Molecular Mechanisms of Microglial Motility: Changes in Ageing and Alzheimer’s Disease


Abstract
1. Introduction
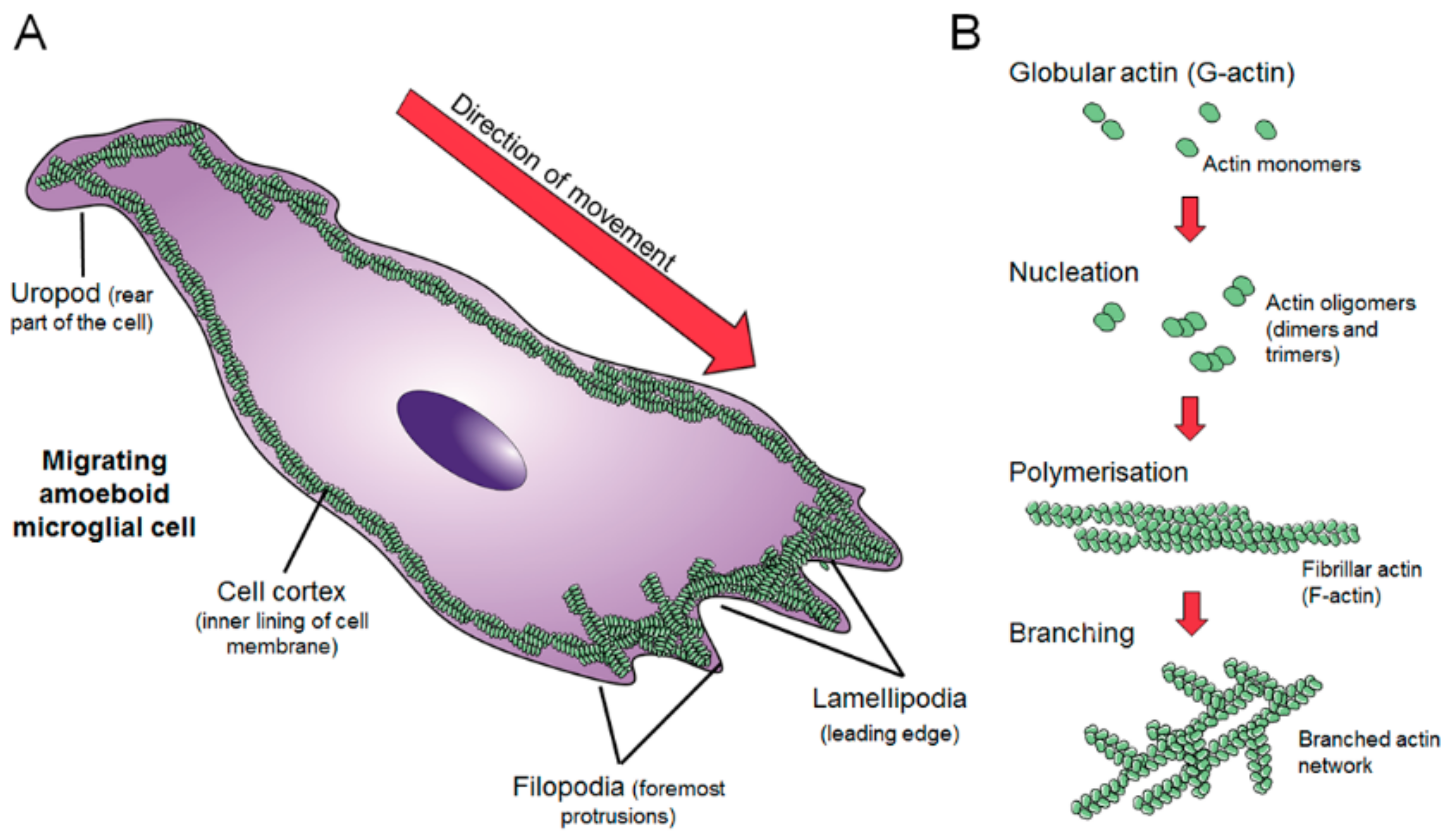
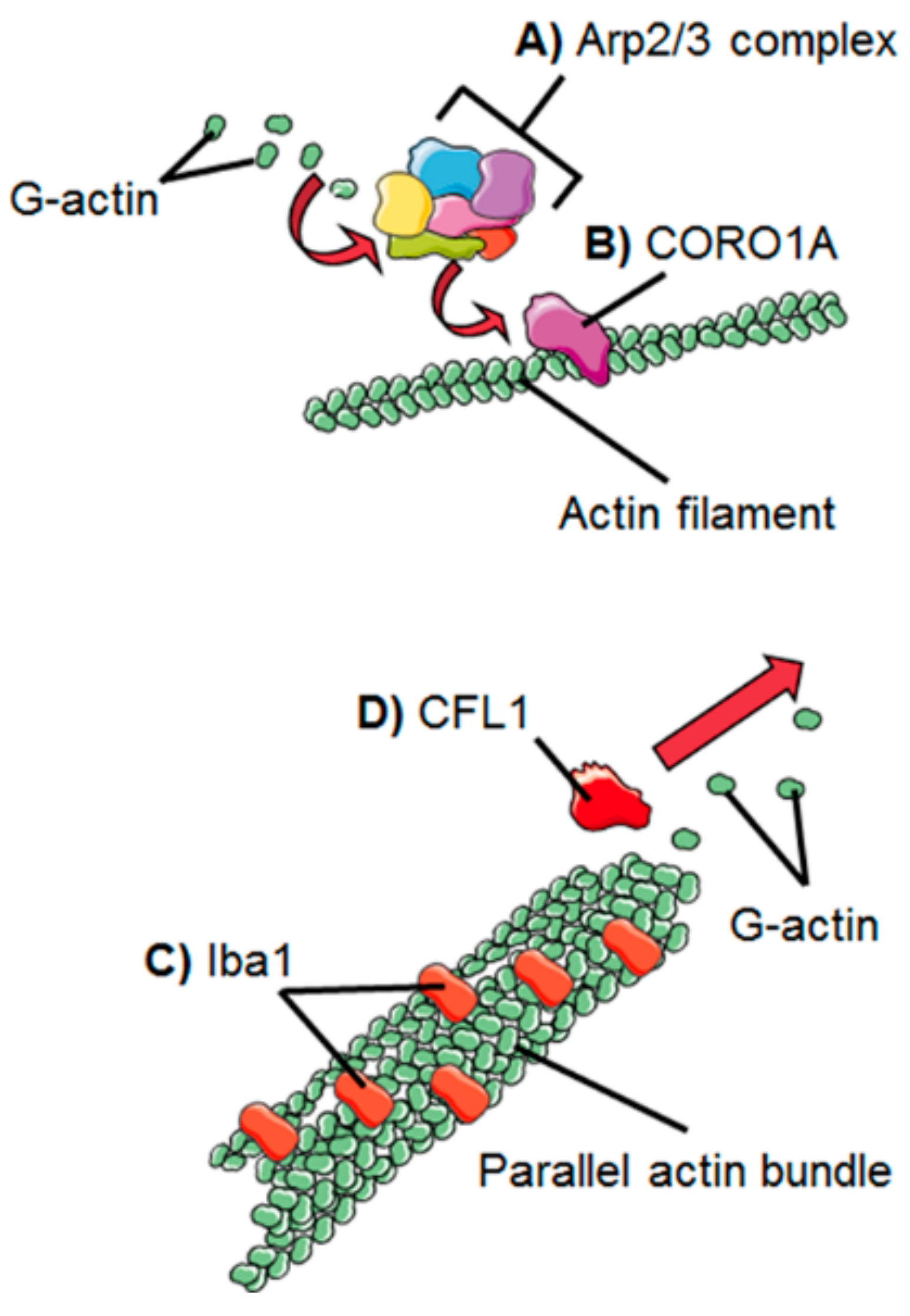
2. Dynamics of the Microglial Actin Cytoskeleton
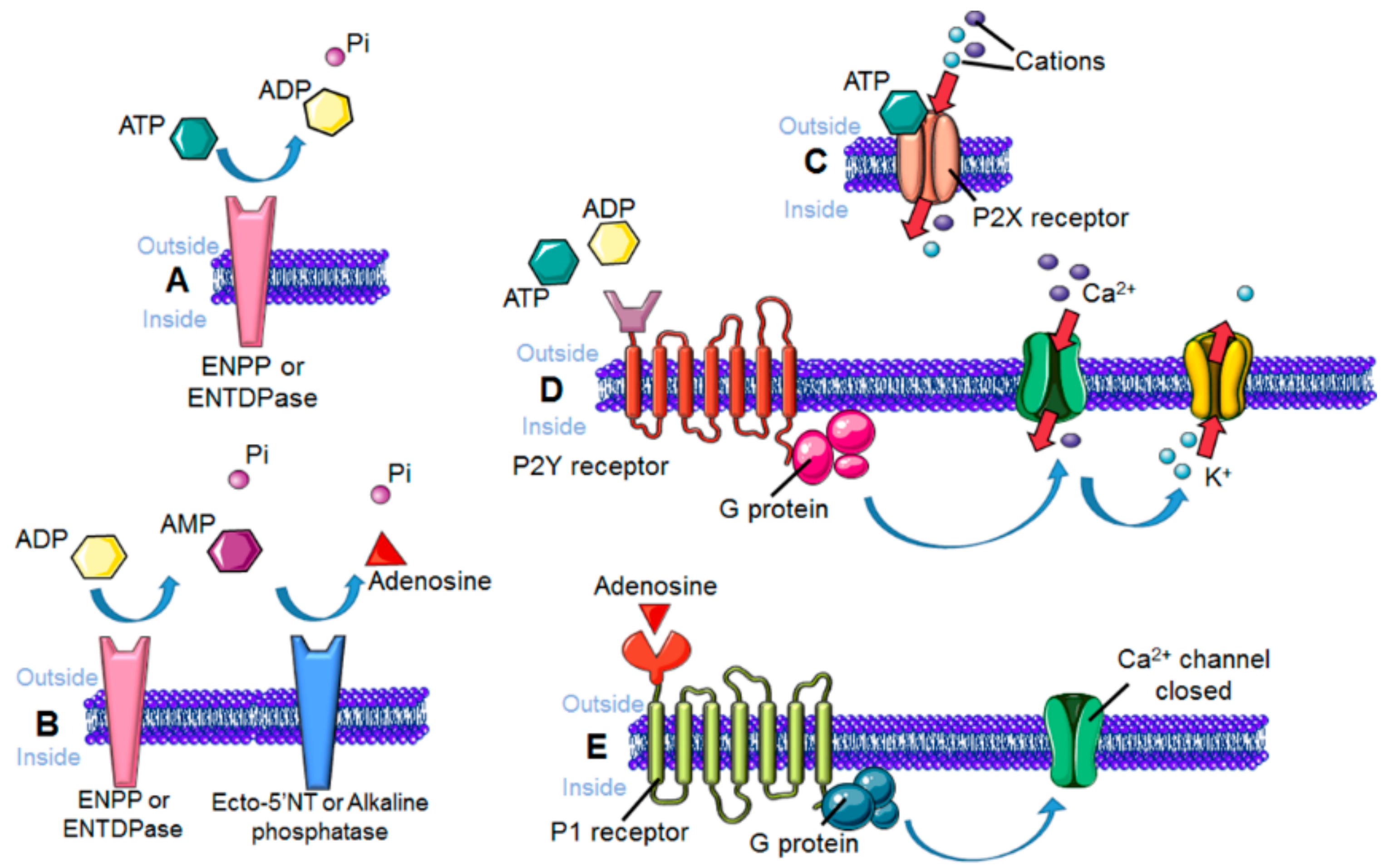
3. Sensing the Environment: Role of the Chemotactic Membrane Receptors
4. Microglial Morphology and Motility in Ageing
5. Age-Related Alterations in Microglial Motility-Related Proteins and Pathways
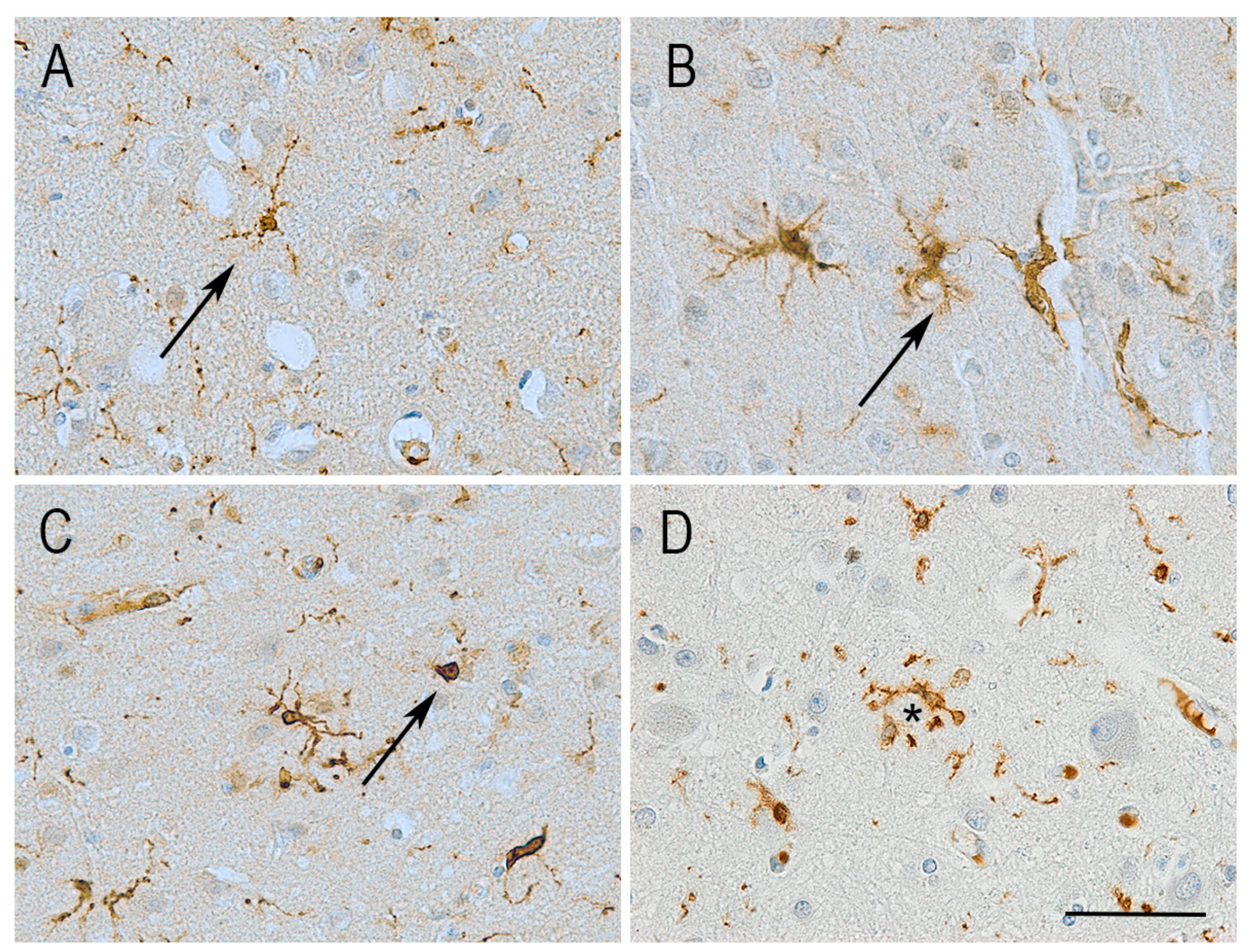
6. Microglial Morphology and Motility in Alzheimer’s Disease
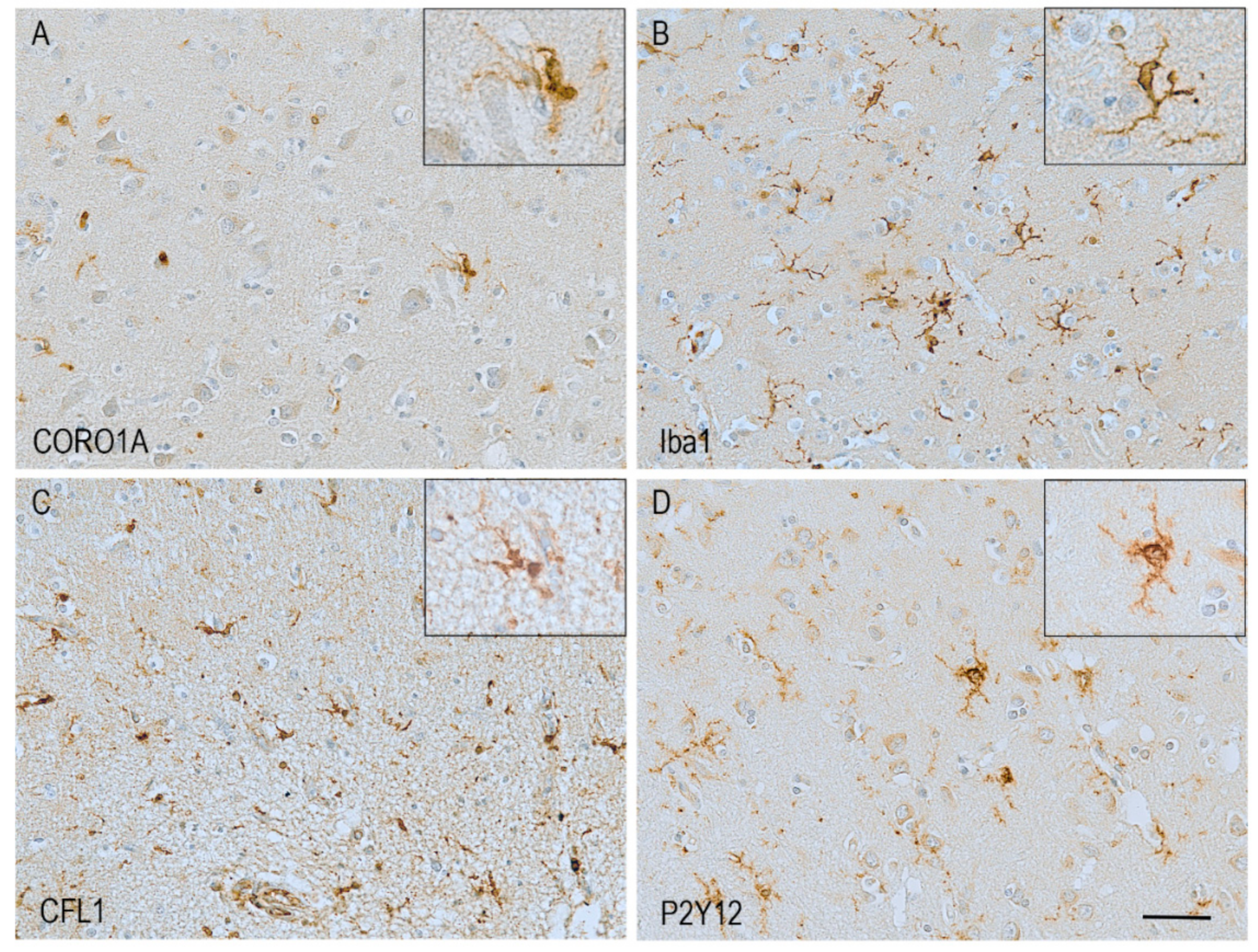
7. Pathological Changes in Actin-Related Proteins in Alzheimer’s Disease
8. Changes in Microglial Membrane Chemotactic Receptors in Alzheimer’s disease
9. Conclusions
Funding
Conflicts of Interest
References
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E. Identification of a unique tgf-[beta]-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Bilbo, S.D.; Schwarz, J.M. The immune system and developmental programming of brain and behavior. Front. Neuroendocrinol. 2012, 33, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E. Microglia in alzheimer brain: A neuropathological perspective. Int. J. Alzheimer’s Dis. 2012, 2012, 6. [Google Scholar] [CrossRef]
- Griffin, W.; Stanley, L.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.; White, C.; Araoz, C. Brain interleukin 1 and s-100 immunoreactivity are elevated in down syndrome and alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef]
- Roy, A.; Fung, Y.K.; Liu, X.; Pahan, K. Up-regulation of microglial cd11b expression by nitric oxide. J. Biol. Chem. 2006, 281, 14971–14980. [Google Scholar] [CrossRef]
- Melissa, A.; Lee, S.C. Human brain parenchymal microglia express cd14 and cd45 and are productively infected by hiv-1 in hiv-1 encephalitis. Brain Pathol. 2002, 12, 442–455. [Google Scholar]
- Mizuno, T.; Doi, Y.; Mizoguchi, H.; Jin, S.; Noda, M.; Sonobe, Y.; Takeuchi, H.; Suzumura, A. Interleukin-34 selectively enhances the neuroprotective effects of microglia to attenuate oligomeric amyloid-β neurotoxicity. Am. J. Pathol. 2011, 179, 2016–2027. [Google Scholar] [CrossRef]
- Minett, T.; Classey, J.; Matthews, F.E.; Fahrenhold, M.; Taga, M.; Brayne, C.; Ince, P.G.; Nicoll, J.A.; Boche, D. Microglial immunophenotype in dementia with Alzheimer’s pathology. J. Neuroinflamm. 2016, 13, 135. [Google Scholar] [CrossRef] [PubMed]
- Sterka, D.; Marriott, I. Characterization of nucleotide-binding oligomerization domain (nod) protein expression in primary murine microglia. J. Neuroimmunol. 2006, 179, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (damps) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [PubMed]
- Šišková, Z.; Page, A.; O’Connor, V.; Perry, V.H. Degenerating synaptic boutons in prion disease: Microglia activation without synaptic stripping. Am. J. Pathol. 2009, 175, 1610–1621. [Google Scholar] [CrossRef] [PubMed]
- Torres-Platas, S.G.; Comeau, S.; Rachalski, A.; Dal Bo, G.; Cruceanu, C.; Turecki, G.; Giros, B.; Mechawar, N. Morphometric characterization of microglial phenotypes in human cerebral cortex. J. Neuroinflamm. 2014, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Garden, G.A.; Möller, T. Microglia biology in health and disease. J. Neuroimmune Pharmacol. 2006, 1, 127–137. [Google Scholar] [CrossRef]
- Madry, C.; Attwell, D. Receptors, ion channels, and signaling mechanisms underlying microglial dynamics. J. Biol. Chem. 2015, 290, 12443–12450. [Google Scholar] [CrossRef]
