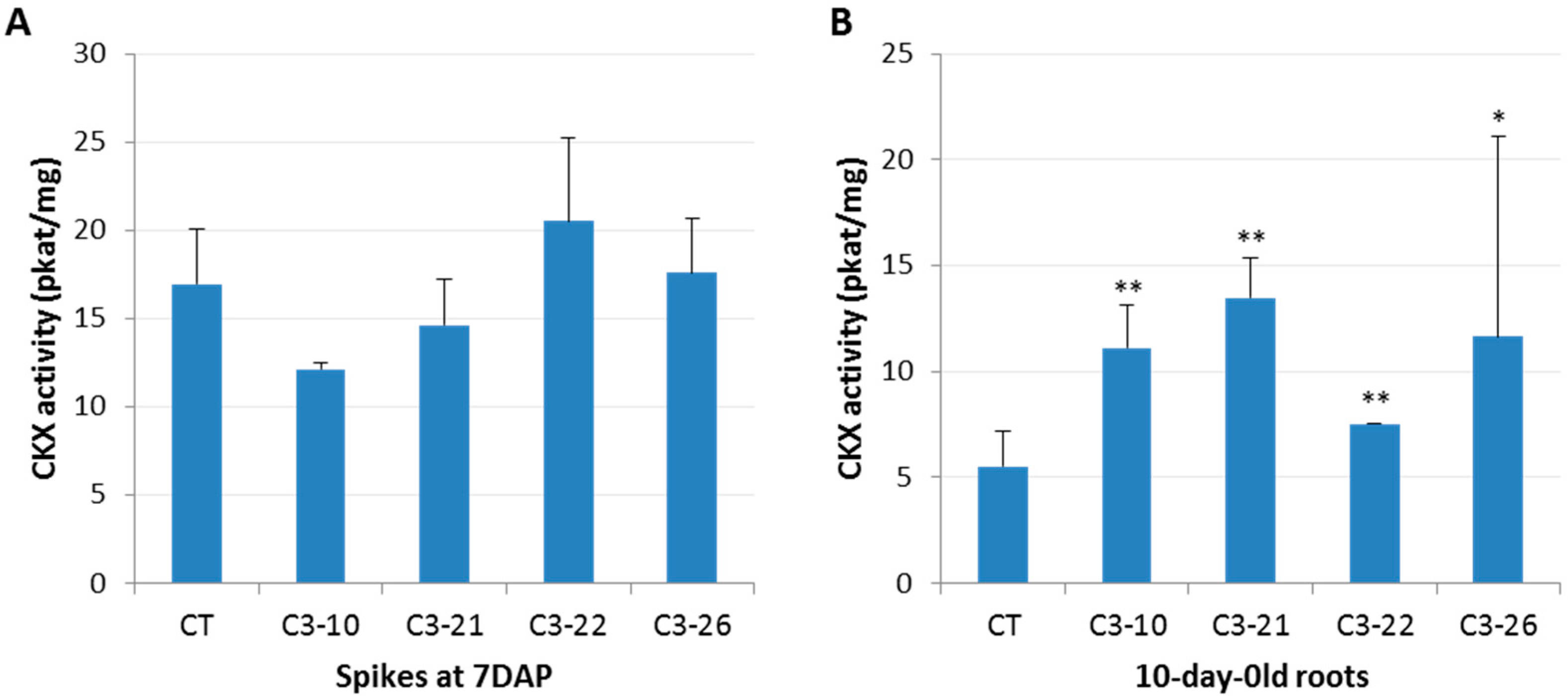
3.2. CKX Activity in the Spikes and Roots
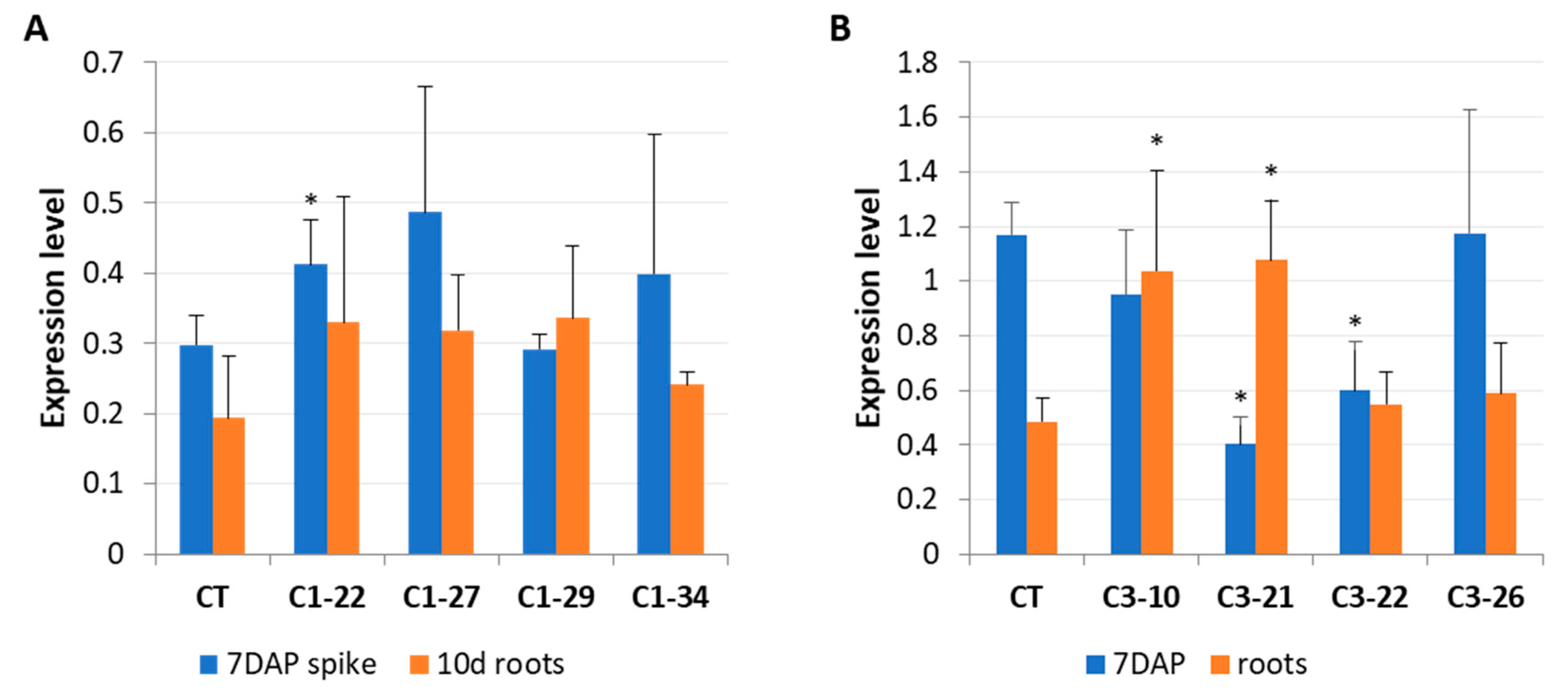
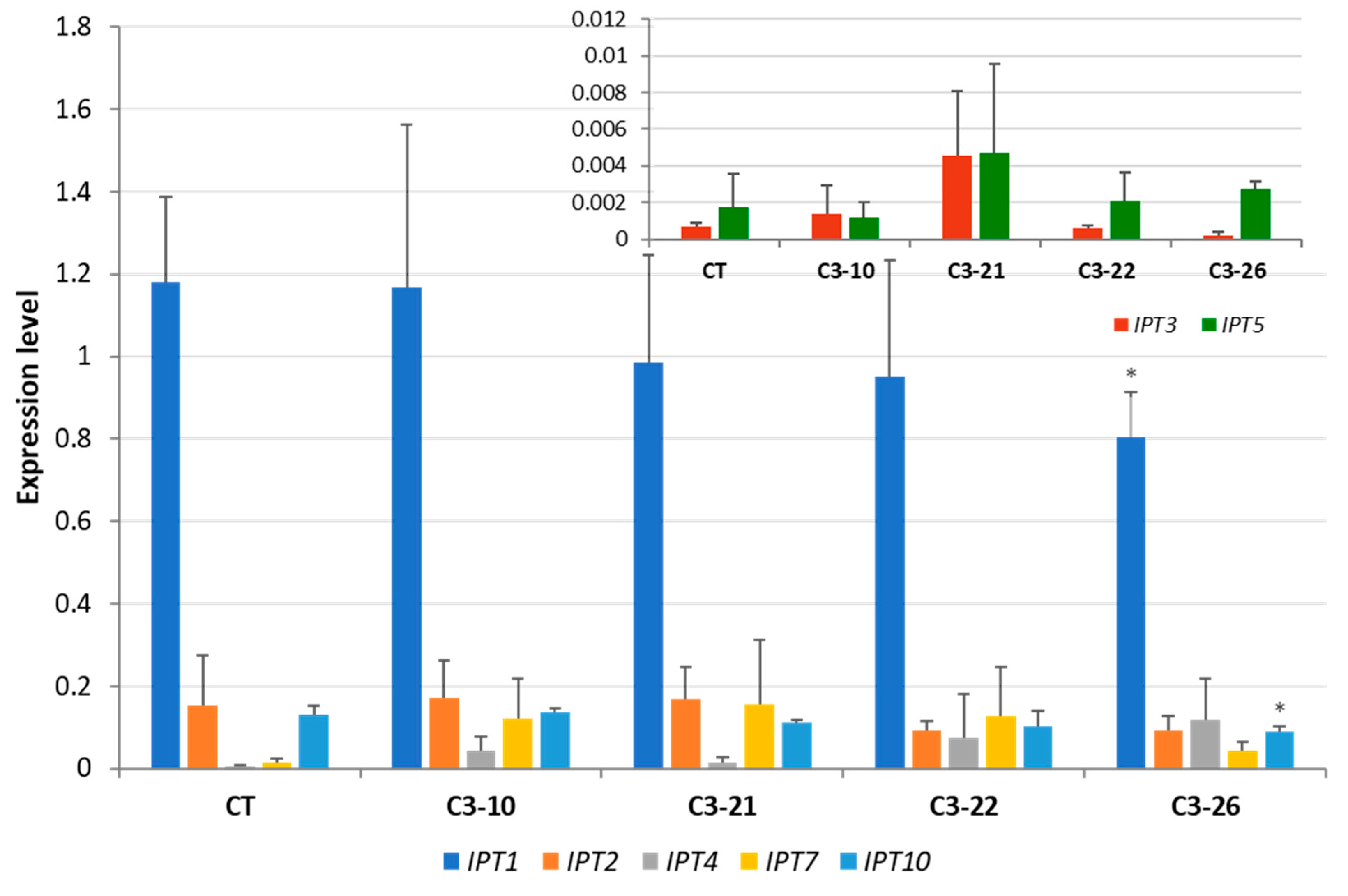
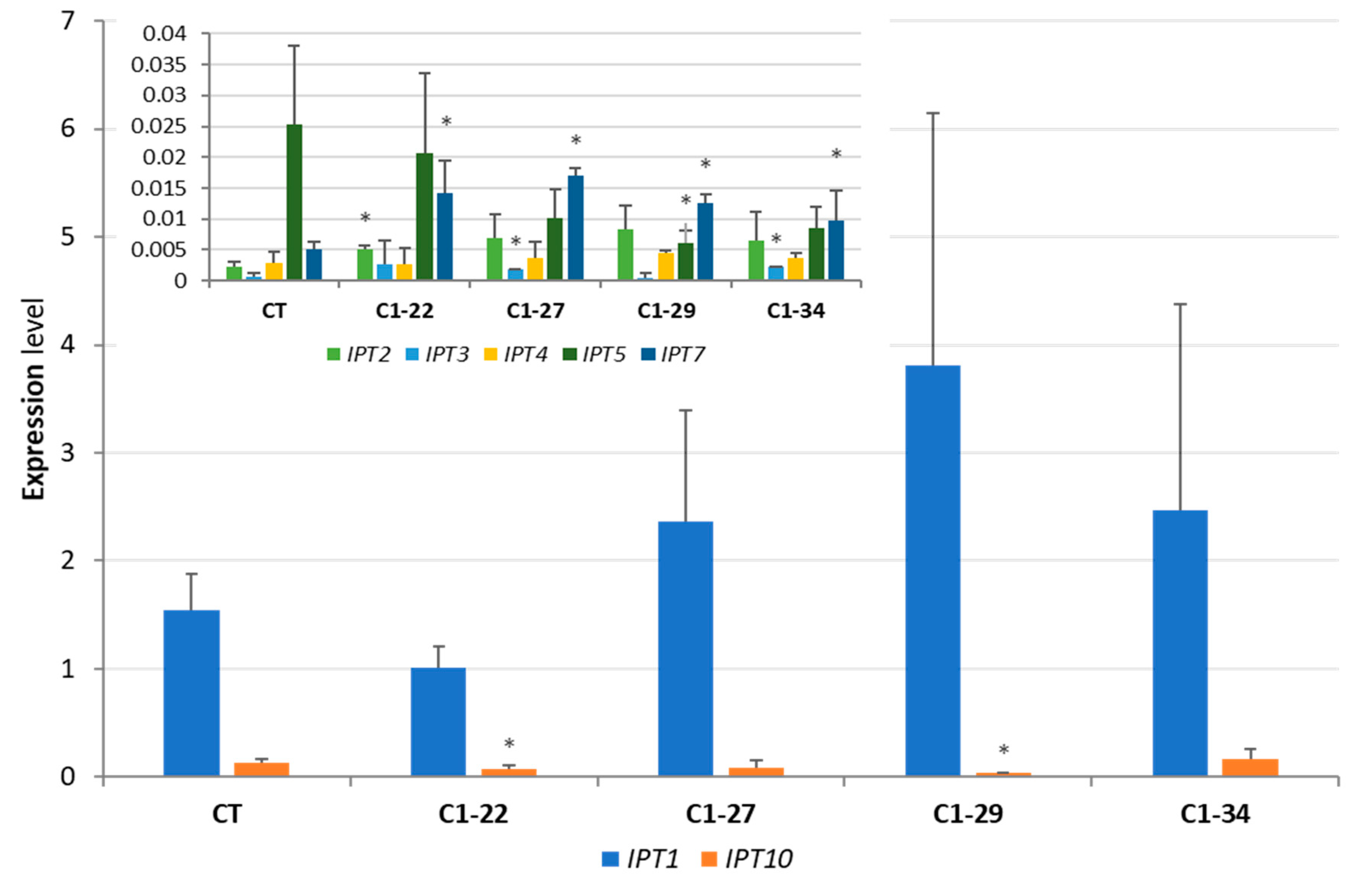
