ER-Targeted Beclin 1 Supports Autophagosome Biogenesis in the Absence of ULK1 and ULK2 Kinases

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Stable Cell Lines, Drugs and Autophagy Induction
2.2. Plasmid Construction
2.3. Transient and Stable Transfection
2.4. Immunofluorescence and Imaging
2.5. Immunofluorescence Image Quantification
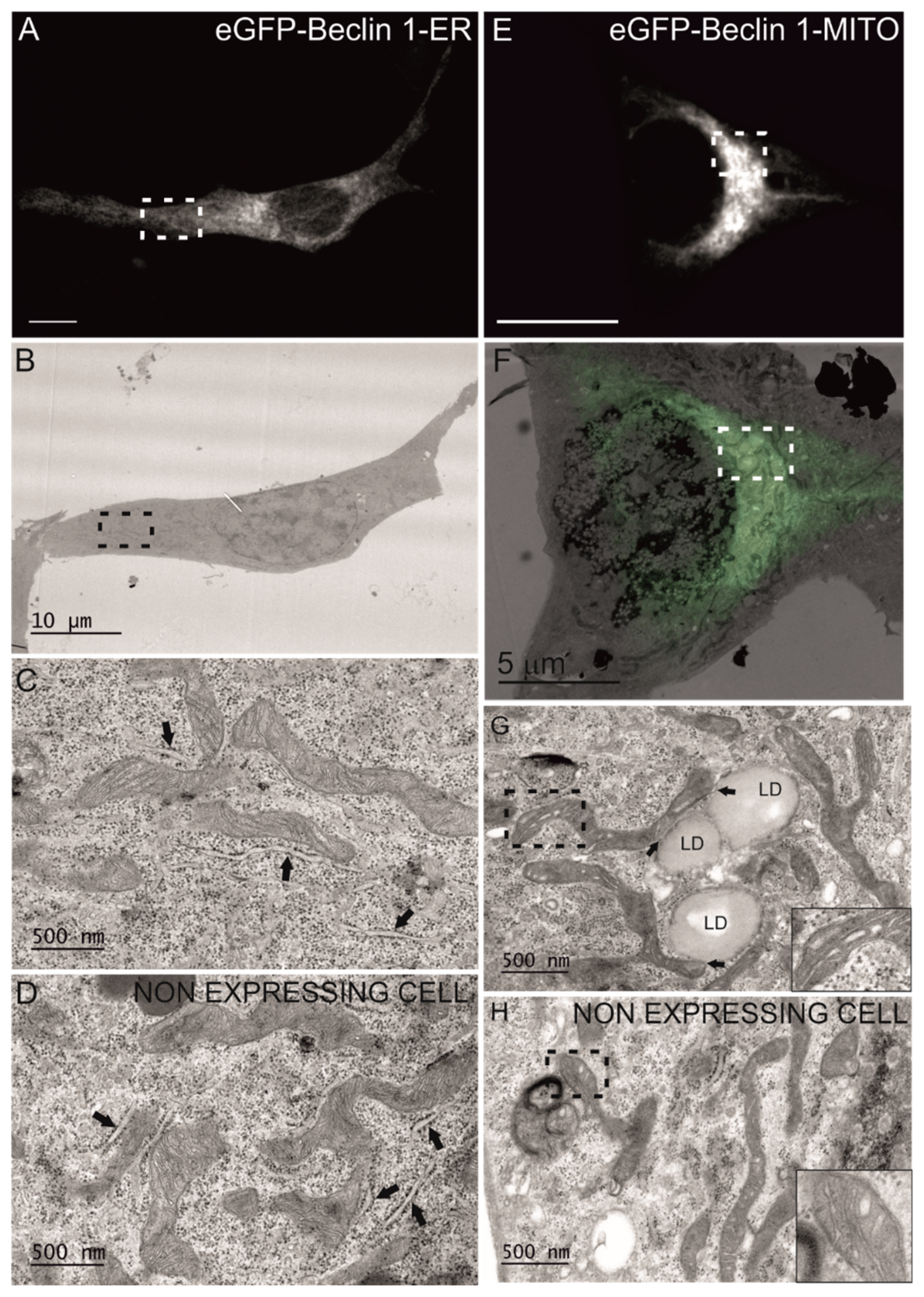
