Improved Oxygen Supply to Multicellular Spheroids Using A Gas-permeable Plate and Embedded Hydrogel Beads

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Gas-Permeable Plate
2.3. Hydrogel Beads
2.4. Methylcellulose (MC) Medium
2.5. Spheroid Production
2.6. Paraffin Sectioning and Hematoxylin-Eosin Staining
2.7. Pimonidazole Labeling
2.8. Frozen Sectioning and Immunostaining
2.9. DNA Quantification
2.10. Specific Rates of Glucose Consumption and Lactate Production
2.11. RNA Extraction and cDNA Synthesis
2.12. qPCR
2.13. CYP3A4 Activity Assay
2.14. Statistical Analysis
3. Results
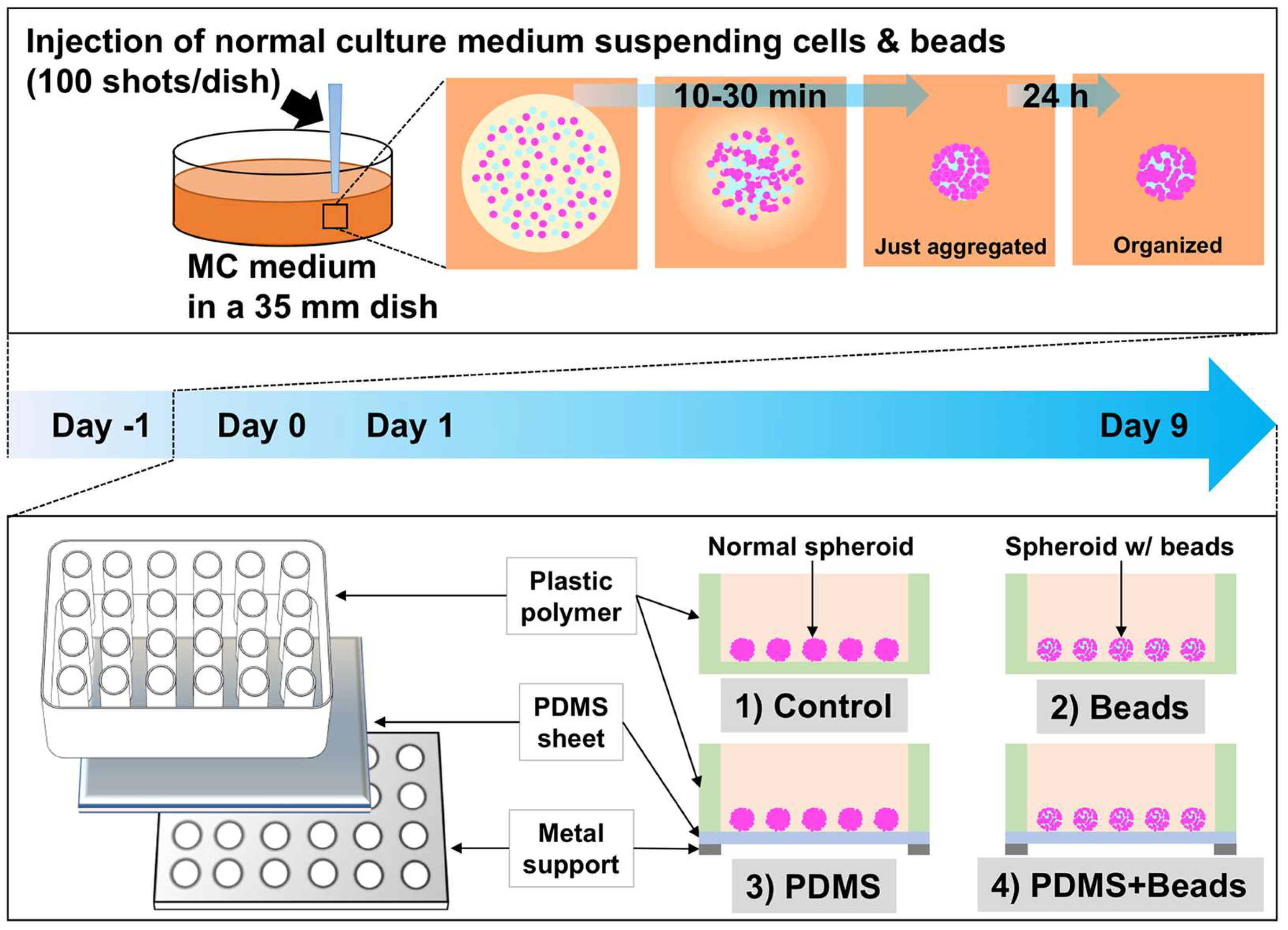
3.1. Spheroid Formation and Culture Conditions
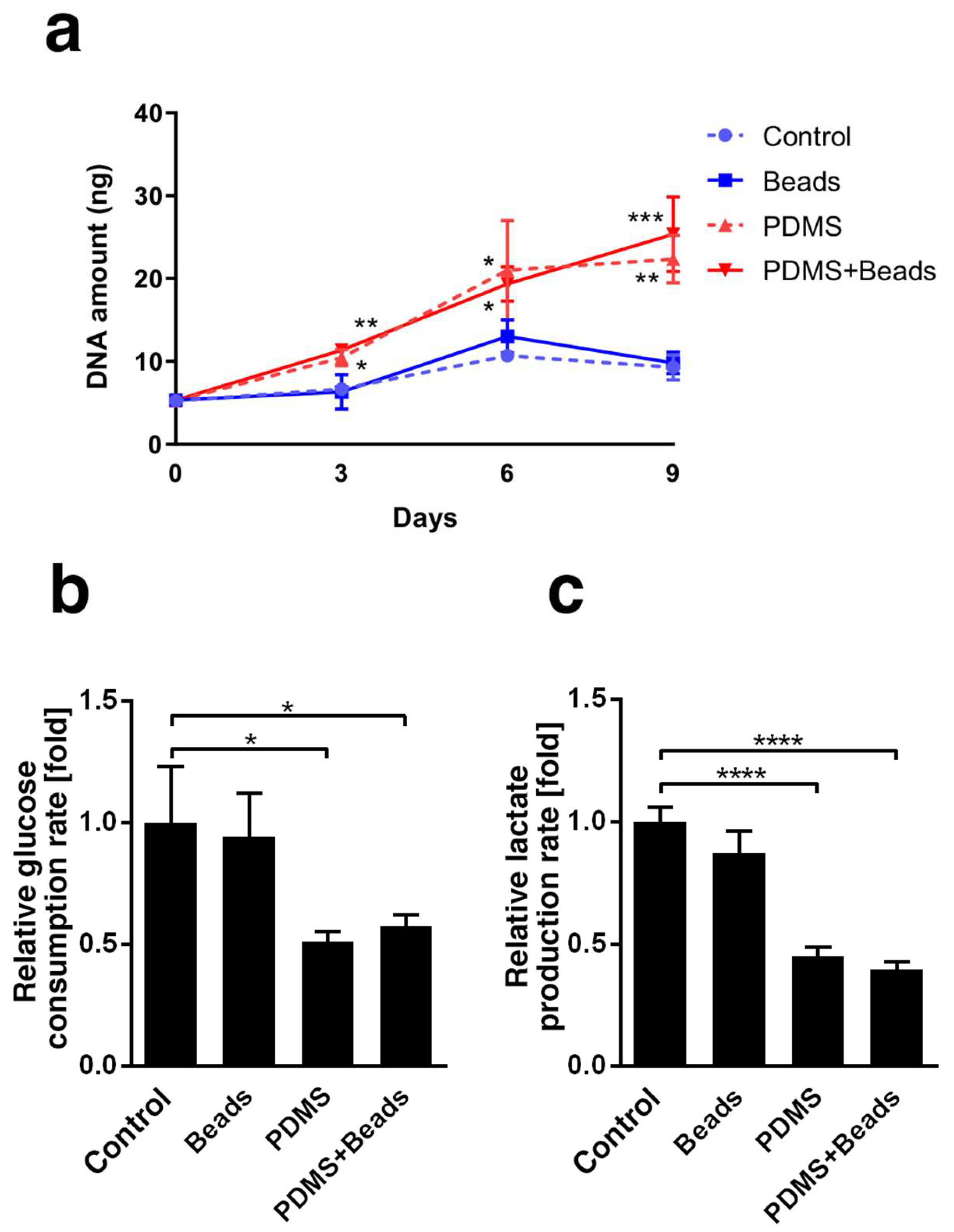
3.2. Differences in Spheroid Cell Growth and Energy Metabolism
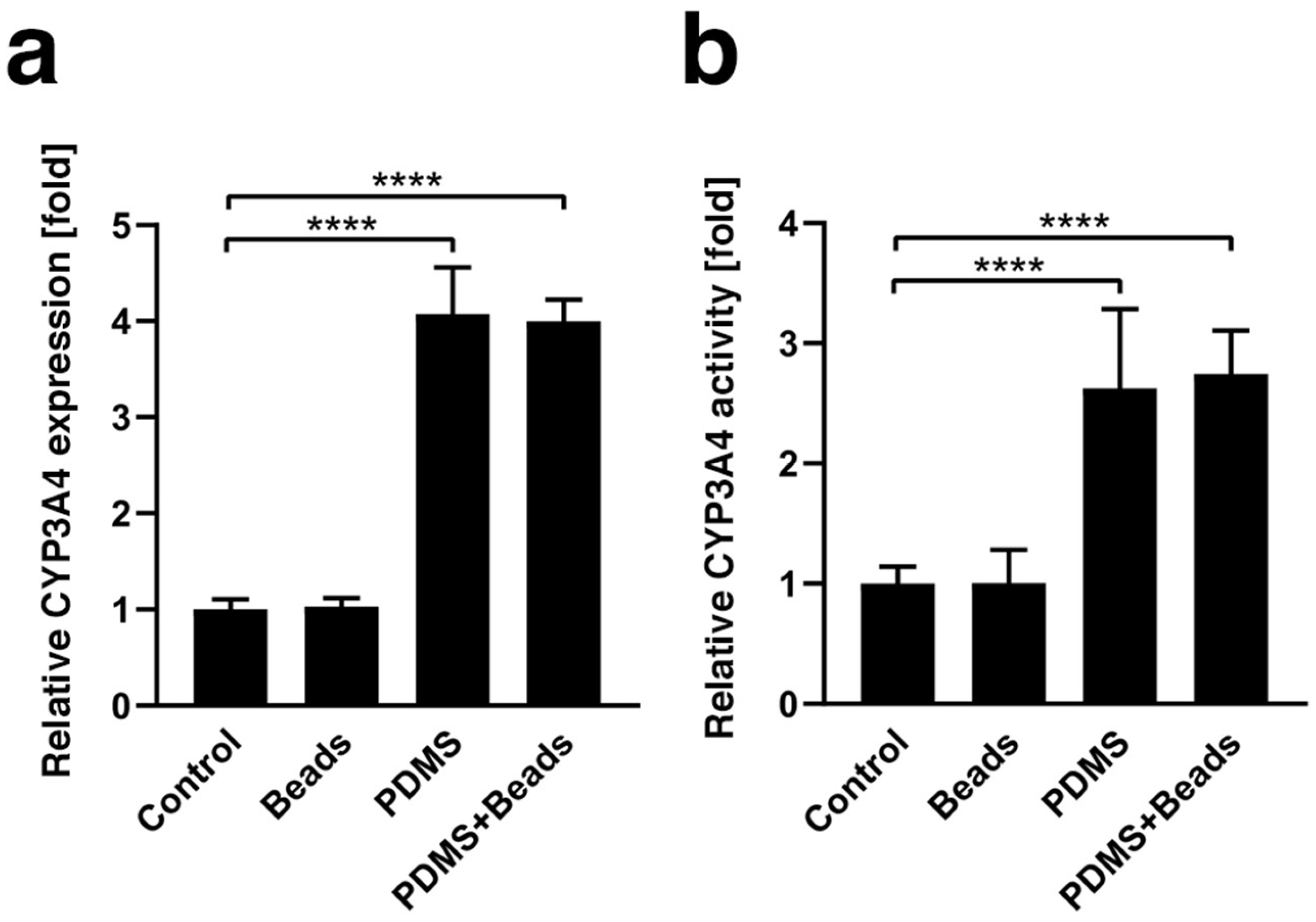
3.3. Enhancement of CYP3A4 Gene Expression and Enzyme Activity
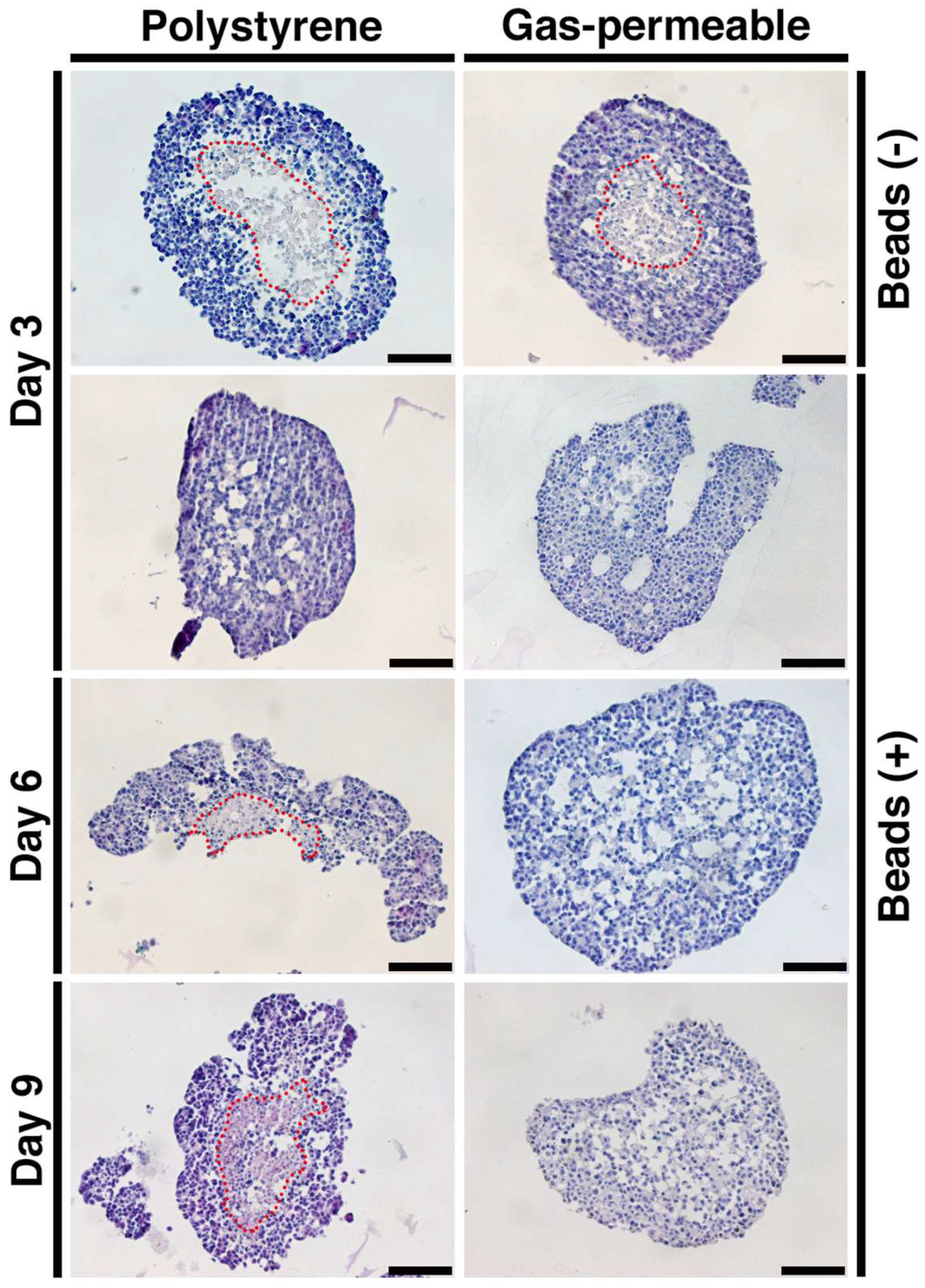
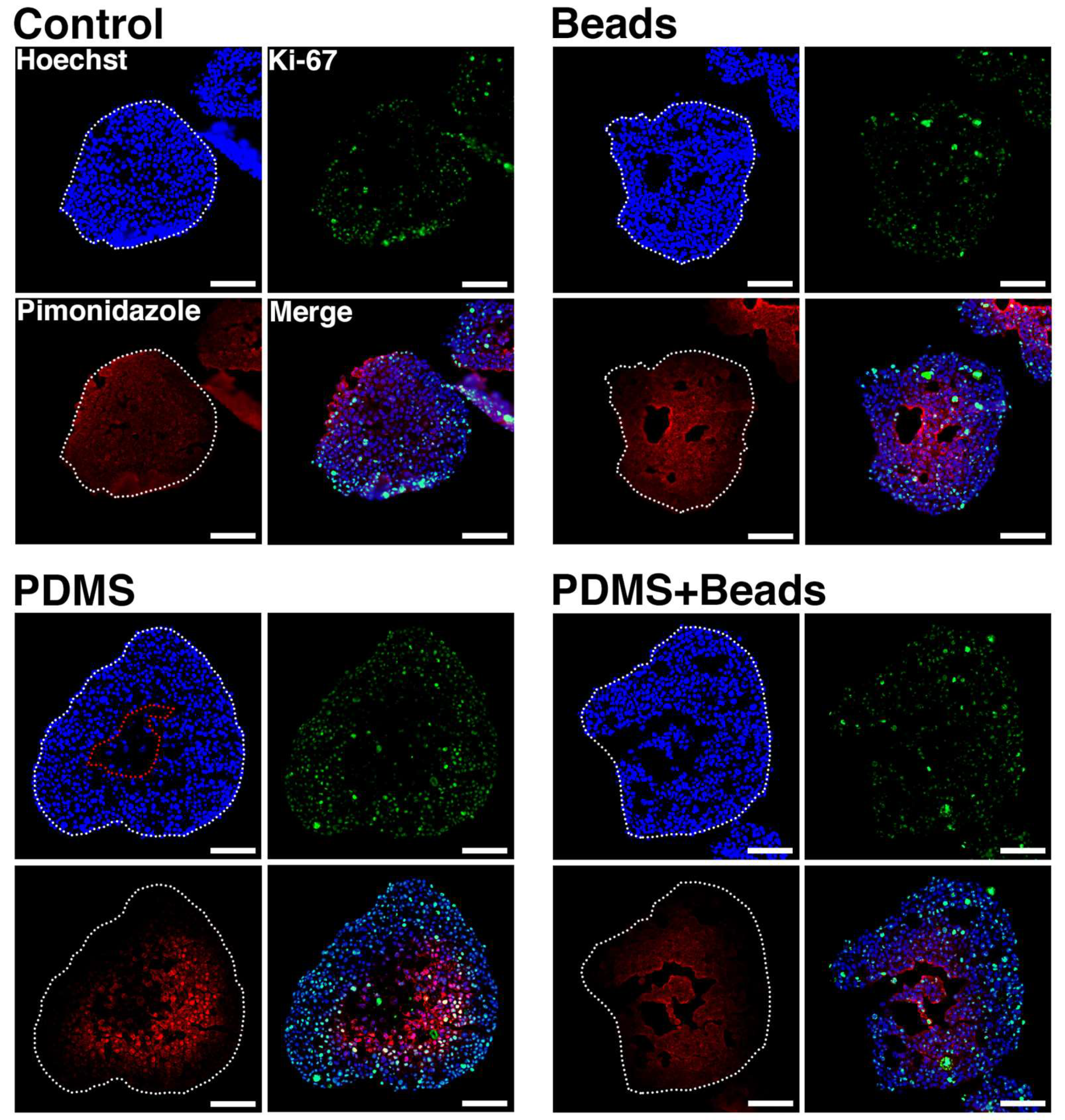
3.4. Prevention of Spheroid Core Necrosis
3.5. Spheroid Oxygen Distribution
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Siolas, D.; Hannon, G.J. Patient-derived tumor xenografts: Transforming clinical samples into mouse models. Cancer Res. 2013, 73, 5315–5319. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; Merlino, G.; Van Dyke, T. Preclinical mouse cancer models: A maze of opportunities and challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, A.; Mauro, F.; Zupi, G. Changes of phenotypic characteristics of variants derived from Lewis lung carcinoma during long-term in vitro growth. Clin. Exp. Metastasis 1984, 2, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Hausser, H.J.; Brenner, R.E. Phenotypic instability of Saos-2 cells in long-term culture. Biochem. Biophys. Res. Commun. 2005, 333, 216–222. [Google Scholar] [CrossRef]
- Kasai, F.; Hirayama, N.; Ozawa, M.; Iemura, M.; Kohara, A. Changes of heterogeneous cell populations in the Ishikawa cell line during long-term culture: Proposal for an in vitro clonal evolution model of tumor cells. Genomics 2016, 107, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Endo, H.; Okuyama, H.; Ishikawa, O.; Iishi, H.; Tsujii, M.; Ohue, M.; Inoue, M. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 6235–6240. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef] [PubMed]
- Muraro, M.G.; Muenst, S.; Mele, V.; Quagliata, L.; Iezzi, G.; Tzankov, A.; Weber, W.P.; Spagnoli, G.C.; Soysal, S.D. Ex-vivo assessment of drug response on breast cancer primary tissue with preserved microenvironments. Oncoimmunology 2017, 6, e1331798. [Google Scholar] [CrossRef]
- Nishikawa, M.; Kojima, N.; Komori, K.; Yamamoto, T.; Fujii, T.; Sakai, Y. Enhanced maintenance and functions of rat hepatocytes induced by combination of on-site oxygenation and coculture with fibroblasts. J. Biotechnol. 2008, 133, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Yamamoto, T.; Kojima, N.; Kikuo, K.; Fujii, T.; Sakai, Y. Stable immobilization of rat hepatocytes as hemispheroids onto collagen-conjugated poly-dimethylsiloxane (PDMS) surfaces: Importance of direct oxygenation through PDMS for both formation and function. Biotechnol. Bioeng. 2008, 99, 1472–1481. [Google Scholar] [CrossRef] [PubMed]
- Kojima, N.; Takeuchi, S.; Sakai, Y. Fabrication of microchannel networks in multicellular spheroids. Sens. Actuators B Chem. 2014, 198, 249–254. [Google Scholar] [CrossRef]
- Motoyama, W.; Sayo, K.; Mihara, H.; Aoki, S.; Kojima, N. Induction of hepatic tissues in multicellular spheroids composed of murine fetal hepatic cells and embedded hydrogel beads. Regen. Ther. 2016, 3, 7–10. [Google Scholar] [CrossRef][Green Version]