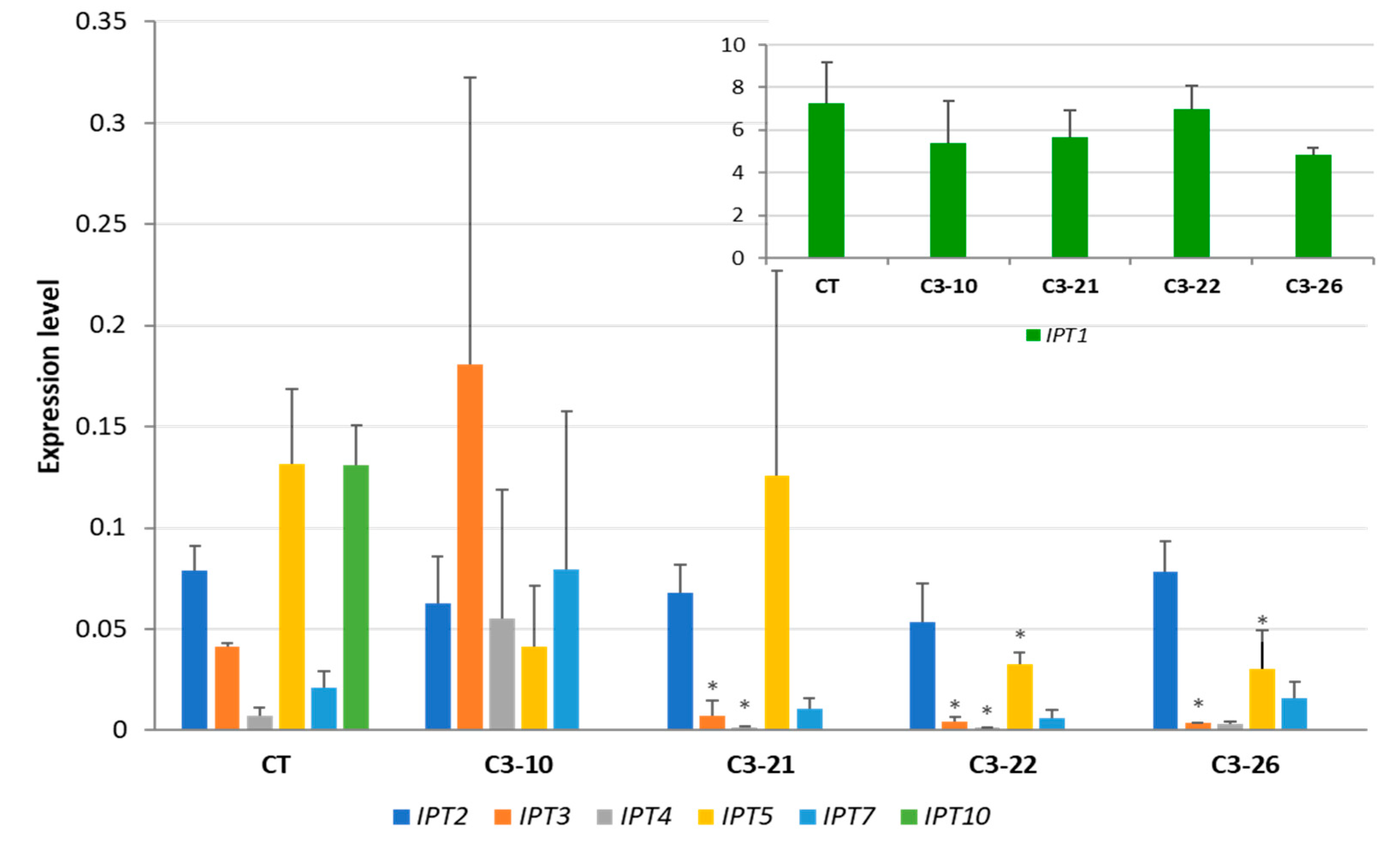
3.3. Expression of IPT Genes in the ckx1 and ckx3 Mutant Lines
3.4. Transcriptome Analysis of the ckx1 and ckx3 Mutant Lines
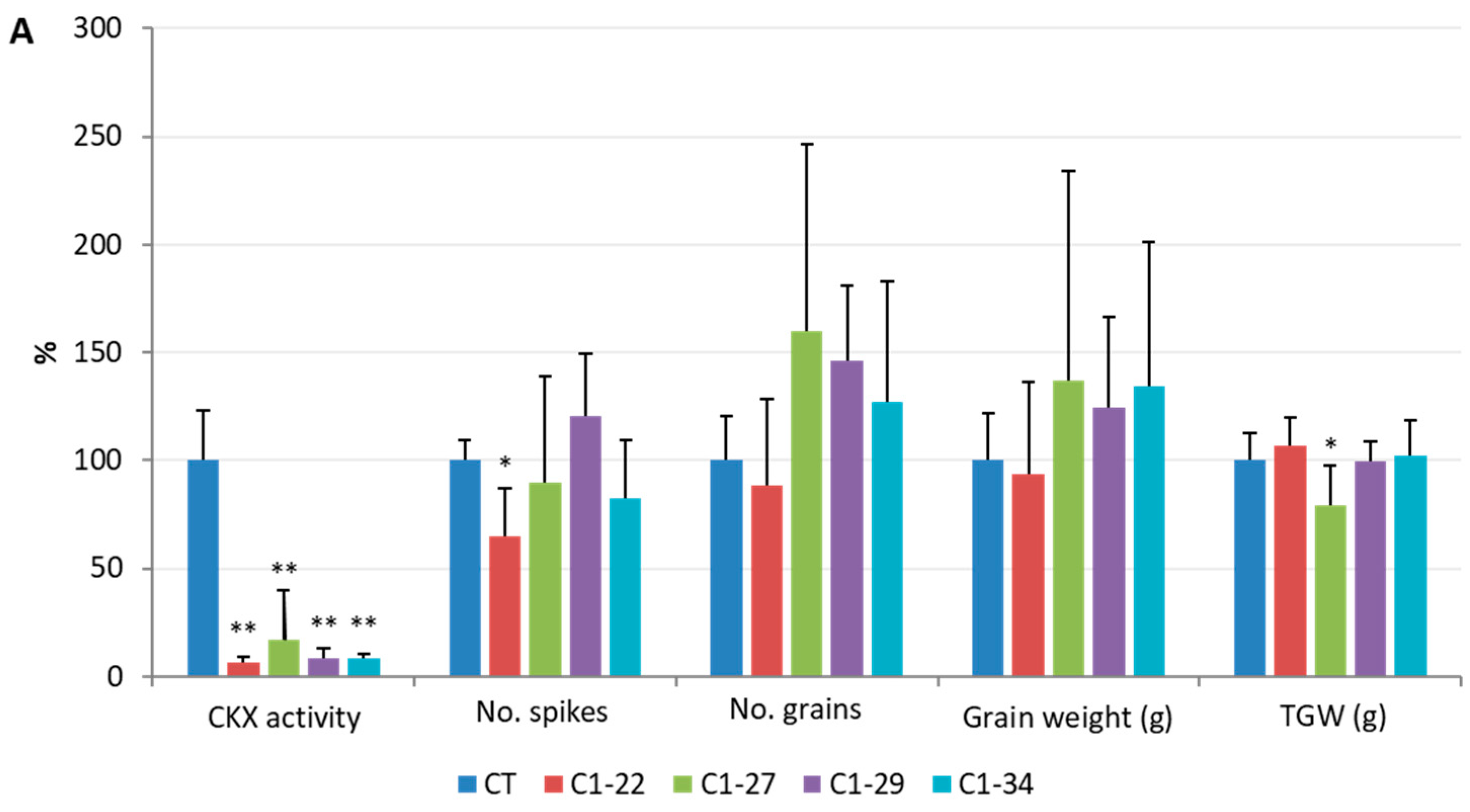