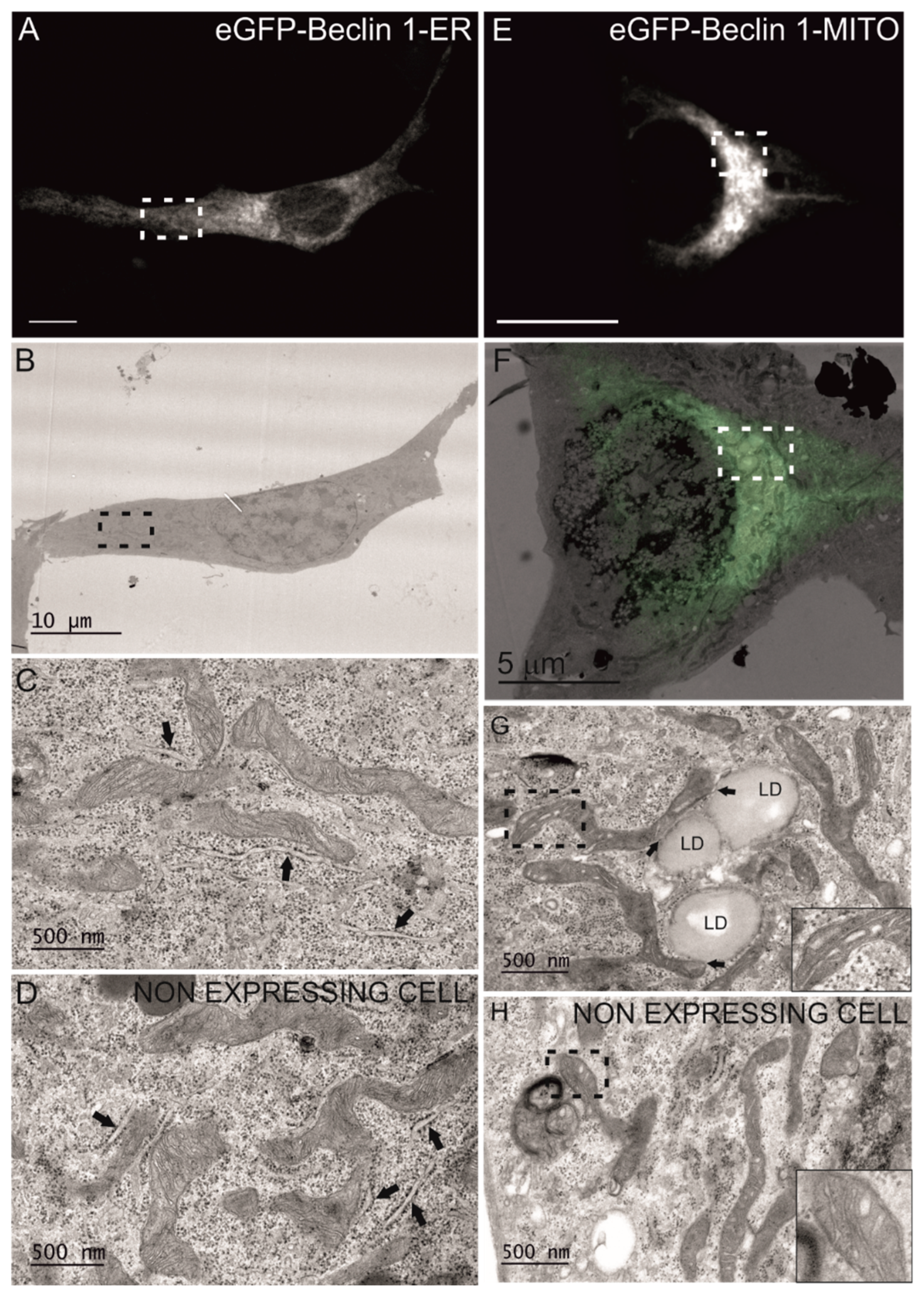
2.6. Correlative Light Electron Microscopy (CLEM)
2.7. Western Blotting
