Intermittent Hypoxia Prevents Myocardial Mitochondrial Ca2+ Overload and Cell Death during Ischemia/Reperfusion: The Role of Reactive Oxygen Species

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of Neonatal Rat Cardiomyocytes
2.3. IH Exposures
2.4. Ischemia and Reperfusion (I/R) Injury
2.5. Analysis of Cell Death by Flow Cytometry
2.6. Cell Viability Assay
2.7. Imaging of Intracellular Reactive Oxygen Species (ROS)
2.8. Flow Cytometric Analysis for Oxidative Stress Detection
2.9. Quantitative Real-Time PCR
2.10. Measurement of Total Antioxidant Capacity and Catalase and Glutathione Peroxidase Activity
2.11. Immunocytofluorescence Staining
2.12. Imaging of Intracellular Ca2+ Concentrations ([Ca2+]i)
2.13. Imaging of Mitochondrial Ca2+ Concentration ([Ca2+]m)
2.14. Measurement of Mitochondrial Membrane Potential Using Flow Cytometry
2.15. In Situ Labeling of Activated Caspase-3
2.16. Statistics
3. Results
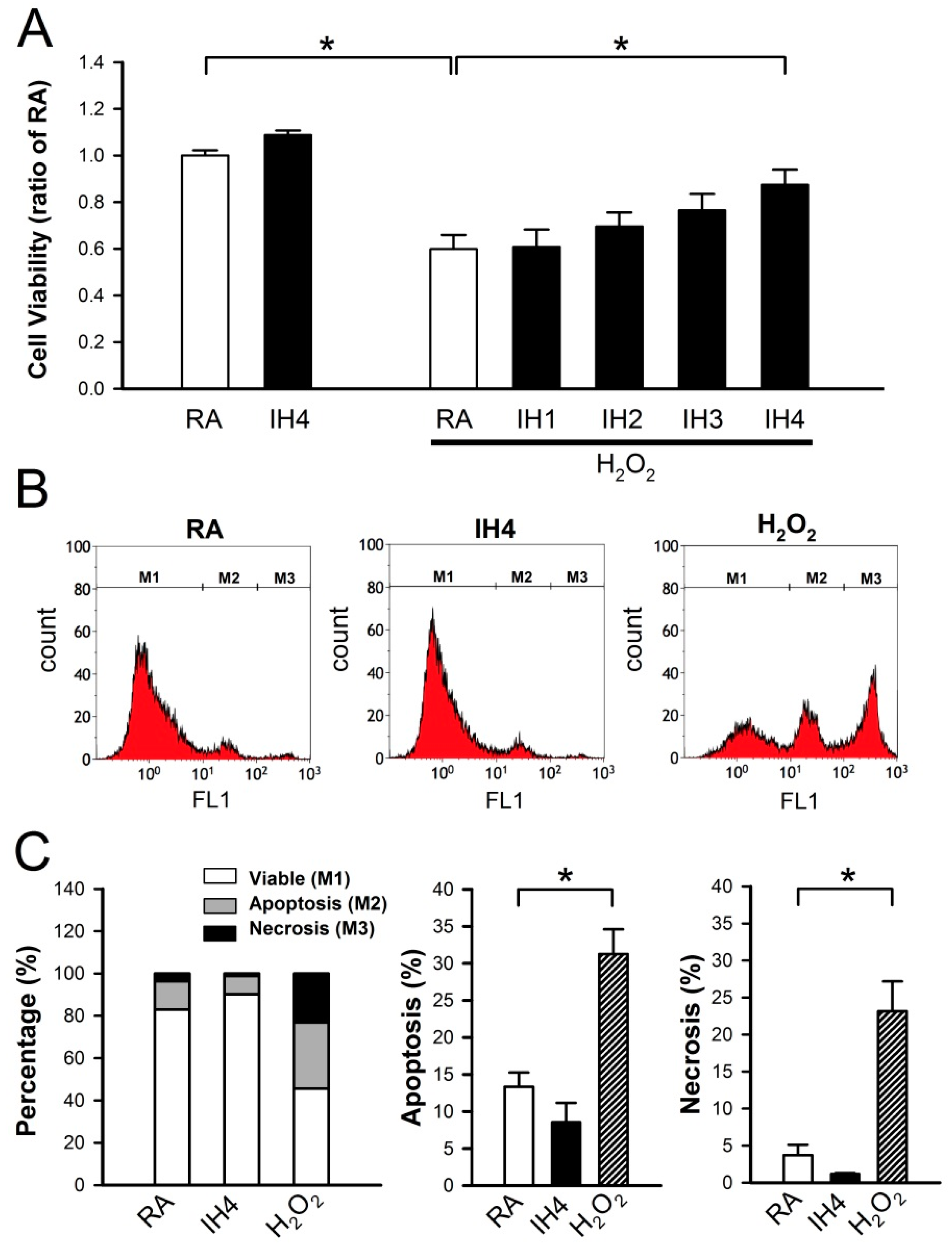
3.1. Effects of IH on Cell Death
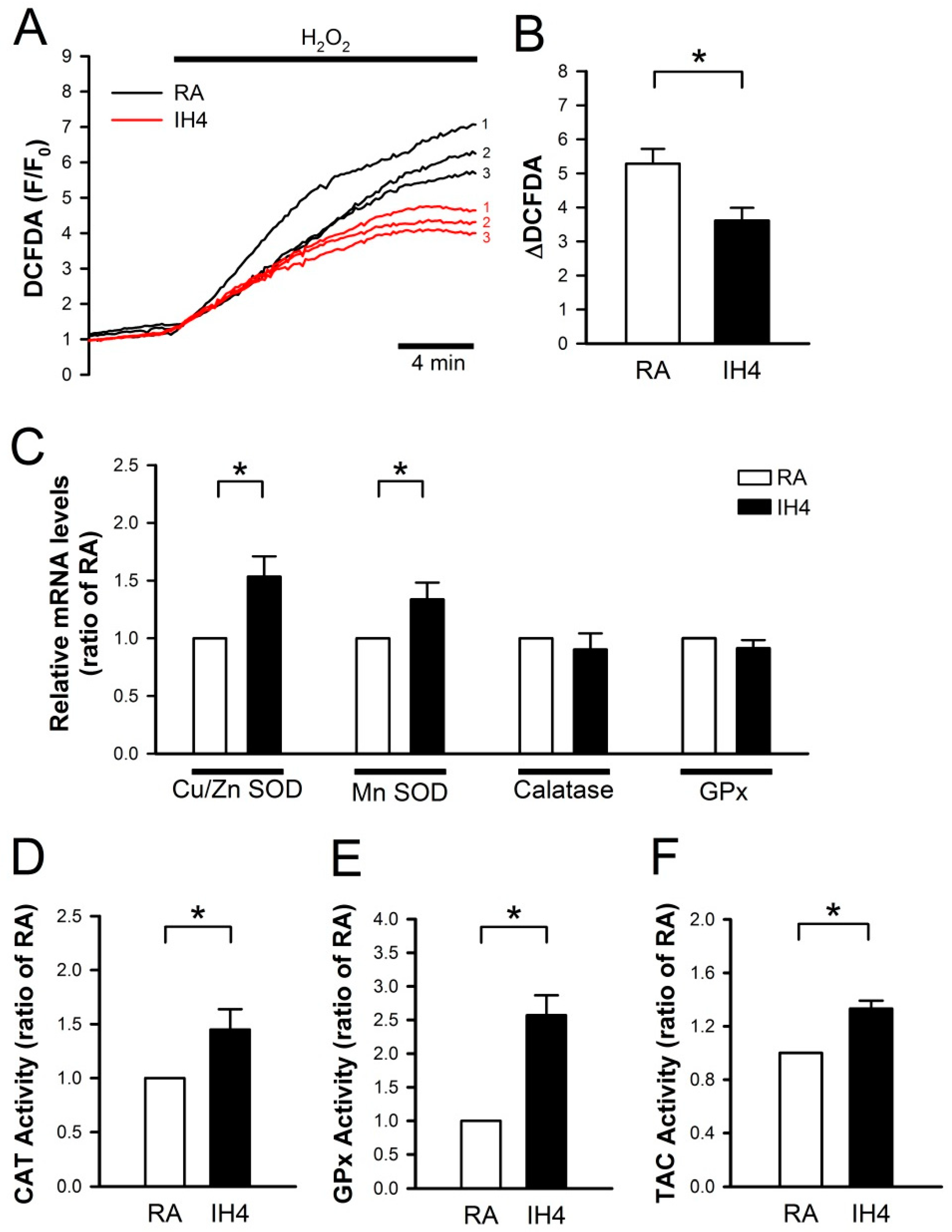
3.2. Effects of IH on the Endogenous Antioxidant Defense in Cardiomyocytes
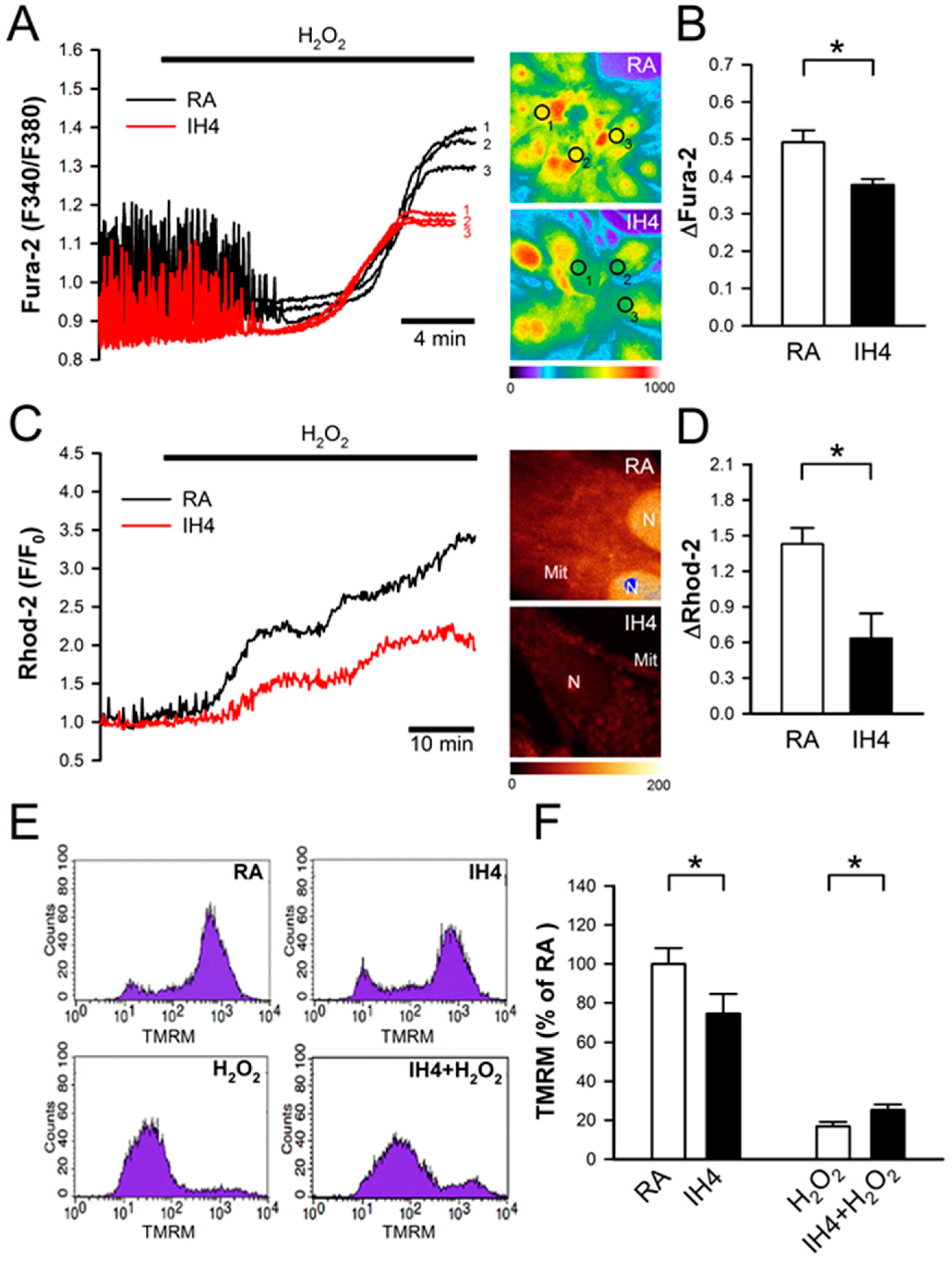
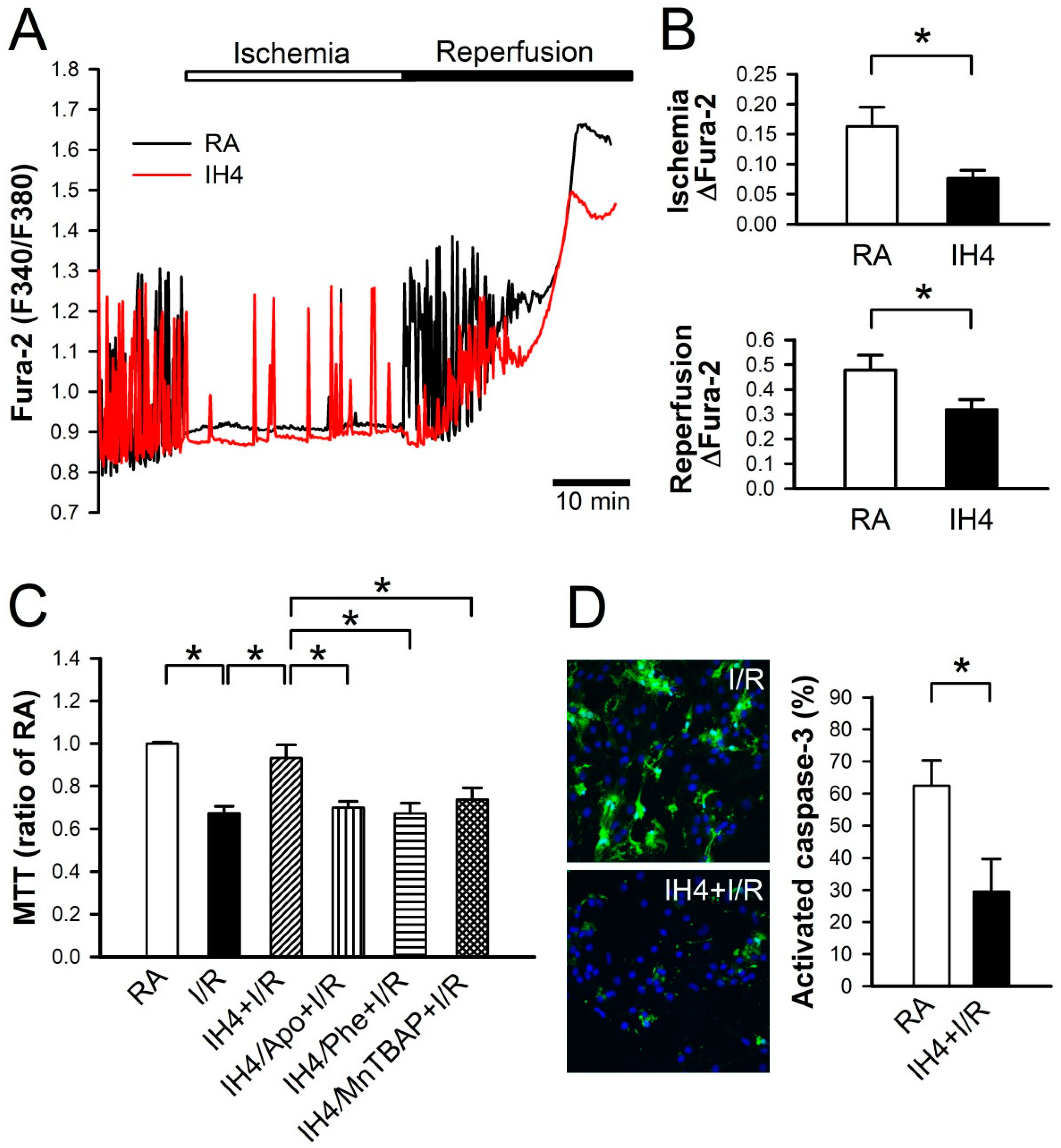
3.3. Effects of IH on Oxidative Stress-Induced Intracellular Ca2+ Disturbance and Mitochondrial Membrane Depolarization
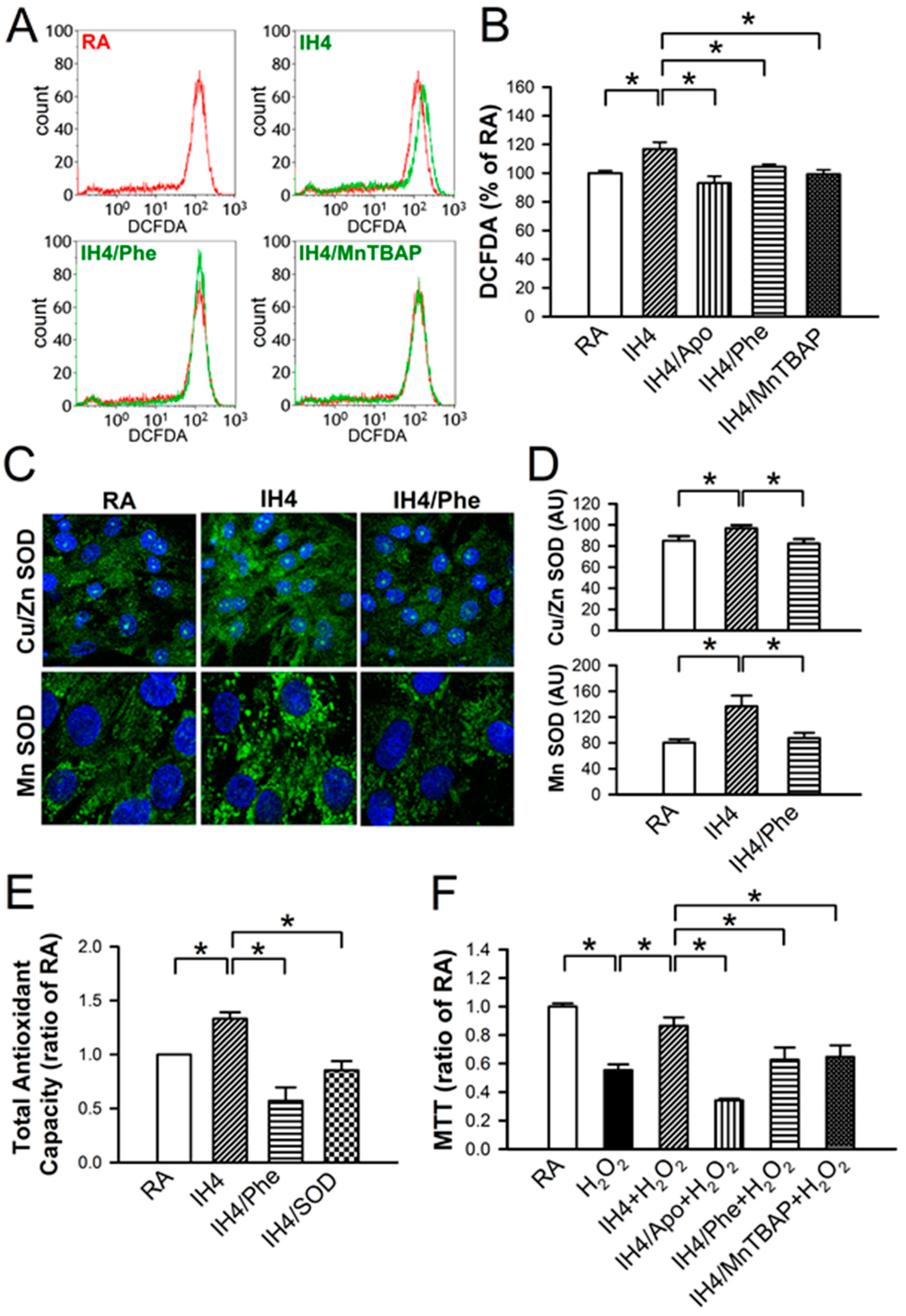
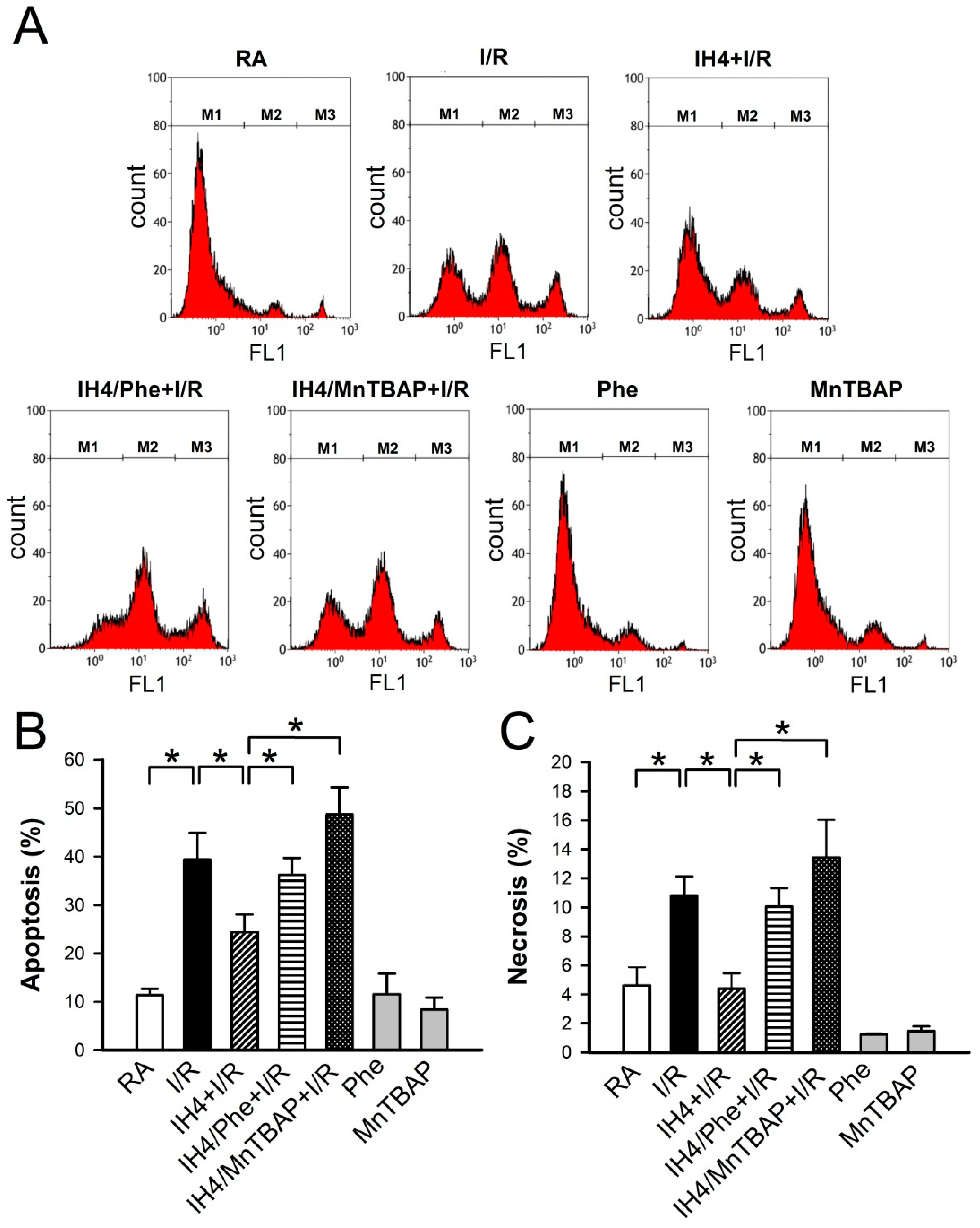
3.4. Involvement of ROS Generation in the IH-Increased Antioxidant Defense and Cardioprotective Effects
