Nucleolar and Ribosomal Dysfunction—A Common Pathomechanism in Childhood Progerias?

{kind=link}
{kind=link}
Abstract
1. Ageing and Progeria
2. Aging and the Nucleolus
3. Nucleolar Size as a Hallmark of Aging
4. Hutchinson–Gilford Progeria Syndrome (HGPS)
5. Cockayne Syndrome (CS)
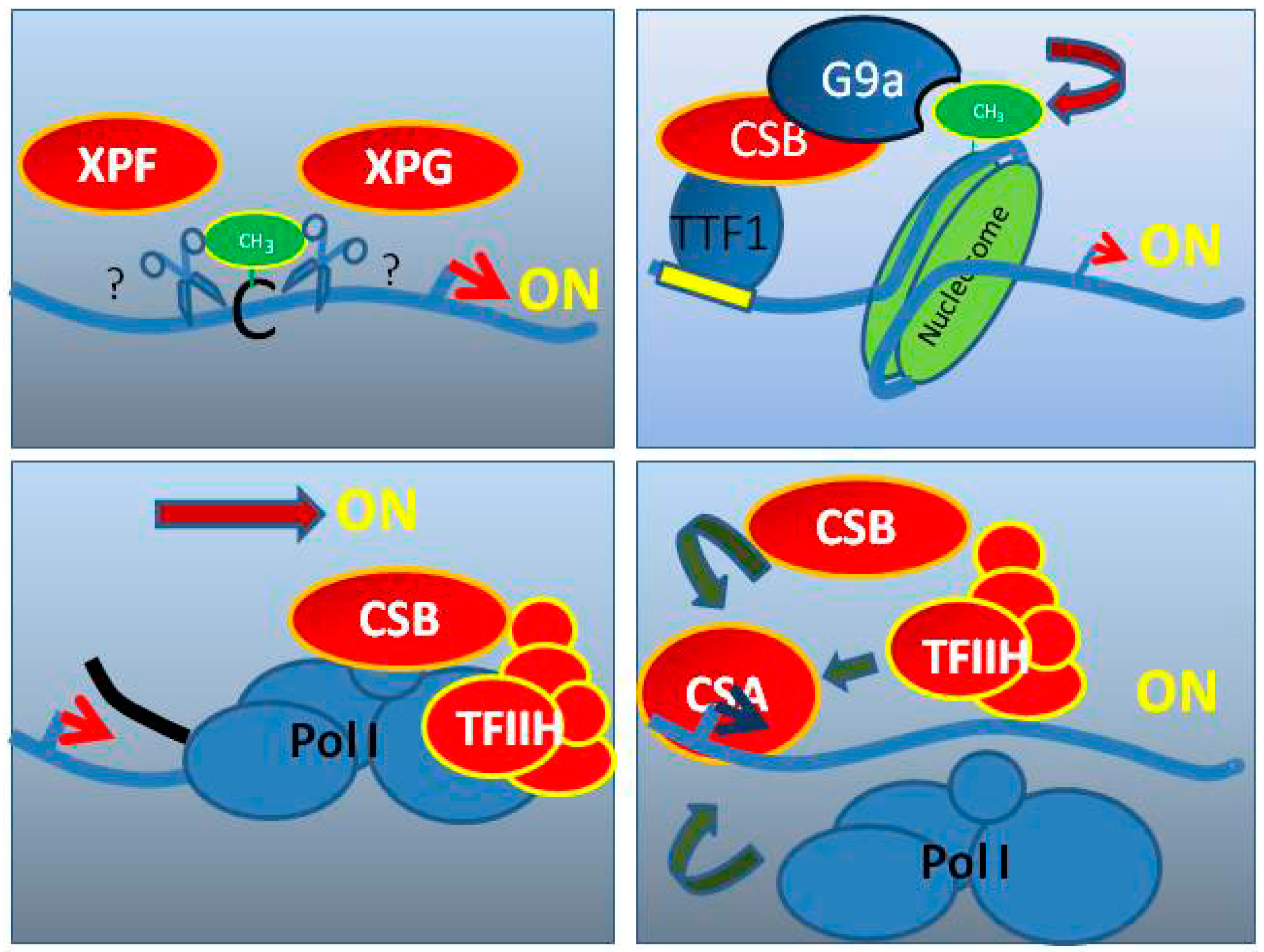
6. CS Factors Are Involved in RNA Pol I Transcription
6.1. TFIIH in RNA Pol I Transcription
6.2. CSA and CSB in RNA Pol I Transcription
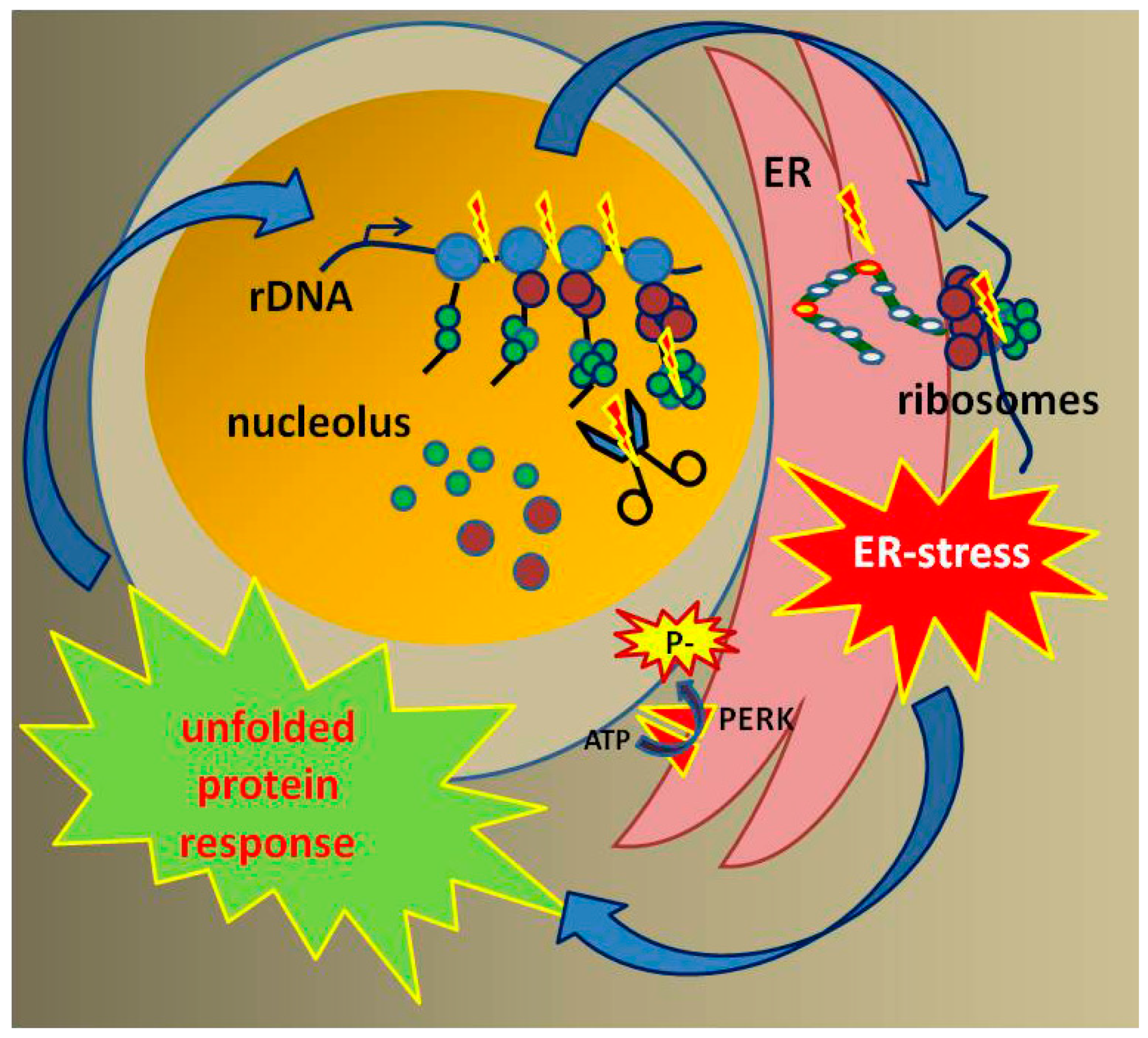
7. Loss of RNA Pol I Transcription Leads to Mitochondrial Dysfunction
8. Loss of Proteostasis in CS
9. Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Salthouse, T.A. When does age-related cognitive decline begin? Neurobiol. Aging 2009, 30, 507–514. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Oshima, J.; Sidorova, J.M.; Monnat, R.J., Jr. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 2017, 33, 105–114. [Google Scholar] [CrossRef]
- Lautrup, S.; Caponio, D.; Cheung, H.H.; Piccoli, C.; Stevnsner, T.; Chan, W.Y.; Fang, E.F. Studying Werner syndrome to elucidate mechanisms and therapeutics of human aging and age-related diseases. Biogerontology 2019, 20, 255–269. [Google Scholar] [CrossRef]
- Shamanna, R.A.; Croteau, D.L.; Lee, J.H.; Bohr, V.A. Recent Advances in Understanding Werner Syndrome. F1000Research 2017, 6, 1779. [Google Scholar] [CrossRef]
- Epstein, C.J.; Martin, G.M.; Motulsky, A.G. Werner’s syndrome; caricature of aging. A genetic model for the study of degenerative diseases. Trans. Assoc. Am. Physicians 1965, 78, 73–81. [Google Scholar]
- Campisi, J.; Robert, L. Cell senescence: role in aging and age-related diseases. Interdiscip. Top. Gerontol. 2014, 39, 45–61. [Google Scholar] [CrossRef]
- Bassler, J.; Hurt, E. Eukaryotic Ribosome Assembly. Annu. Rev. Biochem. 2018, 88. [Google Scholar] [CrossRef]
- Andersen, J.S.; Lyon, C.E.; Fox, A.H.; Leung, A.K.; Lam, Y.W.; Steen, H.; Mann, M.; Lamond, A.I. Directed proteomic analysis of the human nucleolus. Curr. Biol. 2002, 12, 1–11. [Google Scholar] [CrossRef]
