Current Status of Patient-Derived Ovarian Cancer Models



Abstract
1. Introduction
2. Cancer Cell Lines
2.1. General Overview
2.2. Ovarian Cancer Cell Lines
3. Patient-Derived Xenografts (PDXs)
3.1. General Overview
3.2. PDX for Ovarian Cancer
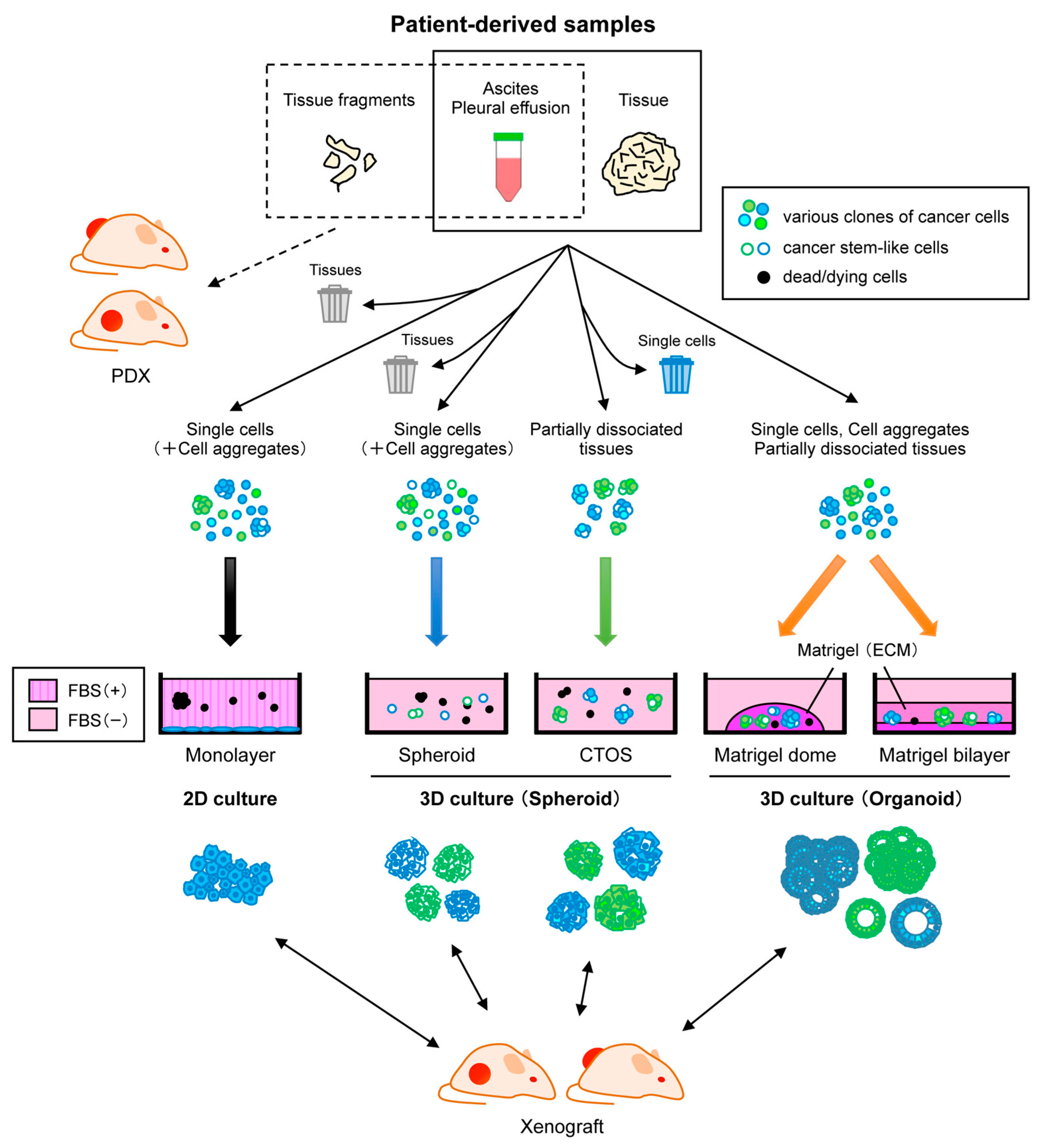
4. Patient-Derived Cells
4.1. Two-Dimensional Adherent Culture
4.1.1. General Overview
4.1.2. Ovarian Cancer Cell Lines
4.2. Three-Dimensional Culture
4.2.1. Spheroid Culture
General Overview
Ovarian Cancer Spheroids
Non-Single Cell-Based Approaches
4.2.2. Organoid Culture
General Overview
Organoid-Based Carcinogenesis Model
Primary Tumor Organoid
Ovarian Cancer Organoid
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Koshiyama, M.; Matsumura, N.; Konishi, I. Recent Concepts of Ovarian Carcinogenesis: Type I and Type II. Biomed Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.-M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noé, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Ayhan, A.; Mao, T.-L.; Seckin, T.; Wu, C.-H.; Guan, B.; Ogawa, H.; Futagami, M.; Mizukami, H.; Yokoyama, Y.; Kurman, R.J.; et al. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int. J. Gynecol. Cancer. 2012, 22, 1310–1315. [Google Scholar] [CrossRef]
- Sato, N.; Tsunoda, H.; Nishida, M.; Morishita, Y.; Takimoto, Y.; Kubo, T.; Noguchi, M. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: Possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res 2000, 60, 7052–7056. [Google Scholar]
- Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [PubMed]
- Hunter, S.M.; Anglesio, M.S.; Ryland, G.L.; Sharma, R.; Chiew, Y.-E.; Rowley, S.M.; Doyle, M.A.; Li, J.; Gilks, C.B.; Moss, P.; et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget 2015, 6, 37663–37677. [Google Scholar] [CrossRef]
- MacKenzie, R.; Kommoss, S.; Winterhoff, B.J.; Kipp, B.R.; García, J.J.; Voss, J.; Halling, K.; Karnezis, A.; Senz, J.; Yang, W.; et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer. 2015, 15, 200. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Wang, T.-L.; Shih, I.-M.; Mao, T.-L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A.; Vogelstein, B.; et al. Frequent Mutations of Chromatin Remodeling Gene ARID1A in Ovarian Clear Cell Carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.-C.; Ayhan, A.; Maeda, D.; Kim, K.-R.; Clarke, B.A.; Shaw, P.; Chui, M.H.; Rosen, B.; Shih, I.-M.; Wang, T.-L. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other major types of gynecologic malignancy. J. Pathol. 2014, 232, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Tanaka, N.; Ohira, M.; Itami, M.; Hippo, Y.; Nagase, H. Identification of novel mutations in Japanese ovarian clear cell carcinoma patients using optimized targeted NGS for clinical diagnosis. Gynecol. Oncol. 2017, 144, 377–383. [Google Scholar] [CrossRef]
- McConechy, M.K.; Ding, J.; Senz, J.; Yang, W.; Melnyk, N.; Tone, A.A.; Prentice, L.M.; Wiegand, K.C.; McAlpine, J.N.; Shah, S.P.; et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Modern Pathol. 2014, 27, 128. [Google Scholar] [CrossRef]