- Khurana, B.; Khurana, T.; Khaire, N.; Noegel, A.A. Functions of lim proteins in cell polarity and chemotactic motility. EMBO J. 2002, 21, 5331–5342. [Google Scholar] [CrossRef]
- Hefendehl, J.K.; Neher, J.J.; Sühs, R.B.; Kohsaka, S.; Skodras, A.; Jucker, M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 2014, 13, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Wickstead, B.; Gull, K. The evolution of the cytoskeleton. J. Cell Biol. 2011, 194, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, F.C.; Bosman, F.T. The cytoskeleton and disease. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2004, 204, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S. Actin and microtubules in cell motility: Which one is in control? Traffic 2004, 5, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Kruse, K.; Joanny, J.-F.; Jülicher, F.; Prost, J.; Sekimoto, K. Asters, vortices, and rotating spirals in active gels of polar filaments. Phys. Rev. Lett. 2004, 92, 078101. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef]
- Lai, F.P.; Szczodrak, M.; Block, J.; Faix, J.; Breitsprecher, D.; Mannherz, H.G.; Stradal, T.E.; Dunn, G.A.; Small, J.V.; Rottner, K. Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 2008, 27, 982–992. [Google Scholar] [CrossRef]
- Gupton, S.L.; Gertler, F.B. Filopodia: The fingers that do the walking. Sci. STKE 2007, 2007, re5. [Google Scholar] [CrossRef]
- Hind, L.E.; Vincent, W.J.; Huttenlocher, A. Leading from the back: The role of the uropod in neutrophil polarization and migration. Dev. Cell. 2016, 38, 161–169. [Google Scholar] [CrossRef]
- Vinzenz, M.; Nemethova, M.; Schur, F.; Mueller, J.; Narita, A.; Urban, E.; Winkler, C.; Schmeiser, C.; Koestler, S.A.; Rottner, K.; et al. Actin branching in the initiation and maintenance of lamellipodia. J. Cell Sci. 2012, 125, 2775–2785. [Google Scholar] [CrossRef]
- Welch, M.D.; DePace, A.H.; Verma, S.; Iwamatsu, A.; Mitchison, T.J. The human arp2/3 complex is composed of evolutionarily conserved subunits and is localized to cellular regions of dynamic actin filament assembly. J. Cell Biol. 1997, 138, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Marchand, J.-B.; Kaiser, D.A.; Pollard, T.D.; Higgs, H.N. Interaction of wasp/scar proteins with actin and vertebrate arp2/3 complex. Nat. Cell Biol. 2001, 3, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.D. How wasp-family proteins and the arp2/3 complex convert intracellular signals into cytoskeletal structures. Curr. Opin. Cell Biol. 2000, 12, 91–96. [Google Scholar] [CrossRef]
- Rouiller, I.; Xu, X.-P.; Amann, K.J.; Egile, C.; Nickell, S.; Nicastro, D.; Li, R.; Pollard, T.D.; Volkmann, N.; Hanein, D. The structural basis of actin filament branching by the arp2/3 complex. J. Cell Biol. 2008, 180, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Neer, E.J.; Schmidt, C.J.; Nambudripad, R.; Smith, T.F. The ancient regulatory-protein family of wd-repeat proteins. Nature 1994, 371, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Min, J. Structure and function of wd40 domain proteins. Protein Cell 2011, 2, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Humphries, C.L.; Balcer, H.I.; D’Agostino, J.L.; Winsor, B.; Drubin, D.G.; Barnes, G.; Andrews, B.J.; Goode, B.L. Direct regulation of arp2/3 complex activity and function by the actin binding protein coronin. J. Cell Biol. 2002, 159, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Shaw, G.; Sharma, V.P.; Yang, C.; McGowan, E.; Dickson, D.W. Actin-binding proteins coronin-1a and iba-1 are effective microglial markers for immunohistochemistry. J. Histochem. Cytochem. 2007, 55, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-L.; Needham, K.M.; May, J.R.; Nolen, B.J. Mechanism of a concentration-dependent switch between activation and inhibition of arp2/3 complex by coronin. J. Biol. Chem. 2011, 286, 17039–17046. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- DeMali, K.A.; Wennerberg, K.; Burridge, K. Integrin signaling to the actin cytoskeleton. Curr. Opin. Cell Biol. 2003, 15, 572–582. [Google Scholar] [CrossRef]