- Bensaddek, D.; Nicolas, A.; Lamond, A.I. Quantitative proteomic analysis of the human nucleolus. In The Nucleolus; Humana Press: New York, NY, USA, 2016. [Google Scholar]
- Kennedy, B.K.; Gotta, M.; Sinclair, D.A.; Mills, K.; McNabb, D.S.; Murthy, M.; Pak, S.M.; Laroche, T.; Gasser, S.M.; Guarente, L. Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 1997, 89, 381–391. [Google Scholar] [CrossRef]
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 1997, 91, 1033–1042. [Google Scholar] [CrossRef]
- Penzo, M.; Montanaro, L.; Trere, D.; Derenzini, M. The Ribosome Biogenesis-Cancer Connection. Cells 2019, 8, 55. [Google Scholar] [CrossRef]
- Tiku, V.; Antebi, A. Nucleolar Function in Lifespan Regulation. Trends Cell Biol. 2018, 28, 662–672. [Google Scholar] [CrossRef]
- Antikainen, H.; Driscoll, M.; Haspel, G.; Dobrowolski, R. TOR-mediated regulation of metabolism in aging. Aging Cell 2017, 16, 1219–1233. [Google Scholar] [CrossRef]
- Finkel, T. The metabolic regulation of aging. Nat. Med. 2015, 21, 1416–1423. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef]
- Tiku, V.; Jain, C.; Raz, Y.; Nakamura, S.; Heestand, B.; Liu, W.; Spath, M.; Suchiman, H.E.D.; Muller, R.U.; Slagboom, P.E.; et al. Small nucleoli are a cellular hallmark of longevity. Nat. Commun. 2017, 8, 16083. [Google Scholar] [CrossRef]
- Frank, D.J.; Roth, M.B. ncl-1 is required for the regulation of cell size and ribosomal RNA synthesis in Caenorhabditis elegans. J. Cell Biol. 1998, 140, 1321–1329. [Google Scholar] [CrossRef]
- Hedgecock, E.M.; Herman, R.K. The ncl-1 gene and genetic mosaics of Caenorhabditis elegans. Genetics 1995, 141, 989–1006. [Google Scholar]
- Buchwalter, A.; Hetzer, M.W. Nucleolar expansion and elevated protein translation in premature aging. Nat. Commun. 2017, 8, 328. [Google Scholar] [CrossRef]
- Gonzalo, S.; Kreienkamp, R.; Askjaer, P. Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res. Rev. 2017, 33, 18–29. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef]
- Prokocimer, M.; Barkan, R.; Gruenbaum, Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell 2013, 12, 533–543. [Google Scholar] [CrossRef]