3.5. Effects of IH on the I/R-Induced Cell Death in Cardiomyocytes
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Neubauer, J.A. Invited review: Physiological and pathophysiological responses to intermittent hypoxia. J. Appl. Physiol. 2001, 90, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.M.; Carrizo, S.J.; Vicente, E.; Agusti, A.G. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: An observational study. Lancet 2005, 365, 1046–1053. [Google Scholar] [CrossRef]
- Shahar, E.; Whitney, C.W.; Redline, S.; Lee, E.T.; Newman, A.B.; Javier Nieto, F.; O’Connor, G.T.; Boland, L.L.; Schwartz, J.E.; Samet, J.M. Sleep-disordered breathing and cardiovascular disease: Cross-sectional results of the Sleep Heart Health Study. Am. J. Respir. Crit. Care Med. 2001, 163, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Manalo, D.J.; Wei, G.; Rodriguez, E.R.; Fox-Talbot, K.; Lu, H.; Zweier, J.L.; Semenza, G.L. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation 2003, 108, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Beguin, P.C.; Joyeux-Faure, M.; Godin-Ribuot, D.; Levy, P.; Ribuot, C. Acute intermittent hypoxia improves rat myocardium tolerance to ischemia. J. Appl. Physiol. 2005, 99, 1064–1069. [Google Scholar] [CrossRef]
- Chen, L.; Lu, X.Y.; Li, J.; Fu, J.D.; Zhou, Z.N.; Yang, H.T. Intermittent hypoxia protects cardiomyocytes against ischemia-reperfusion injury-induced alterations in Ca2+ homeostasis and contraction via the sarcoplasmic reticulum and Na+/Ca2+ exchange mechanisms. Am. J. Physiol. Cell Physiol. 