- Zelen, J. The Role of the Cytoskeleton in Platelet Function. Master’s Thesis, Utrecht University, Utrecht, The Netherlands, 2012. [Google Scholar]
- Giannone, G.; Jiang, G.; Sutton, D.H.; Critchley, D.R.; Sheetz, M.P. Talin1 is critical for force-dependent reinforcement of initial integrin–cytoskeleton bonds but not tyrosine kinase activation. J. Cell Biol. 2003, 163, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, K.; Imai, Y.; Kanazawa, H.; Sasaki, Y.; Kohsaka, S. Involvement of iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J. Cell Sci. 2000, 113, 3073. [Google Scholar] [PubMed]
- Bartles, J.R. Parallel actin bundles and their multiple actin-bundling proteins. Curr. Opin. Cell Biol. 2000, 12, 72–78. [Google Scholar] [CrossRef]
- Ohsawa, K.; Imai, Y.; Sasaki, Y.; Kohsaka, S. Microglia/macrophage-specific protein iba1 binds to fimbrin and enhances its actin-bundling activity. J. Neurochem. 2004, 88, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Ohsawa, K.; Kanazawa, H.; Kohsaka, S.; Imai, Y. Iba1 is an actin-cross-linking protein in macrophages/microglia. Biochem. Biophys. Res. Commun. 2001, 286, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin ii takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Janßen, S.; Gudi, V.; Prajeeth, C.K.; Singh, V.; Stahl, K.; Heckers, S.; Skripuletz, T.; Pul, R.; Trebst, C.; Tsiavaliaris, G.; et al. A pivotal role of nonmuscle myosin ii during microglial activation. Exp. Neurol. 2014, 261, 666–676. [Google Scholar] [CrossRef]
- Dai, J.; Sheetz, M.P. Membrane tether formation from blebbing cells. Biophys. J. 1999, 77, 3363–3370. [Google Scholar] [CrossRef]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by lim-kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of actin reorganization by slingshot, a family of phosphatases that dephosphorylate adf/cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef]
- Samstag, Y.; John, I.; Wabnitz, G.H. Cofilin: A redox sensitive mediator of actin dynamics during t-cell activation and migration. Immunol. Rev. 2013, 256, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Alhadidi, Q.; Shah, Z.A. Cofilin mediates lps-induced microglial cell activation and associated neurotoxicity through activation of nf-κb and jak–stat pathway. Mol. Neurobiol. 2018, 55, 1676–1691. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.-c.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct rna sequencing. Nat. Neurosci. 2013, 16, 1896. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and cx3cr1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Garton, K.J.; Gough, P.J.; Blobel, C.P.; Murphy, G.; Greaves, D.R.; Dempsey, P.J.; Raines, E.W. Tumor necrosis factor-α-converting enzyme (adam17) mediates the cleavage and shedding of fractalkine (cx3cl1). J. Biol. Chem. 2001, 276, 37993–38001. [Google Scholar] [PubMed]
- Paolicelli, R.C.; Bisht, K.; Tremblay, M.-È. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell. Neurosci. 2014, 8, 129. [Google Scholar] [CrossRef]