- Gordon, L.B.; Rothman, F.G.; Lopez-Otin, C.; Misteli, T. Progeria: A paradigm for translational medicine. Cell 2014, 156, 400–407. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2019, 129, 531–545. [Google Scholar] [CrossRef]
- Hansen, M.; Taubert, S.; Crawford, D.; Libina, N.; Lee, S.J.; Kenyon, C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 2007, 6, 95–110. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef]
- Osorio, F.G.; Obaya, A.J.; Lopez-Otin, C.; Freije, J.M. Accelerated ageing: From mechanism to therapy through animal models. Transgenic Res. 2009, 18, 7–15. [Google Scholar] [CrossRef]
- Ferri, D.; Orioli, D.; Botta, E. Heterogeneity and overlaps in nucleotide excision repair (NER) disorders. Clin. Genet. 2019. [Google Scholar] [CrossRef]
- Nance, M.A.; Berry, S.A. Cockayne syndrome: Review of 140 cases. Am. J. Med Genet. 1992, 42, 68–84. [Google Scholar] [CrossRef]
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013, 134, 161–170. [Google Scholar] [CrossRef]
- Theil, A.F.; Hoeijmakers, J.H.; Vermeulen, W. TTDA: Big impact of a small protein. Exp. Cell Res. 2014, 329, 61–68. [Google Scholar] [CrossRef]
- Brooks, P.J. Blinded by the UV light: how the focus on transcription-coupled NER has distracted from understanding the mechanisms of Cockayne syndrome neurologic disease. DNA Repair 2013, 12, 656–671. [Google Scholar] [CrossRef]
- Bohr, V.; Anson, R.M.; Mazur, S.; Dianov, G. Oxidative DNA damage processing and changes with aging. Toxicol. Lett. 1998, 102–103, 47–52. [Google Scholar] [CrossRef]
- D’Errico, M.; Pascucci, B.; Iorio, E.; Van Houten, B.; Dogliotti, E. The role of CSA and CSB protein in the oxidative stress response. Mech. Ageing Dev. 2013, 134, 261–269. [Google Scholar] [CrossRef]
- Sampath, H. Oxidative DNA damage in disease--insights gained from base excision repair glycosylase-deficient mouse models. Environ. Mol. Mutagenesis 2014, 55, 689–703. [Google Scholar] [CrossRef]
- Khobta, A.; Epe, B. Repair of oxidatively generated DNA damage in Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 253–260. [Google Scholar] [CrossRef]