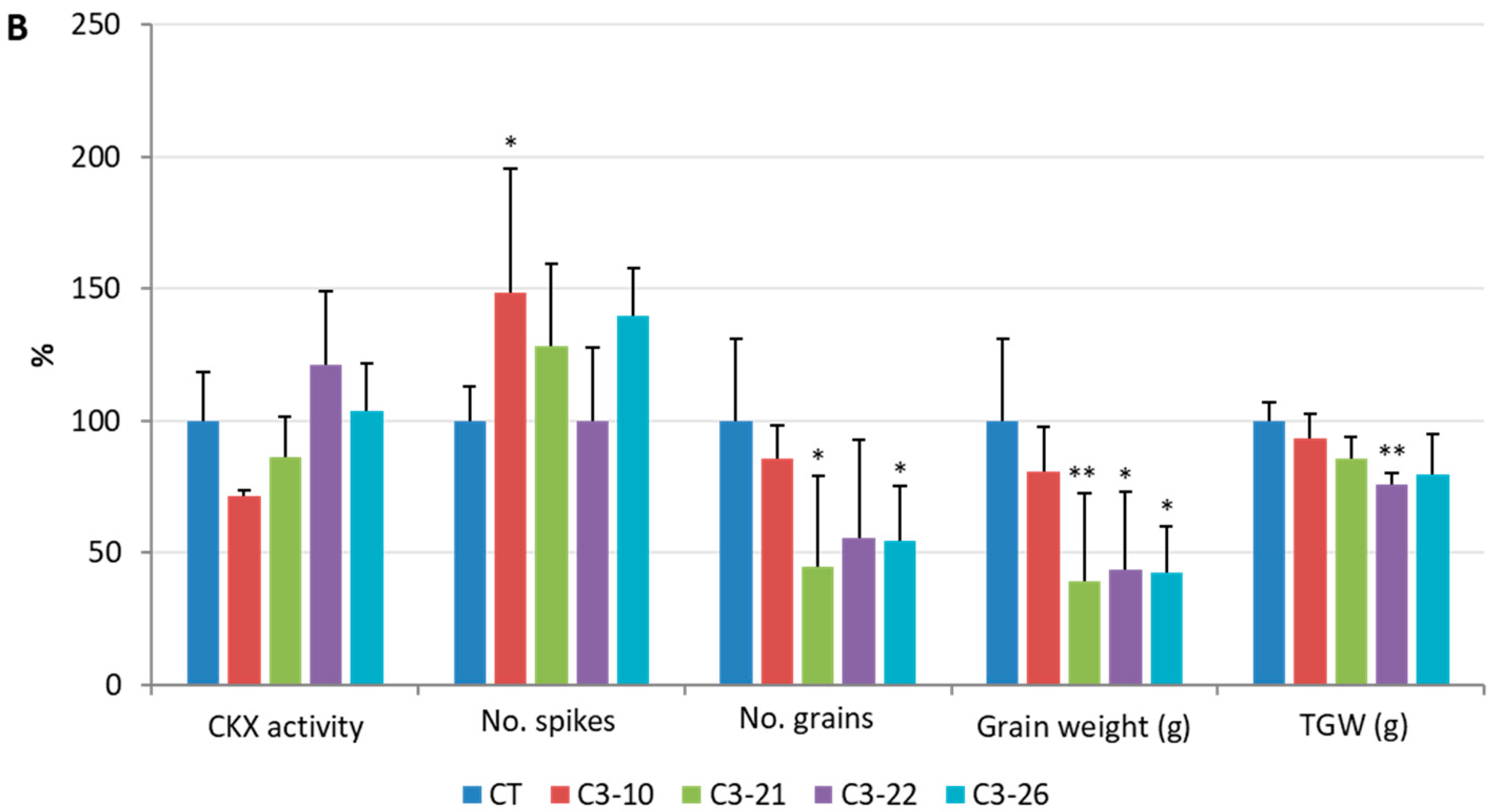
3.5. Analysis of Yield-Related Traits in the ckx1 and ckx3 Mutant Lines
4. Discussion
4.1. Cross-Regulation of Cytokinin Gene Family Members
4.2. Knockout of HvCKX1 and HvCKX3 Did Not Enhance Grain Yields but Affected the Root Phenotype
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Available online: http://www.fao.org/faostat/en/#home (accessed on 30 April 2019).