2006, 290, C1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.Z.; Xie, Y.; Chen, L.; Yang, H.T.; Zhou, Z.N. Intermittent high altitude hypoxia inhibits opening of mitochondrial permeability transition pores against reperfusion injury. J. Mol. Cell Cardiol. 2006, 40, 96–106. [Google Scholar] [CrossRef]
- Guo, H.C.; Zhang, Z.; Zhang, L.N.; Xiong, C.; Feng, C.; Liu, Q.; Liu, X.; Shi, X.L.; Wang, Y.L. Chronic intermittent hypobaric hypoxia protects the heart against ischemia/reperfusion injury through upregulation of antioxidant enzymes in adult guinea pigs. Acta Pharmacol. Sin. 2009, 30, 947–955. [Google Scholar] [CrossRef]
- Yeung, H.M.; Kravtsov, G.M.; Ng, K.M.; Wong, T.M.; Fung, M.L. Chronic intermittent hypoxia alters Ca2+ handling in rat cardiomyocytes by augmented Na+/Ca2+ exchange and ryanodine receptor activities in ischemia-reperfusion. Am. J. Physiol. Cell Physiol. 2007, 292, C2046–2056. [Google Scholar] [CrossRef]
- Mallet, R.T.; Manukhina, E.B.; Ruelas, S.S.; Caffrey, J.L.; Downey, H.F. Cardioprotection by intermittent hypoxia conditioning: Evidence, mechanisms, and therapeutic potential. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H216–H232. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Grueter, C.E.; Colbran, R.J.; Anderson, M.E. CaMKII, an emerging molecular driver for calcium homeostasis, arrhythmias, and cardiac dysfunction. J. Mol. Med. 2007, 85, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Maulik, N.; Sato, M.; Ray, P.S. Reactive oxygen species function as second messenger during ischemic preconditioning of heart. Mol. Cell Biochem. 