- Kojima, N.; Takeuchi, S.; Sakai, Y. Engineering of pseudoislets: Effect on insulin secretion activity by cell number, cell population, and microchannel networks. Transplant. Proc. 2014, 45, 1161–1165. [Google Scholar] [CrossRef]
- Kojima, N.; Takeuchi, S.; Sakai, Y. Rapid aggregation of heterogeneous cells and multiple-sized microspheres in methylcellulose medium. Biomaterials 2012, 33, 4508–4514. [Google Scholar] [CrossRef] [PubMed]
- Tao, F.; Mihara, H.; Kojima, N. Generation of Hepatic Tissue Structures Using Multicellular Spheroid Culture. In Hepatic Stem Cells; Springer: Berlin/Heidelberg, Germany, 2019; pp. 157–165. [Google Scholar]
- Merkel, T.; Bondar, V.; Nagai, K.; Freeman, B.; Pinnau, I. Gas sorption, diffusion, and permeation in poly (dimethylsiloxane). J. Polym. Sci. B 2000, 38, 415–434. [Google Scholar] [CrossRef]
- Nahmias, Y.; Kramvis, Y.; Barbe, L.; Casali, M.; Berthiaume, F.; Yarmush, M.L. A novel formulation of oxygen-carrying matrix enhances liver-specific function of cultured hepatocytes. FASEB J. 2006, 20, 2531–2533. [Google Scholar] [CrossRef]
- Arteel, G.E.; Thurman, R.G.; Yates, J.M.; Raleigh, J.A. Evidence that hypoxia markers detect oxygen gradients in liver: Pimonidazole and retrograde perfusion of rat liver. Br. J. Cancer 1995, 72, 889–895. [Google Scholar] [CrossRef]