- Bortesi, L.; Fischer, R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol. Adv. 2015, 33, 41–52. [Google Scholar] [CrossRef]
- Lawrenson, T.; Shorinola, O.; Stacey, N.; Li, C.D.; Ostergaard, L.; Patron, N.; Uauy, C.; Harwood, W. Induction of targeted, heritable mutations in barley and Brassica oleracea using RNA-guided Cas9 nuclease. Genome Biol. 2015, 16. [Google Scholar] [CrossRef]
- Kapusi, E.; Corcuera-Gómez, M.; Melnik, S.; Stoger, E. Heritable Genomic Fragment Deletions and Small Indels in the Putative ENGase Gene Induced by CRISPR/Cas9 in Barley. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef]
- Gasparis, S.; Kala, M.; Przyborowski, M.; Lyznik, L.A.; Orczyk, W.; Nadolska-Orczyk, A. A simple and efficient CRISPR/Cas9 platform for induction of single and multiple, heritable mutations in barley (Hordeum vulgare L.). Plant Methods 2018, 14, 111. [Google Scholar] [CrossRef]
- Holubova, K.; Hensel, G.; Vojta, P.; Tarkowski, P.; Bergougnoux, V.; Galuszka, P. Modification of Barley Plant Productivity Through Regulation of Cytokinin Content by Reverse-Genetics Approaches. Front. Plant Sci. 2018, 9, 1676. [Google Scholar] [CrossRef]
- Zhu, C.F.; Bortesi, L.; Baysal, C.; Twyman, R.M.; Fischer, R.; Capell, T.; Schillberg, S.; Christou, P. Characteristics of Genome Editing Mutations in Cereal Crops. Trends Plant Sci. 2017, 22, 38–52. [Google Scholar] [CrossRef]
- Nadolska-Orczyk, A.; Rajchel, I.K.; Orczyk, W.; Gasparis, S. Major genes determining yield-related traits in wheat and barley. Theor. Appl. Genet. 2017, 130, 1081–1098. [Google Scholar] [CrossRef]
- Jameson, P.E.; Song, J. Cytokinin: A key driver of seed yield. J. Exp. Bot. 2016, 67, 593–606. [Google Scholar] [CrossRef]
- Vankova, R. Cytokinin Regulation of Plant Growth and Stress Responses. In Phytohormones: A Window to Metabolism, Signaling and Biotechnological Applications; Tran, L.-S.P., Pal, S., Eds.; Springer: New York, NY, USA, 2014; pp. 55–79. [Google Scholar]
- Vidhyasekaran, P. Plant Hormone Signaling Systems in Plant Innate Immunity; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar]
- Ahammed, G.J. Plant Hormones under Challenging Environmental Factors; Springer: Dordrecht, The Netherlands, 2016. [Google Scholar]
- Cortleven, A.; Leuendorf, J.E.; Frank, M.; Pezzetta, D.; Bolt, S.; Schmülling, T. Cytokinin action in response to abiotic and biotic stresses in plants. Plant Cell Environ. 2019, 42, 998–1018. [Google Scholar] [CrossRef]
- Werner, T.; Kollmer, I.; Bartrina, I.; Holst, K.; Schmulling, T. New insights into the biology of cytokinin degradation. Plant Biol. 2006, 8, 371–381. [Google Scholar] [CrossRef]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Li, S.; Zhao, B.; Yuan, D.; Duan, M.; Qian, Q.; Tang, L.; Wang, B.; Liu, X.; Zhang, J.; Wang, J.; et al. Rice zinc finger protein DST enhances grain production through controlling Gn1a/OsCKX2 expression. Proc. Natl. Acad. Sci. USA 2013, 110, 3167–3172. [Google Scholar] [CrossRef]
- Bartrina, I.; Otto, E.; Strnad, M.; Werner, T.; Schmulling, T. Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell 2011, 23, 69–80. [Google Scholar] [CrossRef]
- Zalewski, W.; Galuszka, P.; Gasparis, S.; Orczyk, W.; Nadolska-Orczyk, A. Silencing of the HvCKX1 gene decreases the cytokinin oxidase/dehydrogenase level in barley and leads to higher plant productivity. J. Exp. Bot. 2010, 61, 1839–1851. [Google Scholar] [CrossRef]
- Zalewski, W.; Orczyk, W.; Gasparis, S.; Nadolska-Orczyk, A. HvCKX2 gene silencing by biolistic or Agrobacterium-mediated transformation in barley leads to different phenotypes. BMC Plant Biol. 2012, 12, 206. [Google Scholar] [CrossRef]
- Zhao, J.; Bai, W.; Zeng, Q.; Song, S.; Zhang, M.; Li, X.; Hou, L.; Xiao, Y.; Luo, M.; Li, D.; et al. Moderately enhancing cytokinin level by down-regulation of GhCKX expression in cotton concurrently increases fiber and seed yield. Mol. Breed. 2015, 35, 60. [Google Scholar] [CrossRef]
- Li, M.; Li, X.; Zhou, Z.; Wu, P.; Fang, M.; Pan, X.; Lin, Q.; Luo, W.; Wu, G.; Li, H. Reassessment of the Four Yield-related Genes Gn1a, DEP1, GS3, and IPA1 in Rice Using a CRISPR/Cas9 System. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef]
- Werner, T.; Motyka, V.; Laucou, V.; Smets, R.; Van Onckelen, H.; Schmulling, T. Cytokinin-deficient transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell 2003, 15, 2532–2550. [Google Scholar] [CrossRef]
- Mrizova, K.; Jiskrova, E.; Vyroubalova, S.; Novak, O.; Ohnoutkova, L.; Pospisilova, H.; Frebort, I.; Harwood, W.A.; Galuszka, P. Overexpression of cytokinin dehydrogenase genes in barley (Hordeum vulgare cv. Golden Promise) fundamentally affects morphology and fertility. PLoS ONE 2013, 8, e79029. [Google Scholar] [CrossRef]
- Zalewski, W.; Gasparis, S.; Boczkowska, M.; Rajchel, I.K.; Kala, M.; Orczyk, W.; Nadolska-Orczyk, A. Expression Patterns of HvCKX Genes Indicate Their Role in Growth and Reproductive Development of Barley. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Xiao, A.; Cheng, Z.; Kong, L.; Zhu, Z.; Lin, S.; Gao, G.; Zhang, B. CasOT: A genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 2014, 30, 1180–1182. [Google Scholar] [CrossRef]
- BRACT. Available online: https://www.jic.ac.uk/technologies/genomic-services/bract/ (accessed on 30 April 2019).