1999, 196, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Forbes, R.A.; Steenbergen, C.; Murphy, E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ. Res. 2001, 88, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell Signal. 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Karlstad, J.; Sun, Y.; Singh, B.B. Ca2+ signaling: An outlook on the characterization of Ca2+ channels and their importance in cellular functions. Adv. Exp. Med. Biol. 2012, 740, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.; Gonzalez-Candia, A.; Rodriguez, J.; Carrasco-Pozo, C.; Canas, D.; Garcia-Herrera, C.; Herrera, E.A.; Castillo, R.L. Mechanisms of Cardiovascular Protection Associated with Intermittent Hypobaric Hypoxia Exposure in a Rat Model: Role of Oxidative Stress. Int. J. Mol. Sci. 2018, 19, 366. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.R.; Lien, C.F.; Jeng, J.R.; Hsieh, J.C.; Chang, C.W.; Lin, J.H.; Yang, K.T. Intermittent Hypoxia Inhibits Na+-H+ Exchange-Mediated Acid Extrusion Via Intracellular Na+ Accumulation in Cardiomyocytes. Cell Physiol. Biochem. 2018, 46, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Lien, C.F.; Lee, W.S.; Wang, I.C.; Chen, T.I.; Chen, T.L.; Yang, K.T. Intermittent hypoxia-generated ROS contributes to intracellular zinc regulation that limits ischemia/reperfusion injury in adult rat cardiomyocyte. J. Mol. Cell Cardiol. 2018, 118, 122–132. [Google Scholar] [CrossRef]