- Liang, K.J.; Lee, J.E.; Wang, Y.D.; Ma, W.; Fontainhas, A.M.; Fariss, R.N.; Wong, W.T. Regulation of dynamic behavior of retinal microglia by cx3cr1 signaling. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4444–4451. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.-N.; Linden, J. International union of pharmacology. Xxv. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar]
- Haselkorn, M.L.; Shellington, D.K.; Jackson, E.K.; Vagni, V.A.; Janesko-Feldman, K.; Dubey, R.K.; Gillespie, D.G.; Cheng, D.; Bell, M.J.; Jenkins, L.W. Adenosine a1 receptor activation as a brake on the microglial response after experimental traumatic brain injury in mice. J. Neurotrauma 2010, 27, 901–910. [Google Scholar] [CrossRef]
- Orr, A.G.; Orr, A.L.; Li, X.-J.; Gross, R.E.; Traynelis, S.F. Adenosine a 2a receptor mediates microglial process retraction. Nat. Neurosci. 2009, 12, 872. [Google Scholar] [CrossRef] [PubMed]
- Koscsó, B.; Csóka, B.; Selmeczy, Z.; Himer, L.; Pacher, P.; Virág, L.; Haskó, G. Adenosine augments il-10 production by microglial cells through an a2b adenosine receptor-mediated process. J. Immunol. 2012, 188, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Hammarberg, C.; Schulte, G.; Fredholm, B.B. Evidence for functional adenosine a3 receptors in microglia cells. J. Neurochem. 2003, 86, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Junger, W.G. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 2011, 11, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Ohsawa, K.; Inoue, K.; Kohsaka, S. Purinergic receptors in microglia: Functional modal shifts of microglia mediated by p2 and p1 receptors. Glia 2013, 61, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Luongo, L.; Guida, F.; Imperatore, R.; Napolitano, F.; Gatta, L.; Cristino, L.; Giordano, C.; Siniscalco, D.; Di Marzo, V.; Bellini, G. The a1 adenosine receptor as a new player in microglia physiology. Glia 2014, 62, 122–132. [Google Scholar] [CrossRef]
- Ohsawa, K.; Sanagi, T.; Nakamura, Y.; Suzuki, E.; Inoue, K.; Kohsaka, S. Adenosine a3 receptor is involved in adp-induced microglial process extension and migration. J. Neurochem. 2012, 121, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Ralevic, V.; Burnstock, G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998, 50, 413–492. [Google Scholar]
- Burnstock, G. The therapeutic potential of purinergic signalling. Biochem. Pharmacol. 2018, 151, 157–165. [Google Scholar] [CrossRef]
- Sperlágh, B.; Illes, P. Purinergic modulation of microglial cell activation. Purinergic Signal. 2007, 3, 117–127. [Google Scholar] [CrossRef]
- Del Puerto, A.; Wandosell, F.; Garrido, J.J. Neuronal and glial purinergic receptors functions in neuron development and brain disease. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Light, A.R.; Wu, Y.; Hughen, R.W.; Guthrie, P.B. Purinergic receptors activating rapid intracellular ca2+ increases in microglia. Neuron Glia Biol. 2005, 2, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, K.; Irino, Y.; Nakamura, Y.; Akazawa, C.; Inoue, K.; Kohsaka, S. Involvement of p2x4 and p2y12 receptors in atp-induced microglial chemotaxis. Glia 2007, 55, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Lovewell, R.R.; Hayes, S.M.; O’Toole, G.A.; Berwin, B. Pseudomonas aeruginosa flagellar motility activates the phagocyte pi3k/akt pathway to induce phagocytic engulfment. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L698–L707. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.L.; Adams, D.G.; Vaillancourt, R.R.; Heimark, R.L. Signal transduction from n-cadherin increases bcl-2 regulation of the phosphatidylinositol 3-kinase/akt pathway by homophilic adhesion and actin cytoskeletal organization. J. Biol. Chem. 2002, 277, 32905–32914. [Google Scholar] [CrossRef]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of microglia by amyloid β requires p2x7 receptor expression. J. Immunol. 2009, 182, 4378–4385. [Google Scholar] [CrossRef]