- Tamura, D.; DiGiovanna, J.J.; Khan, S.G.; Kraemer, K.H. Living with xeroderma pigmentosum: comprehensive photoprotection for highly photosensitive patients. Photodermatol. Photoimmunol. Photomed. 2014, 30, 146–152. [Google Scholar] [CrossRef]
- Horibata, K.; Iwamoto, Y.; Kuraoka, I.; Jaspers, N.G.; Kurimasa, A.; Oshimura, M.; Ichihashi, M.; Tanaka, K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 15410–15415. [Google Scholar] [CrossRef]
- Nardo, T.; Oneda, R.; Spivak, G.; Vaz, B.; Mortier, L.; Thomas, P.; Orioli, D.; Laugel, V.; Stary, A.; Hanawalt, P.C.; et al. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 6209–6214. [Google Scholar] [CrossRef]
- Itoh, T.; Fujiwara, Y.; Ono, T.; Yamaizumi, M. UVs syndrome, a new general category of photosensitive disorder with defective DNA repair, is distinct from xeroderma pigmentosum variant and rodent complementation group I. Am. J. Hum. Genet. 1995, 56, 1267–1276. [Google Scholar]
- Soltys, D.T.; Rocha, C.R.; Lerner, L.K.; de Souza, T.A.; Munford, V.; Cabral, F.; Nardo, T.; Stefanini, M.; Sarasin, A.; Cabral-Neto, J.B.; et al. Novel XPG (ERCC5) mutations affect DNA repair and cell survival after ultraviolet but not oxidative stress. Hum. Mutat. 2013, 34, 481–489. [Google Scholar] [CrossRef]
- Yu, Y.; Cui, Y.; Niedernhofer, L.J.; Wang, Y. Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage. Chem. Res. Toxicol. 2016, 29, 2008–2039. [Google Scholar] [CrossRef]
- Alupei, M.C.; Maity, P.; Esser, P.R.; Krikki, I.; Tuorto, F.; Parlato, R.; Penzo, M.; Schelling, A.; Laugel, V.; Montanaro, L.; et al. Loss of Proteostasis Is a Pathomechanism in Cockayne Syndrome. Cell Rep. 2018, 23, 1612–1619. [Google Scholar] [CrossRef]
- Iben, S.; Tschochner, H.; Bier, M.; Hoogstraten, D.; Hozak, P.; Egly, J.M.; Grummt, I. TFIIH plays an essential role in RNA polymerase I transcription. Cell 2002, 109, 297–306. [Google Scholar] [CrossRef]
- Hoogstraten, D.; Nigg, A.L.; Heath, H.; Mullenders, L.H.; van Driel, R.; Hoeijmakers, J.H.; Vermeulen, W.; Houtsmuller, A.B. Rapid switching of TFIIH between RNA polymerase I and II transcription and DNA repair in vivo. Mol. Cell 2002, 10, 1163–1174. [Google Scholar] [CrossRef]