- Navarrete-Opazo, A.; Mitchell, G.S. Therapeutic potential of intermittent hypoxia: A matter of dose. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Hua, H.; Guo, X.; Jia, Z.; Zhang, Y.; Maslov, L.N.; Zhang, X.; Ma, H. PGC-1alpha Participates in the Protective Effect of Chronic Intermittent Hypobaric Hypoxia on Cardiomyocytes. Cell Physiol. Biochem. 2018, 50, 1891–1902. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.K.; Yang, J.; Biju, M.P.; Joseph, S.K.; Johnson, R.S.; Haase, V.H.; Freedman, B.D.; Turka, L.A. Hypoxia inducible factor 1 alpha regulates T cell receptor signal transduction. Proc. Natl. Acad Sci. USA 2005, 102, 17071–17076. [Google Scholar] [CrossRef]
- Formisano, L.; Guida, N.; Valsecchi, V.; Cantile, M.; Cuomo, O.; Vinciguerra, A.; Laudati, G.; Pignataro, G.; Sirabella, R.; Di Renzo, G.; et al. Sp3/REST/HDAC1/HDAC2 Complex Represses and Sp1/HIF-1/p300 Complex Activates ncx1 Gene Transcription, in Brain Ischemia and in Ischemic Brain Preconditioning, by Epigenetic Mechanism. J. Neurosci. 2015, 35, 7332–7348. [Google Scholar] [CrossRef]
- Chen, T.I.; Hsu, Y.C.; Lien, C.F.; Lin, J.H.; Chiu, H.W.; Yang, K.T. Non-lethal levels of oxidative stress in response to short-term intermittent hypoxia enhance Ca2+ handling in neonatal rat cardiomyocytes. Cell Physiol. Biochem. 2014, 33, 513–527. [Google Scholar] [CrossRef]