- Verhoef, P.A.; Estacion, M.; Schilling, W.; Dubyak, G.R. P2x7 receptor-dependent blebbing and the activation of rho-effector kinases, caspases, and il-1β release. J. Immunol. 2003, 170, 5728–5738. [Google Scholar] [CrossRef]
- León-Otegui, M.; Gómez-Villafuertes, R.; Díaz-Hernández, J.I.; Díaz-Hernández, M.; Miras-Portugal, M.T.; Gualix, J. Opposite effects of p2x7 and p2y2 nucleotide receptors on α-secretase-dependent app processing in neuro-2a cells. FEBS Lett. 2011, 585, 2255–2262. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Boeynaems, J.-M.; Barnard, E.A.; Boyer, J.L.; Kennedy, C.; Knight, G.E.; Fumagalli, M.; Gachet, C.; Jacobson, K.A. International union of pharmacology lviii: Update on the p2y g protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 2006, 58, 281–341. [Google Scholar] [CrossRef]
- De Simone, R.; Niturad, C.E.; De Nuccio, C.; Ajmone-Cat, M.A.; Visentin, S.; Minghetti, L. Tgf-β and lps modulate adp-induced migration of microglial cells through p2y1 and p2y12 receptor expression. J. Neurochem. 2010, 115, 450–459. [Google Scholar] [CrossRef]
- Del Puerto, A.; Díaz-Hernández, J.-I.; Tapia, M.; Gomez-Villafuertes, R.; Benitez, M.J.; Zhang, J.; Miras-Portugal, M.T.; Wandosell, F.; Díaz-Hernández, M.; Garrido, J.J. Adenylate cyclase 5 coordinates the action of adp, p2y1, p2y13 and atp-gated p2x7 receptors on axonal elongation. J. Cell Sci. 2012, 125, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ghai, P.; Wu, H.; Wang, C.; Field, J.; Zhou, G.-L. Mammalian adenylyl cyclase-associated protein 1 (cap1) regulates cofilin function, the actin cytoskeleton, and cell adhesion. J. Biol. Chem. 2013, 288, 20966–20977. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Schlichter, L.C. Selective activation of kca3.1 and crac channels by p2y2 receptors promotes ca2+ signaling, store refilling and migration of rat microglial cells. PLoS ONE 2013, 8, e62345. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, W.; Liu, Y.; Xu, P.; Li, Z.; Wu, R.; Shi, X.; Tang, Y. P2y6 receptor-mediated microglial phagocytosis in radiation-induced brain injury. Mol. Neurobiol. 2016, 53, 3552–3564. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B. A unique microglia type associated with restricting development of alzheimer’s disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Madry, C.; Kyrargyri, V.; Arancibia-Carcamo, I.L.; Jolivet, R.; Kohsaka, S.; Bryan, R.M.; Attwell, D. Microglial ramification, surveillance, and interleukin-1beta release are regulated by the two-pore domain k+ channel thik-1. Neuron 2018, 97, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Sandhir, R.; Onyszchuk, G.; Berman, N.E.J. Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Exp. Neurol. 2008, 213, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2011, 10, 263–276. [Google Scholar] [CrossRef]
- Choi, J.H.; Lee, C.H.; Hwang, I.K.; Won, M.H.; Seong, J.K.; Yoon, Y.S.; Lee, H.S.; Lee, I.S. Age-related changes in ionized calcium-binding adapter molecule 1 immunoreactivity and protein level in the gerbil hippocampal ca1 region. J. Vet. Med Sci. 2007, 69, 1131–1136. [Google Scholar] [CrossRef]