2.8. Affinity Purification Mass Spectrometry (AP–MS)
2.9. Statistical Analysis
3. Results
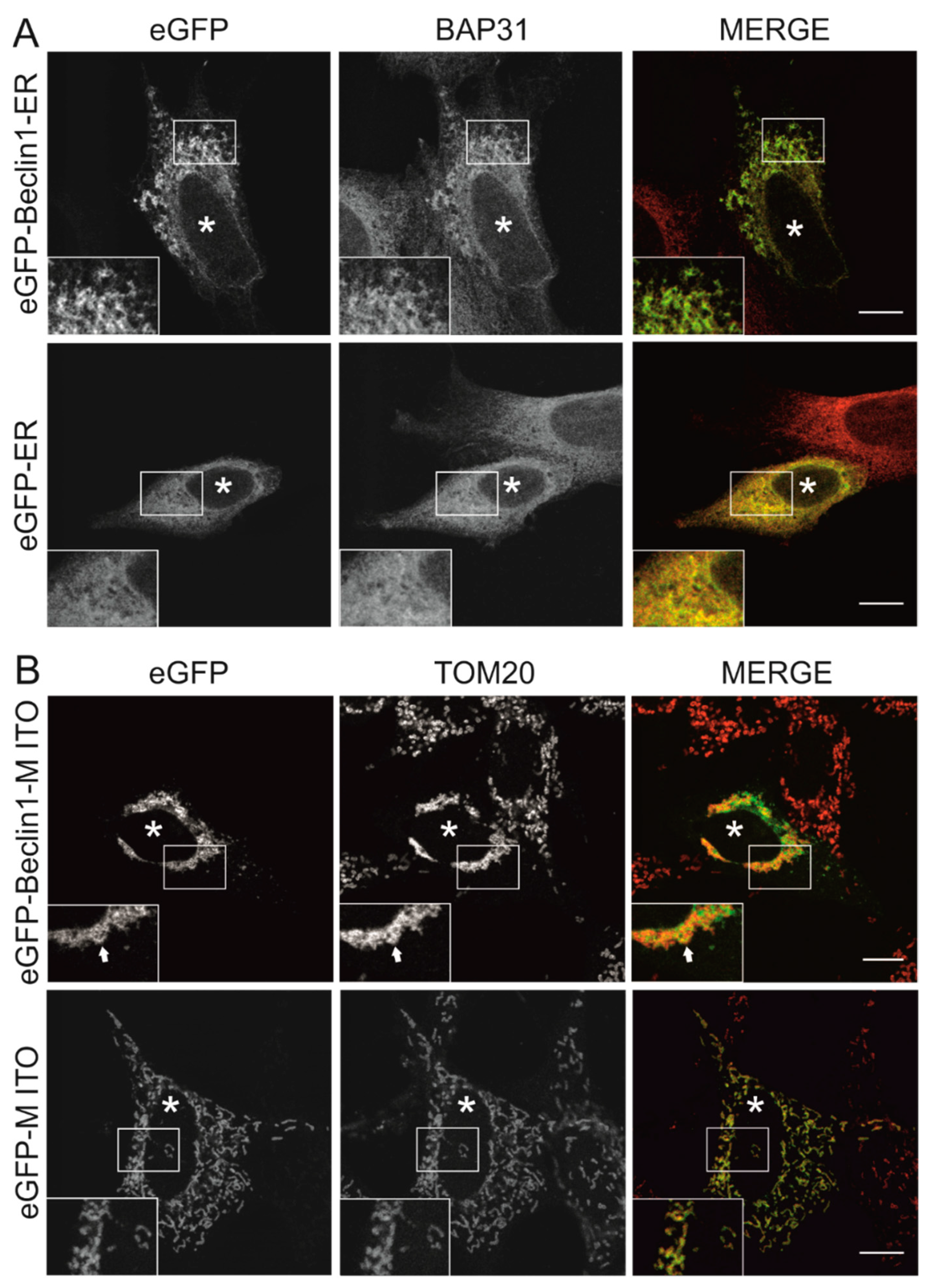