- Pectasides, D.; Fountzilas, G.; Aravantinos, G.; Kalofonos, C.; Efstathiou, H.; Farmakis, D.; Skarlos, D.; Pavlidis, N.; Economopoulos, T.; Dimopoulos, M.A. Advanced stage clear-cell epithelial ovarian cancer: The Hellenic cooperative oncology group experience. Gynecol. Oncol. 2006, 102, 285–291. [Google Scholar] [CrossRef]
- Hess, V.; A’Hern, R.; Nasiri, N.; King, D.M.; Blake, P.R.; Barton, D.P.; Shepherd, J.H.; Ind, T.; Bridges, J.; Harrington, K.; et al. Mucinous Epithelial Ovarian Cancer: A Separate Entity Requiring Specific Treatment. J. Clin. Oncol. 2004, 22, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Monk, B.J.; Minion, L.E.; Coleman, R.L. Anti-angiogenic agents in ovarian cancer: Past, present, and future. Ann. Oncol. 2016, 27, i33–i39. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Greshock, J.; Bachman, K.E.; Degenhardt, Y.Y.; Jing, J.; Wen, Y.H.; Eastman, S.; McNeil, E.; Moy, C.; Wegrzyn, R.; Auger, K.; et al. Molecular Target Class Is Predictive of In vitro Response Profile. Cancer Res 2010, 70, 3677–3686. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modeling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Masters, J.R.W. Human cancer cell lines: Fact and fantasy. Nat. Rev. Mol. Cell Biol. 2000, 1, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J. Trial watch: Phase III and submission failures: 2007–2010. Nat. Rev. Drug Discov. 2011, 10, 87. [Google Scholar] [CrossRef]
- Arrowsmith, J.; Miller, P. Trial watch: Phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Discov. 2013, 12, 569. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; Van Ijcken, W.F.J.; Heine, A.A.J.; Smid, M.; et al. Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of In Vitro Morphological Subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Kim, M.K.; Lyle, L.T.; Bunch, K.P.; House, C.D.; Ning, F.; Noonan, A.M.; Annunziata, C.M. Characterization of ovarian cancer cell lines as in vivo models for preclinical studies. Gynecol. Oncol. 2016, 142, 332–340. [Google Scholar] [CrossRef]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- Cho, Y.B.; Hong, H.K.; Choi, Y.-L.; Oh, E.; Joo, K.M.; Jin, J.; Nam, D.-H.; Ko, Y.-H.; Lee, W.Y. Colorectal cancer patient–derived xenografted tumors maintain characteristic features of the original tumors. J. Surg. Res. 2014, 187, 502–509. [Google Scholar] [CrossRef]
- Guenot, D.; Guerin, E.; Pencreach, E.; Schneider, A.; Neuville, A.; Chenard, M.-P.; Duluc, I.; Du Manoir, S.; Brigand, C.; Oudet, P.; et al. Primary tumour genetic alterations and intra-tumoral heterogeneity are maintained in xenografts of human colon cancers showing chromosome instability. J. Pathol. 2006, 208, 643–652. [Google Scholar] [CrossRef]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer. 2017, 17, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A Renewable Tissue Resource of Phenotypically Stable, Biologically and Ethnically Diverse, Patient-Derived Human Breast Cancer Xenograft Models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Roudier, M.; et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease and Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, X.; Liu, P.; Li, M.; Luo, F. Patient-derived xenograft mouse models: A high fidelity tool for individualized medicine. Oncol. Lett. 2019, 17, 3–10. [Google Scholar] [CrossRef]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo murine-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef]