- Raleigh, J.A.; Calkins-Adams, D.P.; Rinker, L.H.; Ballenger, C.A.; Weissler, M.C.; Fowler, W.C., Jr.; Novotny, D.B.; Varia, M.A. Hypoxia and vascular endothelial growth factor expression in human squamous cell carcinomas using pimonidazole as a hypoxia marker. Cancer Res. 1998, 58, 3765–3768. [Google Scholar]
- Sobhanifar, S.; Aquino-Parsons, C.; Stanbridge, E.J.; Olive, P. Reduced expression of hypoxia-inducible factor-1alpha in perinecrotic regions of solid tumors. Cancer Res. 2005, 65, 7259–7266. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, J.; Schwab, U.; Lemke, H.; Stein, H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int. J. Cancer 1983, 31, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Biel, N.M.; Siemann, D.W. Targeting the Angiopoietin-2/Tie-2 axis in conjunction with VEGF signal interference. Cancer Lett. 2016, 380, 525–533. [Google Scholar] [CrossRef]
- Nishida-Aoki, N.; Gujral, T.S. Emerging approaches to study cell-cell interactions in tumor microenvironment. Oncotarget 2019, 10, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.; Ebner, R.; Kunz-Schughart, L.A. Experimental anti-tumor therapy in 3-D: Spheroids–old hat or new challenge? Int. J. Radiat. Biol. 2007, 83, 849–871. [Google Scholar] [CrossRef] [PubMed]