- Guo, H.C.; Guo, F.; Zhang, L.N.; Zhang, R.; Chen, Q.; Li, J.X.; Yin, J.; Wang, Y.L. Enhancement of Na/K pump activity by chronic intermittent hypobaric hypoxia protected against reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, 2280–2287. [Google Scholar] [CrossRef]
- Ding, H.L.; Zhu, H.F.; Dong, J.W.; Zhu, W.Z.; Zhou, Z.N. Intermittent hypoxia protects the rat heart against ischemia/reperfusion injury by activating protein kinase C. Life Sci. 2004, 75, 2587–2603. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.J.; Li, Q.; Ma, H.J.; Guan, Y.; Shi, M.; Yang, J.; Li, D.P.; Zhang, Y. Chronic intermittent hypobaric hypoxia ameliorates ischemia/reperfusion-induced calcium overload in heart via Na/Ca2+ exchanger in developing rats. Cell Physiol. Biochem. 2014, 34, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, J.; Goncalves, I.O.; Lumini-Oliveira, J.; Marques-Aleixo, I.; Passos, E.; Rocha-Rodrigues, S.; Machado, N.G.; Moreira, A.C.; Rizo, D.; Viscor, G.; et al. Modulation of cardiac mitochondrial permeability transition and apoptotic signaling by endurance training and intermittent hypobaric hypoxia. Int. J. Cardiol. 2014, 173, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Rokita, A.G.; Anderson, M.E.; Maier, L.S. Redox regulation of sodium and calcium handling. Antioxid. Redox Signal. 2013, 18, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.B. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc. Res. 2004, 61, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Estrada, J.A.; Williams, A.G., Jr.; Sun, J.; Gonzalez, L.; Downey, H.F.; Caffrey, J.L.; Mallet, R.T. delta-Opioid receptor (DOR) signaling and reactive oxygen species (ROS) mediate intermittent hypoxia induced protection of canine myocardium. Basic Res. Cardiol. 2016, 111, 17. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Bennett, B.; Zielonka, J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: Mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018, 15, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Kvandova, M.; Majzunova, M.; Dovinova, I. The role of PPARγ in cardiovascular diseases. Physiol. Res. 2016, 65, S343–S363. [Google Scholar] [PubMed]
- Gonchar, O.A.; IMankovska, I.N. Time-dependent effect of severe hypoxia/reoxygenation on oxidative stress level, antioxidant capacity and p53 accumulation in mitochondria of rat heart. Ukr. Biochem. J. 2017, 89, 39–47. [Google Scholar] [CrossRef]
- Wada, T.; Becskei, A. Impact of Methods on the Measurement of mRNA Turnover. Int. J. Mol. Sci. 2017, 18, 2723. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Jian, Z.; Li, J.; Luo, L.; Zhao, L.; Zhou, Y.; Tang, F.; Xiao, Y. Upregulated ATF6 contributes to chronic intermittent hypoxia-afforded protection against myocardial ischemia/reperfusion injury. Int. J. Mol. Med. 2016, 37, 1199–1208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Z.H.; Chen, Y.X.; Zhang, C.M.; Wu, L.; Yu, Z.; Cai, X.L.; Guan, Y.; Zhou, Z.N.; Yang, H.T. Intermittent hypobaric hypoxia improves postischemic recovery of myocardial contractile function via redox signaling during early reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, 1695–1705. [Google Scholar] [CrossRef][Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.