- Morton, J.J.; Bird, G.; Keysar, S.B.; Astling, D.P.; Lyons, T.R.; Anderson, R.T.; Glogowska, M.J.; Estes, P.; Eagles, J.R.; Le, P.N.; et al. XactMice: Humanizing mouse bone marrow enables microenvironment reconstitution in a patient-derived xenograft model of head and neck cancer. Oncogene 2016, 35, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Davy, M.; Mossige, J.; Johannessen, J.V. Heterologous Growth of Human Ovarian Cancer: A New in Vivo Testing System. Acta Obstet. Gynecol. Scand. 1977, 56, 55–59. [Google Scholar] [CrossRef]
- Ward, B.G.; Wallace, K.; Shepherd, J.H.; Balkwill, F.R. Intraperitoneal xenografts of human epithelial ovarian cancer in nude mice. Cancer Res. 1987, 47, 2662–2667. [Google Scholar]
- Fu, X.; Hoffman, R.M. Human ovarian carcinoma metastatic models constructed in nude mice by orthotopic transplantation of histologically-intact patient specimens. Anticancer Res. 1993, 13, 283–286. [Google Scholar]
- Weroha, S.J.; Becker, M.A.; Enderica-Gonzalez, S.; Harrington, S.C.; Oberg, A.L.; Maurer, M.J.; Perkins, S.E.; Al Hilli, M.; Butler, K.A.; McKinstry, S.; et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin. Cancer Res. 2014, 20, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.M.; Shaw, P.A.; Gedye, C.; Bernardini, M.Q.; Neel, B.G.; Ailles, L.E. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. PNAS 2011, 108, 6468–6473. [Google Scholar] [CrossRef]
- Dobbin, Z.C.; Katre, A.A.; Steg, A.D.; Erickson, B.K.; Shah, M.M.; Alvarez, R.D.; Conner, M.G.; Schneider, D.; Chen, D.; Landen, C.N. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 2014, 5, 8750–8764. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Silver, D.F.; Yang, N.-P.; Oflazoglu, E.; Hempling, R.E.; Piver, M.; Repasky, E.A. Characterization of Human Ovarian Carcinomas in a SCID Mouse Model. Gynecol. Oncol. 1999, 72, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Bankert, R.B.; Balu-Iyer, S.V.; Odunsi, K.; Shultz, L.D.; Kelleher, R.J.; Barnas, J.L.; Simpson-Abelson, M.; Parsons, R.; Yokota, S.J. Humanized Mouse Model of Ovarian Cancer Recapitulates Patient Solid Tumor Progression, Ascites Formation, and Metastasis. PLoS ONE 2011, 6, e24420. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Palakurthi, S.; Zeng, Q.; Zhou, S.; Ivanova, E.; Huang, W.; Zervantonakis, I.K.; Selfors, L.M.; Shen, Y.; Pritchard, C.C.; et al. Establishment of Patient-Derived Tumor Xenograft Models of Epithelial Ovarian Cancer for Preclinical Evaluation of Novel Therapeutics. Clin. Cancer Res. 2017, 23, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Press, J.Z.; Kenyon, J.A.; Xue, H.; Miller, M.A.; De Luca, A.; Miller, D.M.; Huntsman, D.G.; Gilks, C.B.; McAlpine, J.N.; Wang, Y. Xenografts of primary human gynecological tumors grown under the renal capsule of NOD/SCID mice show genetic stability during serial transplantation and respond to cytotoxic chemotherapy. Gynecol. Oncol. 