- Hamon, M.; Hanada, S.; Fujii, T.; Sakai, Y. Direct oxygen supply with polydimethylsiloxane (PDMS) membranes induces a spontaneous organization of thick heterogeneous liver tissues from rat fetal liver cells in vitro. Cell Transplant. 2012, 21, 401–410. [Google Scholar] [CrossRef]
- Iwahori, T.; Matsuura, T.; Maehashi, H.; Sugo, K.; Saito, M.; Hosokawa, M.; Chiba, K.; Masaki, T.; Aizaki, H.; Ohkawa, K.; et al. CYP3A4 inducible model for in vitro analysis of human drug metabolism using a bioartificial liver. Hepatology 2003, 37, 665–673. [Google Scholar] [CrossRef]
- Bavli, D.; Prill, S.; Ezra, E.; Levy, G.; Cohen, M.; Vinken, M.; Vanfleteren, J.; Jaeger, M.; Nahmias, Y. Real-time monitoring of metabolic function in liver-on-chip microdevices tracks the dynamics of mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2016, 113, E2231–E2240. [Google Scholar] [CrossRef]
- Ortega-Ribera, M.; Fernández-Iglesias, A.; Illa, X.; Moya, A.; Molina, V.; Maeso-Díaz, R.; Fondevila, C.; Peralta, C.; Bosch, J.; Villa, R. Resemblance of the human liver sinusoid in a fluidic device with biomedical and pharmaceutical applications. Biotechnol. Bioeng. 