-C.; Lien, C.-F.; Lee, W.-S.; Chang, H.-R.; Hsu, Y.-C.; Luo, Y.-P.; Jeng, J.-R.; Hsieh, J.-C.; Yang, K.-T. Intermittent Hypoxia Prevents Myocardial Mitochondrial Ca2+ Overload and Cell Death during Ischemia/Reperfusion: The Role of Reactive Oxygen Species. Cells 2019, 8, 564. https://doi.org/10.3390/cells8060564
Chang J-C, Lien C-F, Lee W-S, Chang H-R, Hsu Y-C, Luo Y-P, Jeng J-R, Hsieh J-C, Yang K-T. Intermittent Hypoxia Prevents Myocardial Mitochondrial Ca2+ Overload and Cell Death during Ischemia/Reperfusion: The Role of Reactive Oxygen Species. Cells. 2019; 8(6):564. https://doi.org/10.3390/cells8060564
Chicago/Turabian StyleChang, Jui-Chih, Chih-Feng Lien, Wen-Sen Lee, Huai-Ren Chang, Yu-Cheng Hsu, Yu-Po Luo, Jing-Ren Jeng, Jen-Che Hsieh, and Kun-Ta Yang. 2019. "Intermittent Hypoxia Prevents Myocardial Mitochondrial Ca2+ Overload and Cell Death during Ischemia/Reperfusion: The Role of Reactive Oxygen Species" Cells 8, no. 6: 564. https://doi.org/10.3390/cells8060564
APA StyleChang, J.-C., Lien, C.-F., Lee, W.-S., Chang, H.-R., Hsu, Y.-C., Luo, Y.-P., Jeng, J.-R., Hsieh, J.-C., & Yang, K.-T. (2019). Intermittent Hypoxia Prevents Myocardial Mitochondrial Ca2+ Overload and Cell Death during Ischemia/Reperfusion: The Role of Reactive Oxygen Species. Cells, 8(6), 564. https://doi.org/10.3390/cells8060564