2008, 110, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Cybulska, P.; Stewart, J.M.; Sayad, A.; Virtanen, C.; Shaw, P.A.; Clarke, B.; Stickle, N.; Bernardini, M.Q.; Neel, B.G. A Genomically Characterized Collection of High-Grade Serous Ovarian Cancer Xenografts for Preclinical Testing. Am. J. Pathol. 2018, 188, 1120–1131. [Google Scholar] [CrossRef]
- Liu, Y.; Chanana, P.; Davila, J.I.; Hou, X.; Zanfagnin, V.; McGehee, C.D.; Goode, E.L.; Polley, E.C.; Haluska, P.; Weroha, S.J.; et al. Gene expression differences between matched pairs of ovarian cancer patient tumors and patient-derived xenografts. Sci. Rep. 2019, 9, 6314. [Google Scholar] [CrossRef]
- Kolfschoten, G.M.; Schlüper, H.M.; Erkelens, C.A.; Pinedo, H.M.; Scheffer, P.G.; Boven, E. Development of a Panel of 15 Human Ovarian Cancer Xenografts for Drug Screening and Determination of the Role of the Glutathione Detoxification System. Gynecol. Oncol. 2000, 76, 362–368. [Google Scholar] [CrossRef]
- Vidal, A.; Muñoz, C.; Guillén, M.-J.; Moretó, J.; Puertas, S.; Martínez-Iniesta, M.; Figueras, A.; Padullés, L.; García-Rodriguez, F.J.; Berdiel-Acer, M.; et al. Lurbinectedin (PM01183), a New DNA Minor Groove Binder, Inhibits Growth of Orthotopic Primary Graft of Cisplatin-Resistant Epithelial Ovarian Cancer. Clin. Cancer Res. 2012, 18, 5399–5411. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Bizzaro, F.; Cesca, M.; Guffanti, F.; Ganzinelli, M.; Decio, A.; Ghilardi, C.; Perego, P.; Fruscio, R.; Buda, A.; et al. Patient-Derived Ovarian Tumor Xenografts Recapitulate Human Clinicopathology and Genetic Alterations. Cancer Res. 2014, 74, 6980–6990. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.D.; Hartley, L.; Cook, M.; Heong, V.; Boehm, E.; McShane, L.; Pyman, J.; McNally, O.; Ananda, S.; Harrell, M.; et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol. Oncol. 2014, 8, 656–668. [Google Scholar] [CrossRef]
- Groeneweg, J.W.; DiGloria, C.M.; Yuan, J.; Richardson, W.S.; Growdon, W.B.; Sathyanarayanan, S.; Foster, R.; Rueda, B.R. Inhibition of Notch Signaling in Combination with Paclitaxel Reduces Platinum-Resistant Ovarian Tumor Growth. Front. Oncol. 2014, 4, 171. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Becker, M.A.; Haluska, P.; Samimi, G. Patient-Derived Xenograft Models to Improve Targeted Therapy in Epithelial Ovarian Cancer Treatment. Front. Oncol. 2013, 3, 295. [Google Scholar] [CrossRef]
- Kortmann, U.; McAlpine, J.N.; Xue, H.; Guan, J.; Ha, G.; Tully, S.; Shafait, S.; Lau, A.; Cranston, A.N.; O’Connor, M.J.; et al. Tumor growth inhibition by olaparib in BRCA2 germline-mutated patient-derived ovarian cancer tissue xenografts. Clin. Cancer Res. 2011, 17, 783–791. [Google Scholar] [CrossRef]
- George, E.; Kim, H.; Krepler, C.; Wenz, B.; Makvandi, M.; Tanyi, J.L.; Brown, E.; Zhang, R.; Brafford, P.; Jean, S.; et al. A patient-derived-xenograft platform to study BRCA-deficient ovarian cancers. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Kwon, J.; Ali, B.; Wang, E.; Patra, S.; Shridhar, V.; Mukherjee, P. Role of Hedgehog Signaling in Ovarian Cancer. Clin. Cancer Res. 2008, 14, 7659–7666. [Google Scholar] [CrossRef]