2018, 115, 2585–2594. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihara, H.; Kugawa, M.; Sayo, K.; Tao, F.; Shinohara, M.; Nishikawa, M.; Sakai, Y.; Akama, T.; Kojima, N. Improved Oxygen Supply to Multicellular Spheroids Using A Gas-permeable Plate and Embedded Hydrogel Beads. Cells 2019, 8, 525. https://doi.org/10.3390/cells8060525
Mihara H, Kugawa M, Sayo K, Tao F, Shinohara M, Nishikawa M, Sakai Y, Akama T, Kojima N. Improved Oxygen Supply to Multicellular Spheroids Using A Gas-permeable Plate and Embedded Hydrogel Beads. Cells. 2019; 8(6):525. https://doi.org/10.3390/cells8060525
Chicago/Turabian StyleMihara, Hirotaka, Mai Kugawa, Kanae Sayo, Fumiya Tao, Marie Shinohara, Masaki Nishikawa, Yasuyuki Sakai, Takeshi Akama, and Nobuhiko Kojima. 2019. "Improved Oxygen Supply to Multicellular Spheroids Using A Gas-permeable Plate and Embedded Hydrogel Beads" Cells 8, no. 6: 525. https://doi.org/10.3390/cells8060525
APA StyleMihara, H., Kugawa, M., Sayo, K., Tao, F., Shinohara, M., Nishikawa, M., Sakai, Y., Akama, T., & Kojima, N. (2019). Improved Oxygen Supply to Multicellular Spheroids Using A Gas-permeable Plate and Embedded Hydrogel Beads. Cells, 8(6), 525. https://doi.org/10.3390/cells8060525