- Harwood, W.A.; Bartlett, J.G.; Alves, S.C.; Perry, M.; Smedley, M.A.; Leyl, N.; Snape, J.W. Barley Transformation Using Agrobacterium-Mediated Techniques. In Transgenic Wheat, Barley and Oats: Production and Characterization Protocols; Jones, H.D., Shewry, P.R., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 137–147. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef]
- Frebort, I.; Sebela, M.; Galuszka, P.; Werner, T.; Schmulling, T.; Pec, P. Cytokinin oxidase/cytokinin dehydrogenase assay: Optimized procedures and applications. Anal. Biochem. 2002, 306, 1–7. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 2014, 37, 1250–1258. [Google Scholar] [CrossRef]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Muller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Hoagland, D.R.; Arnon, D.I. The water-culture method for growing plants without soil. Calif. Agric. Exp. Stn. Circ. 1950, 347, 32. [Google Scholar]
- Travella, S.; Klimm, T.E.; Keller, B. RNA interference-based gene silencing as an efficient tool for functional genomics in hexaploid bread wheat. Plant Physiol. 2006, 142, 6–20. [Google Scholar] [CrossRef]
- Gasparis, S.; Kala, M.; Przyborowski, M.; Orczyk, W.; Nadolska-Orczyk, A. Artificial MicroRNA-Based Specific Gene Silencing of Grain Hardness Genes in Polyploid Cereals Appeared to Be Not Stable Over Transgenic Plant Generations. Front. Plant Sci. 2016, 7, 2017. [Google Scholar] [CrossRef]
- Waterhouse, P.M.; Helliwell, C.A. Exploring plant genomes by RNA-induced gene silencing. Nat. Rev. Genet. 2003, 4, 29–38. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Xie, K.; Minkenberg, B.; Yang, Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl. Acad. Sci. USA 2015, 112, 3570–3575. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, B.; Weeks, D.P.; Spalding, M.H.; Yang, B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 2014, 42, 10903–10914. [Google Scholar] [CrossRef]
- Xing, H.L.; Dong, L.; Wang, Z.P.; Zhang, H.Y.; Han, C.Y.; Liu, B.; Wang, X.C.; Chen, Q.J. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 2014, 14, 327. [Google Scholar] [CrossRef]
- Svitashev, S.; Young, J.K.; Schwartz, C.; Gao, H.; Falco, S.C.; Cigan, A.M. Targeted Mutagenesis, Precise Gene Editing, and Site-Specific Gene Insertion in Maize Using Cas9 and Guide RNA. Plant Physiol. 2015, 169, 931–945. [Google Scholar] [CrossRef]
- Werner, T.; Schmulling, T. Cytokinin action in plant development. Curr. Opin. Plant Biol. 2009, 12, 527–538. [Google Scholar] [CrossRef]
- Song, J.; Jiang, L.; Jameson, P.E. Co-ordinate regulation of cytokinin gene family members during flag leaf and reproductive development in wheat. BMC Plant Biol. 2012, 12, 78. [Google Scholar] [CrossRef]
- Vyroubalova, S.; Vaclavikova, K.; Tureckova, V.; Novak, O.; Smehilova, M.; Hluska, T.; Ohnoutkova, L.; Frebort, I.; Galuszka, P. Characterization of new maize genes putatively involved in cytokinin metabolism and their expression during osmotic stress in relation to cytokinin levels. Plant Physiol. 2009, 151, 433–447. [Google Scholar] [CrossRef]
- Brugiere, N.; Humbert, S.; Rizzo, N.; Bohn, J.; Habben, J.E. A member of the maize isopentenyl transferase gene family, Zea mays isopentenyl transferase 2 (ZmIPT2), encodes a cytokinin biosynthetic enzyme expressed during kernel development. Cytokinin biosynthesis in maize. Plant Mol. Biol. 2008, 67, 215–229. [Google Scholar] [CrossRef]
- Podlesakova, K.; Zalabak, D.; Cudejkova, M.; Plihal, O.; Szucova, L.; Dolezal, K.; Spichal, L.; Strnad, M.; Galuszka, P. Novel cytokinin derivatives do not show negative effects on root growth and proliferation in submicromolar range. PLoS ONE 2012, 7, e39293. [Google Scholar] [CrossRef]
- Ogonowska, H.; Barchacka, K.; Gasparis, S.; Jablonski, B.; Orczyk, W.; Dmochowska-Boguta, M.; Nadolska-Orczyk, A. Specificity of expression of TaCKX family genes in developing plants of wheat and their co-operation within and among organs. PLoS ONE 2019, 14, e0214239. [Google Scholar] [CrossRef]
- Kurakawa, T.; Ueda, N.; Maekawa, M.; Kobayashi, K.; Kojima, M.; Nagato, Y.; Sakakibara, H.; Kyozuka, J. Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 2007, 445, 652–655. [Google Scholar] [CrossRef]
- Martin, R.C.; Mok, M.C.; Mok, D.W. Isolation of a cytokinin gene, ZOG1, encoding zeatin O-glucosyltransferase from Phaseolus lunatus. Proc. Natl. Acad. Sci. USA 1999, 96, 284–289. [Google Scholar] [CrossRef]
- Martin, R.C.; Mok, M.C.; Habben, J.E.; Mok, D.W. A maize cytokinin gene encoding an O-glucosyltransferase specific to cis-zeatin. Proc. Natl. Acad. Sci. USA 2001, 98, 5922–5926. [Google Scholar] [CrossRef]