- Bradsher, J.; Auriol, J.; Proietti de Santis, L.; Iben, S.; Vonesch, J.L.; Grummt, I.; Egly, J.M. CSB is a component of RNA pol I transcription. Mol. Cell 2002, 10, 819–829. [Google Scholar] [CrossRef]
- Schmitz, K.M.; Schmitt, N.; Hoffmann-Rohrer, U.; Schafer, A.; Grummt, I.; Mayer, C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol. Cell 2009, 33, 344–353. [Google Scholar] [CrossRef]
- Koch, S.; Garcia Gonzalez, O.; Assfalg, R.; Schelling, A.; Schafer, P.; Scharffetter-Kochanek, K.; Iben, S. Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth. Cell Cycle 2014, 13, 2029–2037. [Google Scholar] [CrossRef]
- Assfalg, R.; Lebedev, A.; Gonzalez, O.G.; Schelling, A.; Koch, S.; Iben, S. TFIIH is an elongation factor of RNA polymerase I. Nucleic Acids Res. 2012, 40, 650–659. [Google Scholar] [CrossRef]
- Nonnekens, J.; Perez-Fernandez, J.; Theil, A.F.; Gadal, O.; Bonnart, C.; Giglia-Mari, G. Mutations in TFIIH causing trichothiodystrophy are responsible for defects in ribosomal RNA production and processing. Hum. Mol. Genet. 2013, 22, 2881–2893. [Google Scholar] [CrossRef]
- Yuan, X.; Feng, W.; Imhof, A.; Grummt, I.; Zhou, Y. Activation of RNA polymerase I transcription by cockayne syndrome group B protein and histone methyltransferase G9a. Mol. Cell 2007, 27, 585–595. [Google Scholar] [CrossRef]
- Xie, W.; Ling, T.; Zhou, Y.; Feng, W.; Zhu, Q.; Stunnenberg, H.G.; Grummt, I.; Tao, W. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc. Natl. Acad. Sci. USA 2012, 109, 8161–8166. [Google Scholar] [CrossRef]
- Lebedev, A.; Scharffetter-Kochanek, K.; Iben, S. Truncated Cockayne syndrome B protein represses elongation by RNA polymerase I. J. Mol. Biol. 2008, 382, 266–274. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Tseng, A.; Borch Jensen, M.; Scheibye-Alsing, K.; Fang, E.F.; Iyama, T.; Bharti, S.K.; Marosi, K.; Froetscher, L.; Kassahun, H.; et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. USA 2016, 113, 12502–12507. [Google Scholar] [CrossRef]