- Hwang, I.K.; Choong, A.E.; Lee, H.; Hua, A.E.; Ae, L.; Yoo, K.-Y.; Jung, A.E.; Choi, H. Comparison of ionized calcium-binding adapter molecule 1 immunoreactivity of the hippocampal dentate gyrus and ca1 region in adult and aged dogs. Neurochem. Res. 2008, 33, 1309–1315. [Google Scholar] [CrossRef]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Tischer, J.; Krueger, M.; Mueller, W.; Staszewski, O.; Prinz, M.; Streit, W.J.; Bechmann, I. Inhomogeneous distribution of iba-1 characterizes microglial pathology in alzheimer’s disease. Glia 2016, 64, 1562–1572. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.S.; Ma, J.; Jegathees, T.; Goldsbury, C. Microglia show altered morphology and reduced arborization in human brain during aging and alzheimer’s disease. Brain Pathol. 2017, 27, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.M.K.; Currie, M.S.; Padmanabhan, J.; Cohen, H.J. Age-related alterations in actin cytoskeleton and receptor expression in human leukocytes. J. Gerontol. 1992, 47, B37–B44. [Google Scholar] [CrossRef] [PubMed]
- Starodubtseva, M.N. Mechanical properties of cells and ageing. Ageing Res. Rev. 2011, 10, 16–25. [Google Scholar] [CrossRef]
- Sato, Y.; Yamanaka, H.; Toda, T.; Shinohara, Y.; Endo, T. Comparison of hippocampal synaptosome proteins in young-adult and aged rats. Neurosci. Lett. 2005, 382, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Potter, D.A.; Tirnauer, J.S.; Janssen, R.; Croall, D.E.; Hughes, C.N.; Fiacco, K.A.; Mier, J.W.; Maki, M.; Herman, I.M. Calpain regulates actin remodeling during cell spreading. J. Cell Biol. 1998, 141, 647–662. [Google Scholar] [CrossRef]
- Hinman, J.D.; Duce, J.A.; Siman, R.A.; Hollander, W.; Abraham, C.R. Activation of calpain-1 in myelin and microglia in the white matter of the aged rhesus monkey. J. Neurochem. 2004, 89, 430–441. [Google Scholar] [CrossRef]
- Barone, E.; Mosser, S.; Fraering, P.C. Inactivation of brain cofilin-1 by age, alzheimer’s disease and γ-secretase. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2500–2509. [Google Scholar] [CrossRef]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.; Möller, T. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162. [Google Scholar] [CrossRef]
- Mildner, A.; Huang, H.; Radke, J.; Stenzel, W.; Priller, J. P2y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 2017, 65, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Plescher, M.; Seifert, G.; Hansen, J.N.; Bedner, P.; Steinhäuser, C.; Halle, A. Plaque-dependent morphological and electrophysiological heterogeneity of microglia in an alzheimer’s disease mouse model. Glia 2018, 66, 1464–1480. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of alzheimer-related changes. Acta Neuropathol. 1991, 82. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of alzheimer’s disease. Neurology 2012, 42, 631. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Paasila, P.J.; Davies, D.S.; Kril, J.J.; Goldsbury, C.S.; Sutherland, G.T. The relationship between the morphological subtypes of microglia and alzheimer’s disease neuropathology. Brain Pathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with beta-amyloid deposition in mice with alzheimer-like pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef] [PubMed]
- Fulga, T.A.; Elson-Schwab, I.; Khurana, V.; Steinhilb, M.L.; Spires, T.L.; Hyman, B.T.; Feany, M.B. Abnormal bundling and accumulation of f-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2007, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Neuropathology, G. Pathological correlates of late-onset dementia in a multicentre, community-based population in england and wales. Neuropathology group of the medical research council cognitive function and ageing study (mrc cfas). Lancet 2001, 357, 169. [Google Scholar]