- McCann, C.K.; Growdon, W.B.; Kulkarni-Datar, K.; Curley, M.D.; Friel, A.M.; Proctor, J.L.; Sheikh, H.; Deyneko, I.; Ferguson, J.A.; Vathipadiekal, V.; et al. Inhibition of Hedgehog Signaling Antagonizes Serous Ovarian Cancer Growth in a Primary Xenograft Model. PLoS ONE 2011, 6, e28077. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.R.; Zhang, P.; Yang, L.; Hou, X.; Leventakos, K.; Weroha, S.J.; Vasmatzis, G.; Kovtun, I.V. Targeting HER2 in patient-derived xenograft ovarian cancer models sensitizes tumors to chemotherapy. Mol. Oncol. 2019, 13, 132–152. [Google Scholar] [CrossRef] [PubMed]
- Eoh, K.J.; Chung, Y.S.; Lee, S.H.; Park, S.A.; Kim, H.J.; Yang, W.; Lee, I.O.; Lee, J.Y.; Cho, H.; Chay, D.B.; et al. Comparison of Clinical Features and Outcomes in Epithelial Ovarian Cancer according to Tumorigenicity in Patient-Derived Xenograft Models. Cancer Res. Treat 2018, 50, 956–963. [Google Scholar] [CrossRef]
- Dong, R.; Qiang, W.; Guo, H.; Xu, X.; Kim, J.J.; Mazar, A.; Kong, B.; Wei, J.-J. Histologic and molecular analysis of patient derived xenografts of high-grade serous ovarian carcinoma. J. Hematol. Oncol. 2016, 9, 1376. [Google Scholar] [CrossRef] [PubMed]
- McCallum, H.M.; Lowther, G.W. Long-term culture of primary breast cancer in defined medium. Breast Cancer Res. Treat. 1996, 39, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Kurvari, V.; Virmani, A.; Gollahon, L.; Sakaguchi, M.; Westerfield, M.; Kodagoda, D.; Stasny, V.; Cunningham, H.T.; Wistuba, I.I.; et al. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int. J. Cancer 1998, 78, 766–774. [Google Scholar] [CrossRef]
- Zaniolo, K.; Bergeron, M.-A.; Weidmann, C.; Fournier, F.; Droit, A.; Morcos, M.W.; Landreville, S.; Guérin, S.L.; Mouriaux, F.; De La Fouchardière, A. Effects of Long-term Serial Passaging on the Characteristics and Properties of Cell Lines Derived From Uveal Melanoma Primary Tumors. Invest. Opthalmol. Vis. Sci. 2016, 57, 5288. [Google Scholar]
- Ince, T.A.; Sousa, A.D.; Jones, M.A.; Harrell, J.C.; Agoston, E.S.; Krohn, M.; Selfors, L.M.; Liu, W.; Chen, K.; Yong, M.; et al. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat. Commun. 2015, 6, 7419. [Google Scholar] [CrossRef]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Ponti, D. Isolation and In vitro Propagation of Tumorigenic Breast Cancer Cells with Stem/Progenitor Cell Properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, W.; Guo, H.; Zhang, Y.; He, Y.; Lee, S.H.; Song, X.; Li, X.; Guo, Y.; Zhao, Y.; et al. NOTCH1 Signaling Regulates Self-Renewal and Platinum Chemoresistance of Cancer Stem–like Cells in Human Non–Small Cell Lung Cancer. Cancer Res. 2017, 77, 3082–3091. [Google Scholar] [CrossRef]
- A Bapat, S.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.-C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.-M.; Nephew, K.P. Identification and Characterization of Ovarian Cancer-Initiating Cells from Primary Human Tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef]