- Azpurua, J.; Ke, Z.; Chen, I.X.; Zhang, Q.; Ermolenko, D.N.; Zhang, Z.D.; Gorbunova, V.; Seluanov, A. Naked mole-rat has increased translational fidelity compared with the mouse, as well as a unique 28S ribosomal RNA cleavage. Proc. Natl. Acad. Sci. USA 2013, 110, 17350–17355. [Google Scholar] [CrossRef]
- Orgel, L.E. The maintenance of the accuracy of protein synthesis and its relevance to ageing. Proc. Natl. Acad. Sci. USA 1963, 49, 517–521. [Google Scholar] [CrossRef]
- Gallant, J.; Kurland, C.; Parker, J.; Holliday, R.; Rosenberger, R. The error catastrophe theory of aging. Point counterpoint. Exp. Gerontol. 1997, 32, 333–346. [Google Scholar] [CrossRef]
- Luce, M.C.; Bunn, C.L. Decreased accuracy of protein synthesis in extracts from aging human diploid fibroblasts. Exp. Gerontol. 1989, 24, 113–125. [Google Scholar] [CrossRef]
- Mori, N.; Hiruta, K.; Funatsu, Y.; Goto, S. Codon recognition fidelity of ribosomes at the first and second positions does not decrease during aging. Mech. Ageing Dev. 1983, 22, 1–10. [Google Scholar] [CrossRef]
- Ke, Z.; Seluanov, A.; Gorbunova, V. Accurate translation is important for longevity. Aging 2018, 10, 297–298. [Google Scholar] [CrossRef]
- Paolini, N.A.; Attwood, M.; Sondalle, S.B.; Vieira, C.; van Adrichem, A.M.; di Summa, F.M.; O’Donohue, M.F.; Gleizes, P.E.; Rachuri, S.; Briggs, J.W.; et al. A Ribosomopathy Reveals Decoding Defective Ribosomes Driving Human Dysmorphism. Am. J. Hum. Genet. 2017, 100, 506–522. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, T.; Khalid, F.; Iben, S. Nucleolar and Ribosomal Dysfunction—A Common Pathomechanism in Childhood Progerias? Cells 2019, 8, 534. https://doi.org/10.3390/cells8060534
Phan T, Khalid F, Iben S. Nucleolar and Ribosomal Dysfunction—A Common Pathomechanism in Childhood Progerias? Cells. 2019; 8(6):534. https://doi.org/10.3390/cells8060534
Chicago/Turabian StylePhan, Tamara, Fatima Khalid, and Sebastian Iben. 2019. "Nucleolar and Ribosomal Dysfunction—A Common Pathomechanism in Childhood Progerias?" Cells 8, no. 6: 534. https://doi.org/10.3390/cells8060534
APA StylePhan, T., Khalid, F., & Iben, S. (2019). Nucleolar and Ribosomal Dysfunction—A Common Pathomechanism in Childhood Progerias? Cells, 8(6), 534. https://doi.org/10.3390/cells8060534