- Kim, T.; Vidal, G.S.; Djurisic, M. Human lilrb2 is a b-amyloid receptor and its murine homolog pirb regulates synaptic plasticity in an alzheimer’s mode. Science 2013, 341, 1399–1404. [Google Scholar] [CrossRef]
- Rahman, T.; Davies, D.S.; Tannenberg, R.K.; Fok, S.; Shepherd, C.; Dodd, P.R.; Cullen, K.M.; Goldsbury, C. Cofilin rods and aggregates concur with tau pathology and the development of alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 1443–1460. [Google Scholar] [CrossRef] [PubMed]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2 × 7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of alzheimer’s disease. J. Biol. Chem. 2003, 278, 13309–13317. [Google Scholar] [CrossRef] [PubMed]
- McLarnon, J.; JK, R.; DG, W.; HB, C. Upregulated expression of purinergic p2x(7) receptor in alzheimer disease and amyloid-beta peptide-treated microglia and in peptide-injected rat hippocampus. J. Neuropathol. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; McLarnon, J.G. Block of purinergic p2x7 receptor is neuroprotective in an animal model of alzheimer’s disease. Neuroreport 2008, 19, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Amar, M.; Dalle, C.; Youssef, I.; Boucher, C.; Le Duigou, C.; Brückner, M.; Prigent, A.; Sazdovitch, V.; Halle, A. New role of p2x7 receptor in an alzheimer’s disease mouse model. Mol. Psychiatry 2019, 24, 108. [Google Scholar] [CrossRef] [PubMed]
- Solle, M.; Labasi, J.; Perregaux, D.G.; Stam, E.; Petrushova, N.; Koller, B.H.; Griffiths, R.J.; Gabel, C.A. Altered cytokine production in mice lacking p2x7receptors. J. Biol. Chem. 2001, 276, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, N.; Delekate, A.; Breithausen, B.; Keppler, K.; Poll, S.; Schulte, T.; Peter, J.; Plescher, M.; Hansen, J.N.; Blank, N.; et al. P2y1 receptor blockade normalizes network dysfunction and cognition in an alzheimer’s disease model. J. Exp. Med. 2018, 215, 1649–1663. [Google Scholar] [CrossRef]
- Ajit, D.; Woods, L.T.; Camden, J.M.; Thebeau, C.N.; El-Sayed, F.G.; Greeson, G.W.; Erb, L.; Petris, M.J.; Miller, D.C.; Sun, G.Y.; et al. Loss of p2y 2 nucleotide receptors enhances early pathology in the tgcrnd8 mouse model of alzheimer’s disease. Mol. Neurobiol. 2014, 49, 1031–1042. [Google Scholar] [CrossRef]
- Lai, M.K.P.; Tan, M.G.K.; Kirvell, S.; Hobbs, C.; Lee, J.; Esiri, M.M.; Chen, C.P.; Francis, P.T. Selective loss of p2y2 nucleotide receptor immunoreactivity is associated with alzheimer’s disease neuropathology. J. Neural Transm. 2008, 115, 1165–1172. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z. The trem2-apoe pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef]
- Song, W.M.; Colonna, M. The identity and function of microglia in neurodegeneration. Nat. Immunol. 2018, 1. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Physiological Role | Changes in Ageing | Changes in AD and Animal Models of AD |
|---|---|---|---|
| Cytoskeletal Proteins | |||
| Actin | Main constituent of cytoskeletal microfilaments. | Animals: unknown. Humans: Increased F-actin level; decreased stimulus-induced actin polymerization. | Animals: Presence of CFL1-actin rods. Humans: unknown |
| Arp2/3 complex | Controls branching of actin filaments. | Animals: unknown. Humans: Decreased expression of subunits ARPC1A and ARPC1B. | unknown |
| CORO1A | Recruits Arp2/3 to the ends of actin filaments to initiate branching. | Animals: Decreased expression. Humans: Decreased expression. | unknown |
| CAPN1 | Cleaves and degrades cytoskeletal proteins, such as TLN1, ACTN1, FLNC and spectrin. | Animals: Increased expression in aged monkeys and mice. Humans: unknown. | unknown |