- Ishiguro, T.; Sato, A.; Ohata, H.; Ikarashi, Y.; Takahashi, R.U.; Ochiya, T.; Yoshida, M.; Tsuda, H.; Onda, T.; Kato, T.; et al. Establishment and Characterization of an In Vitro Model of Ovarian Cancer Stem-like Cells with an Enhanced Proliferative Capacity. Cancer Res. 2016, 76, 150–160. [Google Scholar] [CrossRef]
- Raghavan, S.; Mehta, P.; Ward, M.R.; Bregenzer, M.E.; Fleck, E.M.A.; Tan, L.; McLean, K.; Buckanovich, R.J.; Mehta, G. Personalized Medicine Based Approach to Model Patterns of Chemoresistance, and Tumor Recurrence Using Ovarian Cancer Stem Cell Spheroids. Clin. Cancer Res. 2017, 23, 6934–6945. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Endo, H.; Okuyama, H.; Ishikawa, O.; Iishi, H.; Tsujii, M.; Ohue, M.; Inoue, M. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. PNAS 2011, 108, 6235–6240. [Google Scholar] [CrossRef]
- Kiyohara, Y.; Yoshino, K.; Kubota, S.; Okuyama, H.; Endo, H.; Ueda, Y.; Kimura, T.; Kimura, T.; Kamiura, S.; Inoue, M. Drug screening and grouping by sensitivity with a panel of primary cultured cancer spheroids derived from endometrial cancer. Cancer Sci. 2016, 107, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Okami, J.; Okuyama, H.; Kumagai, T.; Uchida, J.; Kondo, J.; Takehara, T.; Nishizawa, Y.; Imamura, F.; Higashiyama, M.; et al. Spheroid Culture of Primary Lung Cancer Cells with Neuregulin 1/HER3 Pathway Activation. J. Thorac. Oncol. 2013, 8, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Ekawa, T.; Endo, H.; Yamazaki, K.; Tanaka, N.; Kukita, Y.; Okuyama, H.; Okami, J.; Imamura, F.; Ohue, M.; et al. High-throughput screening in colorectal cancer tissue-originated spheroids. Cancer Sci. 2019, 110, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Higa, A.; Hoshi, H.; Hiyama, G.; Takahashi, N.; Ryufuku, M.; Morisawa, G.; Yanagisawa, Y.; Ito, E.; Imai, J.-I.; et al. Evaluation of anticancer agents using patient-derived tumor organoids characteristically similar to source tissues. Oncol. Rep. 2018, 40, 635–646. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Boretto, M.; Cox, B.; Noben, M.; Hendriks, N.; Fassbender, A.; Roose, H.; Amant, F.; Timmerman, D.; Tomassetti, C.; Vanhie, A.; et al. Development of organoids from mouse and human endometrium showing endometrial epithelium physiology and long-term expandability. Development 2017, 144, 1775–1786. [Google Scholar] [CrossRef]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.-J.; Mangler, M.; Sehouli, J.; et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 2015, 6, 8989. [Google Scholar] [CrossRef] [PubMed]
- DeWard, A.D.; Cramer, J.; Lagasse, E. Cellular heterogeneity in the mouse esophagus implicates the presence of a non-quiescent epithelial stem cell population. Cell Rep. 2014, 9, 701–711. [Google Scholar] [CrossRef]