- Brzobohaty, B.; Moore, I.; Kristoffersen, P.; Bako, L.; Campos, N.; Schell, J.; Palme, K. Release of active cytokinin by a beta-glucosidase localized to the maize root meristem. Science 1993, 262, 1051–1054. [Google Scholar] [CrossRef]
- Kiran, N.S.; Benkova, E.; Rekova, A.; Dubova, J.; Malbeck, J.; Palme, K.; Brzobohaty, B. Retargeting a maize beta-glucosidase to the vacuole--evidence from intact plants that zeatin-O-glucoside is stored in the vacuole. Phytochemistry 2012, 79, 67–77. [Google Scholar] [CrossRef]
- Chandler, J.W.; Werr, W. Cytokinin-auxin crosstalk in cell type specification. Trends Plant Sci. 2015, 20, 291–300. [Google Scholar] [CrossRef]
- Birnbaum, K.D. How many ways are there to make a root? Curr. Opin. Plant Biol. 2016, 34, 61–67. [Google Scholar] [CrossRef]
- Choudhary, S.P.; Yu, J.Q.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S. Benefits of brassinosteroid crosstalk. Trends Plant Sci. 2012, 17, 594–605. [Google Scholar] [CrossRef]
- Zwack, P.J.; Rashotte, A.M. Interactions between cytokinin signalling and abiotic stress responses. J. Exp. Bot. 2015, 66, 4863–4871. [Google Scholar] [CrossRef]
- Horvath, E.; Szalai, G.; Janda, T. Induction of abiotic stress tolerance by salicylic acid signaling. J. Plant Growth Regul. 2007, 26, 290–300. [Google Scholar] [CrossRef]
- Kazan, K. Diverse roles of jasmonates and ethylene in abiotic stress tolerance. Trends Plant Sci. 2015, 20, 219–229. [Google Scholar] [CrossRef]
- O’Brien, J.A.; Benkova, E. Cytokinin cross-talking during biotic and abiotic stress responses. Front. Plant Sci. 2013, 4. [Google Scholar] [CrossRef]
- Park, J.; Lee, Y.; Martinoia, E.; Geisler, M. Plant hormone transporters: What we know and what we would like to know. BMC Biol. 2017, 15, 93. [Google Scholar] [CrossRef]
- Durán-Medina, Y.; Díaz-Ramírez, D.; Marsch-Martínez, N. Cytokinins on the Move. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Y.L.; Gao, L.F.; Zhao, G.Y.; Zhou, R.H.; Zhang, B.S.; Jia, J.Z. TaCKX6-D1, the ortholog of rice OsCKX2, is associated with grain weight in hexaploid wheat. New Phytol. 2012, 195, 574–584. [Google Scholar] [CrossRef]
- Li, M.; Tang, D.; Wang, K.; Wu, X.; Lu, L.; Yu, H.; Gu, M.; Yan, C.; Cheng, Z. Mutations in the F-box gene LARGER PANICLE improve the panicle architecture and enhance the grain yield in rice. Plant Biotechnol. J. 2011, 9, 1002–1013. [Google Scholar] [CrossRef]
- Brugiere, N.; Jiao, S.; Hantke, S.; Zinselmeier, C.; Roessler, J.A.; Niu, X.; Jones, R.J.; Habben, J.E. Cytokinin oxidase gene expression in maize is localized to the vasculature, and is induced by cytokinins, abscisic acid, and abiotic stress. Plant Physiol. 2003, 132, 1228–1240. [Google Scholar] [CrossRef]
- Motyka, V.; Vankova, R.; Capkova, V.; Petrasek, J.; Kaminek, M.; Schmulling, T. Cytokinin-induced upregulation of cytokinin oxidase activity in tobacco includes changes in enzyme glycosylation and secretion. Physiol. Plant. 2003, 117, 11–21. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, Y.; Zhang, M.; Liu, Y.; Kong, L.; Zou, M.; Lu, G.; Cao, J.; Yu, X. Identification, expression, and comparative genomic analysis of the IPT and CKX gene families in Chinese cabbage (Brassica rapa ssp. pekinensis). BMC Genom. 2013, 14, 594. [Google Scholar] [CrossRef]
- Ehness, R.; Roitsch, T. Co-ordinated induction of mRNAs for extracellular invertase and a glucose transporter in Chenopodium rubrum by cytokinins. Plant J. Cell Mol. Biol. 1997, 11, 539–548. [Google Scholar] [CrossRef]
- Godt, D.E.; Roitsch, T. Regulation and tissue-specific distribution of mRNAs for three extracellular invertase isoenzymes of tomato suggests an important function in establishing and maintaining sink metabolism. Plant Physiol. 1997, 115, 273–282. [Google Scholar] [CrossRef]
- Roitsch, T.; Gonzalez, M.C. Function and regulation of plant invertases: Sweet sensations. Trends Plant Sci. 2004, 9, 606–613. [Google Scholar] [CrossRef]
- Pacifici, E.; Polverari, L.; Sabatini, S. Plant hormone cross-talk: The pivot of root growth. J. Exp. Bot. 2015, 66, 1113–1121. [Google Scholar] [CrossRef]
- Werner, T.; Nehnevajova, E.; Kollmer, I.; Novak, O.; Strnad, M.; Kramer, U.; Schmulling, T. Root-specific reduction of cytokinin causes enhanced root growth, drought tolerance, and leaf mineral enrichment in Arabidopsis and tobacco. Plant Cell 2010, 22, 3905–3920. [Google Scholar] [CrossRef]
- Li, X.; Mo, X.; Shou, H.; Wu, P. Cytokinin-Mediated Cell Cycling Arrest of Pericycle Founder Cells in Lateral Root Initiation of Arabidopsis. Plant Cell Physiol. 2006, 47, 1112–1123. [Google Scholar] [CrossRef]
- Hutchison, C.E.; Li, J.; Argueso, C.; Gonzalez, M.; Lee, E.; Lewis, M.W.; Maxwell, B.B.; Perdue, T.D.; Schaller, G.E.; Alonso, J.M.; et al. The Arabidopsis histidine phosphotransfer proteins are redundant positive regulators of cytokinin signaling. Plant Cell 2006, 18, 3073–3087. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| sgRNA Construct | Edited Gene | No. Explants | No. Transgenic Events (%) | No. Independent Transgenic Plants | No. PCR-RE-Detected Mutants |