| TLN1 | Mediates integrin-cytoskeleton bonds, important for cell adhesion. | Animals: unknown. Humans: Decreased expression. | unknown |
| Iba1 | Involved in actin bundling and membrane ruffling. | Animals: Increased in aged gerbils, dogs and mice. Humans: Positively correlated to MMSE score. | Animals: Increased in APPPS1 mouse model. Humans: Positively correlated to MMSE score. |
| NM II | Essential for the contractile properties of the cytoskeleton. | unknown | unknown |
| CFL1 | Depolymerizes and severs actin filaments. | Animals: Increased load of pCFL1, presence of CFL1-actin rods. Humans: unknown. | Animals: Increased load of pCFL1, presence of CFL1-actin rods. Humans: Increased load of pCFL1, presence of CFL1-actin rods. |
| SSH1 | Dephosphorylates CFL-1, inducing activation. | Animals: Decreased activity in aged mice. Humans: unknown. | Animals: Decreased activity in aged mice. Human: unknown. |
| Chemotactic Receptors | |||
| CX3CR1 | Binds to fractalkine, a neuron-secreted chemokine. Involved in baseline and directed motility and used as a marker specific of microglia. | unknown | unknown |
| P1 family | A2A involved in process retraction, A3 in process extension. | unknown | unknown |
| P2X4 | ATP/ADP receptor involved in chemotaxis. | unknown | unknown |
| P2X7 | ATP receptor; regulates IL1 secretion. | Animals: Increased in aged mice. Humans: unknown. | Animals: Upregulated in APPPS1 mice and associated with Aβ plaque load. Increased expression after Aβ42 intrahippocampal injections in WT mice. Decreased Aβ plaques and soluble Aβ and improved memory in APPPS1-P2X7 KO mice. Humans: Increased expression and noted in proximity to Aβ plaques. Upregulated and correlated with Aβ plaque load. |
| P2Y1 | ADP receptor involved in chemotaxis; indirectly influences CFL1 activity. | unknown | Animals: In APPPS1 mice, its blockade improved spatial learning and memory. Humans: unknown. |
| P2Y2 | UTP receptor, regulates levels of intracellular calcium, involved in chemotaxis. | unknown | Animals: CRND8 P2Y2-KO mice had increased soluble Aβ and plaques, and shortened lifespan. Humans: Reduced expression. |
| P2Y4 | ATP receptor involved in pinocytosis. | unknown | Animals: unknown. Humans: No changes found. |
| P2Y6 | UDP receptor, associated with chemotaxis and phagocytosis. | unknown | Animals: unknown. Humans: No changes observed. |
| P2Y12 | ATP/ADP receptor associated with directed motility. | Animals: Increased expression in aged mice. Humans: unknown. | Animals: unknown. Humans: Downregulated in microglia clustered around Aβ plaques. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco-Bocanegra, D.K.; McAuley, C.; Nicoll, J.A.R.; Boche, D. Molecular Mechanisms of Microglial Motility: Changes in Ageing and Alzheimer’s Disease. Cells 2019, 8, 639. https://doi.org/10.3390/cells8060639
Franco-Bocanegra DK, McAuley C, Nicoll JAR, Boche D. Molecular Mechanisms of Microglial Motility: Changes in Ageing and Alzheimer’s Disease. Cells. 2019; 8(6):639. https://doi.org/10.3390/cells8060639
Chicago/Turabian StyleFranco-Bocanegra, Diana K., Ciaran McAuley, James A. R. Nicoll, and Delphine Boche. 2019. "Molecular Mechanisms of Microglial Motility: Changes in Ageing and Alzheimer’s Disease" Cells 8, no. 6: 639. https://doi.org/10.3390/cells8060639
APA StyleFranco-Bocanegra, D. K., McAuley, C., Nicoll, J. A. R., & Boche, D. (2019). Molecular Mechanisms of Microglial Motility: Changes in Ageing and Alzheimer’s Disease. Cells, 8(6), 639. https://doi.org/10.3390/cells8060639