- Bartfeld, S.; Bayram, T.; van de Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, R.; Peters, P.J.; Clevers, H. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 2015, 148, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Huang, S.X.; De Carvalho, A.L.R.T.; Ho, S.-H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.-L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell. Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Aihara, E.; Feng, R.; Engevik, A.; Shroyer, N.F.; Ottemann, K.M.; Worrell, R.T.; Montrose, M.H.; Shivdasani, R.A.; Zavros, Y. The use of murine-derived fundic organoids in studies of gastric physiology. J. Physiol. 2015, 593, 1809–1827. [Google Scholar] [CrossRef]
- Maru, Y.; Orihashi, K.; Hippo, Y. Lentivirus-Based Stable Gene Delivery into Intestinal Organoids. Methods Mol. Biol. 2016, 1422, 13–21. [Google Scholar]
- Onuma, K.; Ochiai, M.; Orihashi, K.; Takahashi, M.; Imai, T.; Nakagama, H.; Hippo, Y. Genetic reconstitution of tumorigenesis in primary intestinal cells. PNAS 2013, 110, 11127–11132. [Google Scholar] [CrossRef]
- Ochiai, M.; Yoshihara, Y.; Maru, Y.; Matsuura, T.; Izumiya, M.; Imai, T.; Hippo, Y. Kras-driven heterotopic tumor development from hepatobiliary organoids. Carcinogenesis 2019. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Morita, M.; Tanaka, R.; Inoue, Y.; Nomura, M.; Sakamoto, Y.; Miura, K.; Ito, S.; Sato, I.; Tanaka, N.; et al. Ex vivo model of non-small cell lung cancer using mouse lung epithelial cells. Oncol. Lett. 2017, 14, 6863–6868. [Google Scholar] [CrossRef]
- Drost, J.; Van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; Van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.P.; Loizou, E.; Livshits, G.; Schatoff, E.M.; Baslan, T.; Manchado, E.; Simon, J.; Romesser, P.B.; Leach, B.; Han, T.; et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 2017, 35, 577–582. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, C.; Hu, Y.; Mu, L.; Huang, K.; Li, Q.; Li, X.; Tao, D.; Qin, J. Sphere-forming assay vs. organoid culture: Determining long-term stemness and the chemoresistant capacity of primary colorectal cancer cells. Int. J. Oncol. 2019, 54, 893–904. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Brink, S.V.D.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term Expansion of Epithelial Organoids From Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.H.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897.e11. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Sachs, N.; De Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef]
- Li, X.; Francies, H.E.; Secrier, M.; Perner, J.; Miremadi, A.; Galeano-Dalmau, N.; Barendt, W.J.; Letchford, L.; Leyden, G.M.; Goffin, E.K.; et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 2018, 9, 2983. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef]
- Girda, E.; Huang, E.C.; Leiserowitz, G.S.; Smith, L.H. The Use of Endometrial Cancer Patient–Derived Organoid Culture for Drug Sensitivity Testing Is Feasible. Int. J. Gynecol. Cancer. 2017, 27, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Van De Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; Van Houdt, W.; Van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a Living Organoid Biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; Van De Haar, J.; Fanchi, L.F.; Slagter, M.; Van Der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Ajima, R.; Luo, C.T.-Y.; Yamaguchi, T.P.; Stappenbeck, T.S. Wnt5a Potentiates TGF-β Signaling to Promote Colonic Crypt Regeneration after Tissue Injury. Science 2012, 338, 108–113. [Google Scholar] [CrossRef]