|---|---|---|---|---|---|
| ckx1-sgRNA | HvCKX1 | 1143 | 137 (12) | 71 | 47 |
| ckx3-sgRNA | HvCKX3 | 931 | 76 (8) | 53 | 37 |
| Gene | Identifier | Description | C1-22 (ckx1) | C3-21 (ckx3) |
|---|---|---|---|---|
| Spikes at Seven DAP | Change in Expression (Fold Change) | |||
| CKX1 | HORVU3Hr1G019850 | Cytokinin oxidase/dehydrogenase | nd * | downregulation (−8.03) |
| CKX2.1 | HORVU3Hr1G027460 | Cytokinin oxidase/dehydrogenase | nd | downregulation (−2.83) |
| CKX2.2 | HORVU3Hr1G027430 | Cytokinin oxidase/dehydrogenase | upregulation (4.39) | downregulation (−5.29) |
| CKX3 | HORVU1Hr1G042360 | Cytokinin oxidase/dehydrogenase | upregulation (7.50) | nd |
| CKX4 | HORVU3Hr1G105360 | Cytokinin oxidase/dehydrogenase | downregulation (−5.00) | downregulation (−5.87) |
| CKX5 | HORVU3Hr1G075920 | Cytokinin oxidase/dehydrogenase | downregulation (−4.70) | downregulation (−2.57) |
| CKX9 | HORVU1Hr1G057860 | Cytokinin oxidase/dehydrogenase | downregulation (−6.70) | upregulation (4.91) |
| IPT1 | HORVU2Hr1G062320 | tRNA isopentenyltransferase | downregulation (−4.16) | downregulation (−5.58) |
| IPT2 | HORVU5Hr1G055220 | Adenylate isopentenyltransferase | downregulation (−4.17) | nd |
| IPT4 | HORVU1Hr1G011480 | Adenylate isopentenyltransferase | upregulation (5.32) | upregulation (3.60) |
| IPT10 | HORVU7Hr1G120070 | tRNA isopentenyltransferase | downregulation (−5.88) | downregulation (−4.81) |
| cZOG1 | HORVU2Hr1G096890 | Putative cis-zeatin O-glucosyltransferase (ortholog of TacZOG1) | nd | upregulation (4.23) |
| ZOG2 | HORVU2Hr1G004720 | Putative zeatin O-glucosyltransferase (ortholog of TaZOG2) | upregulation (4.70) | upregulation (5.64) |
| ZOG3 | HORVU7Hr1G038510 | Putative zeatin O-glucosyltransferase (ortholog of TaZOG3) | upregulation (7.19) | upregulation (2.69) |
| ZOG3 | HORVU2Hr1G075920 | Putative zeatin O-glucosyltransferase (ortholog of TaZOG3) | nd | upregulation (3.73) |
| LOG | HORVU3Hr1G050770 | Putative cytokinin riboside 5’-monophosphate phosphoribohydrolase (ortholog of LOC_Os01g40630.1) | nd | downregulation (−4.46) |
| LOG | HORVU3Hr1G066810 | Putative cytokinin riboside 5’-monophosphate phosphoribohydrolase (ortholog of LOC_Os01g51210.1) | downregulation (−4.00) | nd |
| LOG | HORVU1Hr1G079570 | Putative cytokinin riboside 5’-monophosphate phosphoribohydrolase (ortholog of LOC_Os05g46360.1) | downregulation (−6.04) | downregulation (−7.07) |
| LOG | HORVU4Hr1G005660 | Putative cytokinin riboside 5’-monophosphate phosphoribohydrolase (ortholog of LOC_Os03g49050.1) | downregulation (−6.23) | nd |
| LOG | HORVU1Hr1G094290 | Putative cytokinin riboside 5’-monophosphate phosphoribohydrolase (ortholog of LOC_Os05g51390.1) | downregulation (−7.02) | downregulation (−6.32) |
| LOG | HORVU5Hr1G124750 | Putative cytokinin riboside 5’-monophosphate phosphoribohydrolase (ortholog of LOC_Os03g64070.1) | nd | upregulation (6.77) |
| GLU | HORVU2Hr1G023590 | Β-glucosidase | downregulation (−8.24) | downregulation (−7.67) |
| 10-day-old roots | change in expression (fold change) | |||
| CKX1 | HORVU3Hr1G019850 | Cytokinin oxidase/dehydrogenase | nd | upregulation (0.53) |
| LOG | HORVU2Hr1G089620 | Putative cytokinin riboside 5’-monophosphate phosphoribohydrolase (ortholog of LOC_Os04g43840.1) | nd | downregulation (−3.41) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gasparis, S.; Przyborowski, M.; Kała, M.; Nadolska-Orczyk, A. Knockout of the HvCKX1 or HvCKX3 Gene in Barley (Hordeum vulgare L.) by RNA-Guided Cas9 Nuclease Affects the Regulation of Cytokinin Metabolism and Root Morphology. Cells 2019, 8, 782. https://doi.org/10.3390/cells8080782
Gasparis S, Przyborowski M, Kała M, Nadolska-Orczyk A. Knockout of the HvCKX1 or HvCKX3 Gene in Barley (Hordeum vulgare L.) by RNA-Guided Cas9 Nuclease Affects the Regulation of Cytokinin Metabolism and Root Morphology. Cells. 2019; 8(8):782. https://doi.org/10.3390/cells8080782
Chicago/Turabian StyleGasparis, Sebastian, Mateusz Przyborowski, Maciej Kała, and Anna Nadolska-Orczyk. 2019. "Knockout of the HvCKX1 or HvCKX3 Gene in Barley (Hordeum vulgare L.) by RNA-Guided Cas9 Nuclease Affects the Regulation of Cytokinin Metabolism and Root Morphology" Cells 8, no. 8: 782. https://doi.org/10.3390/cells8080782
APA StyleGasparis, S., Przyborowski, M., Kała, M., & Nadolska-Orczyk, A. (2019). Knockout of the HvCKX1 or HvCKX3 Gene in Barley (Hordeum vulgare L.) by RNA-Guided Cas9 Nuclease Affects the Regulation of Cytokinin Metabolism and Root Morphology. Cells, 8(8), 782. https://doi.org/10.3390/cells8080782