- Miyoshi, H.; Stappenbeck, T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013, 8, 2471–2482. [Google Scholar] [CrossRef]
- Tiriac, H.; Bucobo, J.C.; Tzimas, D.; Grewel, S.; LaComb, J.F.; Rowehl, L.M.; Nagula, S.; Wu, M.; Kim, J.; Sasson, A.; et al. Successful creation of pancreatic cancer organoids by means of EUS-guided fine-needle biopsy sampling for personalized cancer treatment. Gastrointest. Endosc. 2018, 87, 1474–1480. [Google Scholar] [CrossRef]
- Jabs, J.; Zickgraf, F.M.; Park, J.; Wagner, S.; Jiang, X.; Jechow, K.; Kleinheinz, K.; Toprak, U.H.; A Schneider, M.; Meister, M.; et al. Screening drug effects in patient-derived cancer cells links organoid responses to genome alterations. Mol. Syst. Biol. 2017, 13, 955. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef]
- Kopper, O.; De Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Tanaka, N.; Itami, M.; Hippo, Y. Efficient use of patient-derived organoids as a preclinical model for gynecologic tumors. Gynecol. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Onuma, K.; Ochiai, M.; Imai, T.; Hippo, Y. Shortcuts to intestinal carcinogenesis by genetic engineering in organoids. Cancer Sci. 2019, 110, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Krausz, T.; Yamada, S.D.; Lengyel, E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int. J. Cancer 2007, 121, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-S.; Ip, C.K.M.; Tang, M.Y.H.; Sy, S.K.H.; Yung, S.; Chan, T.-M.; Yang, M.; Shum, H.C.; Wong, A.S. Modeling Ovarian Cancer Multicellular Spheroid Behavior in a Dynamic 3D Peritoneal Microdevice. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]


{kind=link}
| Reference | Success | Histological Type | Patient’s Material | PDX | |
|---|---|---|---|---|---|
| Case (n) | Rate (%) | ||||
| [102] | 1 | 100 | N.D. | N.D. | N.T. |
| [111] | 9 | N.D. | SC | Tissue, Ascites, Pleural effusion | Yes |
| [112] | 23 | 80–90 | HGSC, CS | Tissue, Pleural effusion | N.T. |
| [113] | 32 | 65 | MBT, SBT, CCC, EMC, MC, LGSC, HGSC | Tissue, Ascites, Pleural effusion | Yes |
| [114] | 9 (4*, 5#) | 60 (44*, 83#) | BBT, SBT, EMC, MC, HGSC | Tissue | Yes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maru, Y.; Hippo, Y. Current Status of Patient-Derived Ovarian Cancer Models. Cells 2019, 8, 505. https://doi.org/10.3390/cells8050505
Maru Y, Hippo Y. Current Status of Patient-Derived Ovarian Cancer Models. Cells. 2019; 8(5):505. https://doi.org/10.3390/cells8050505
Chicago/Turabian StyleMaru, Yoshiaki, and Yoshitaka Hippo. 2019. "Current Status of Patient-Derived Ovarian Cancer Models" Cells 8, no. 5: 505. https://doi.org/10.3390/cells8050505
APA StyleMaru, Y., & Hippo, Y. (2019). Current Status of Patient-Derived Ovarian Cancer Models. Cells, 8(5), 505. https://doi.org/10.3390/cells8050505