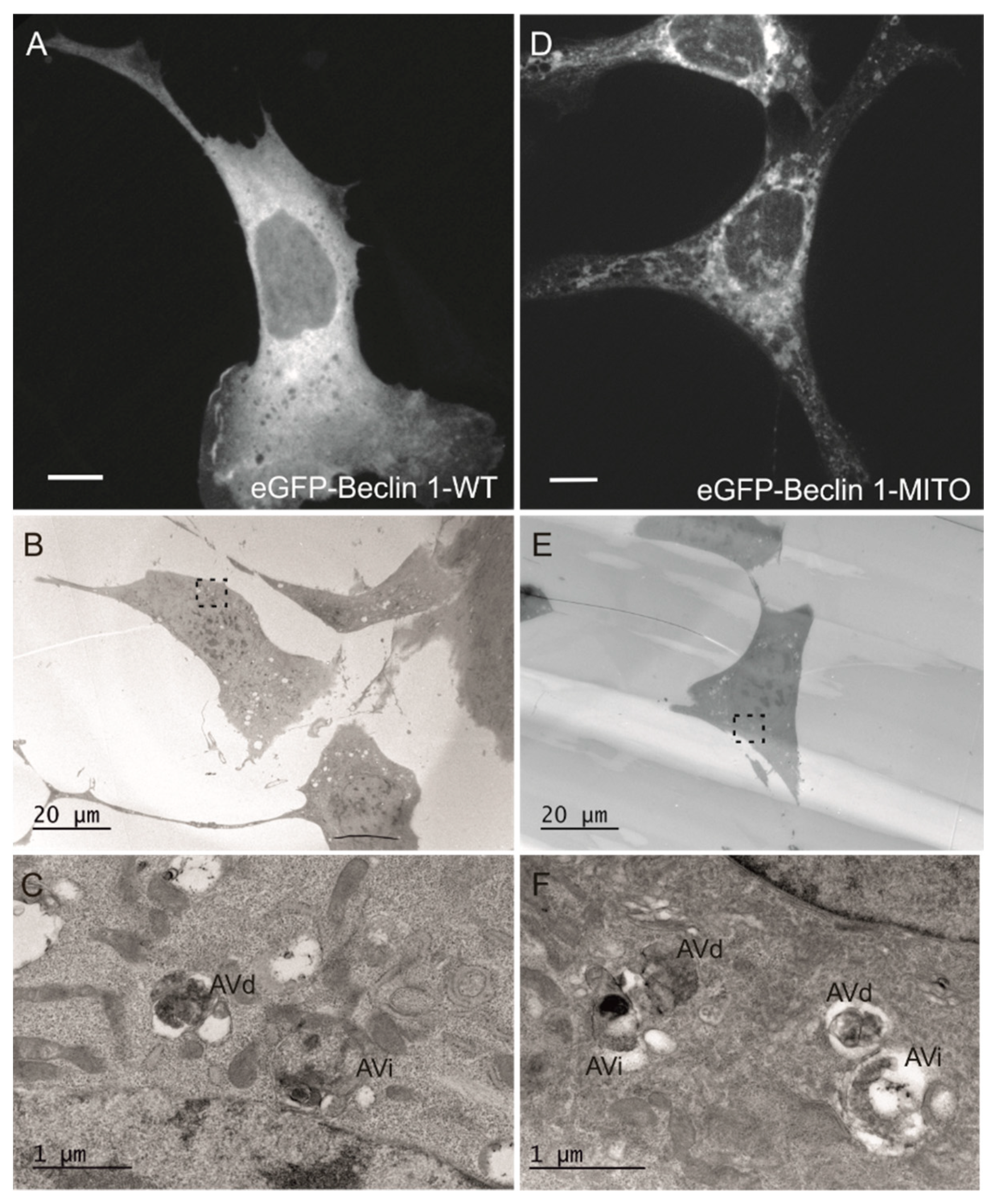
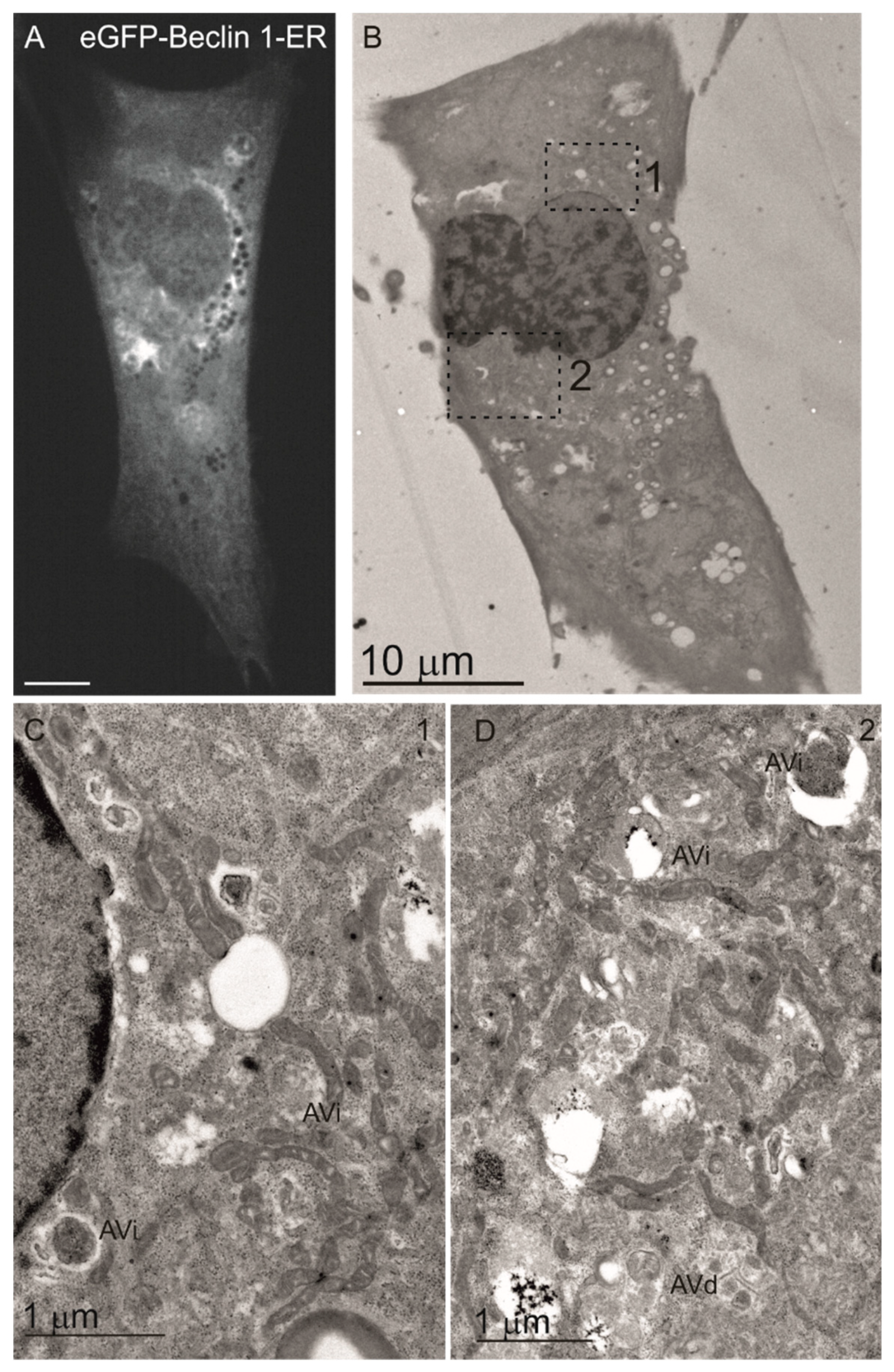
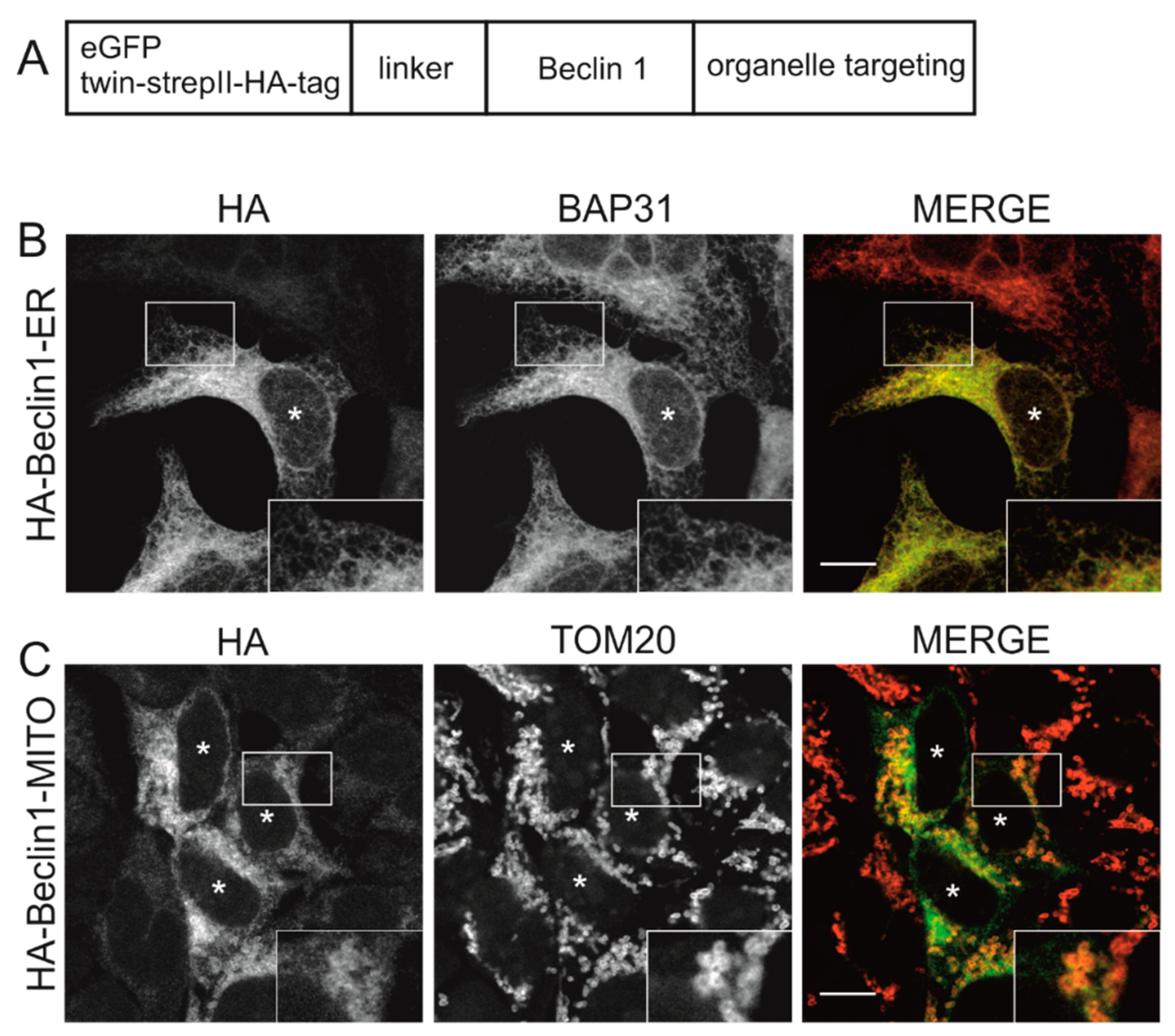
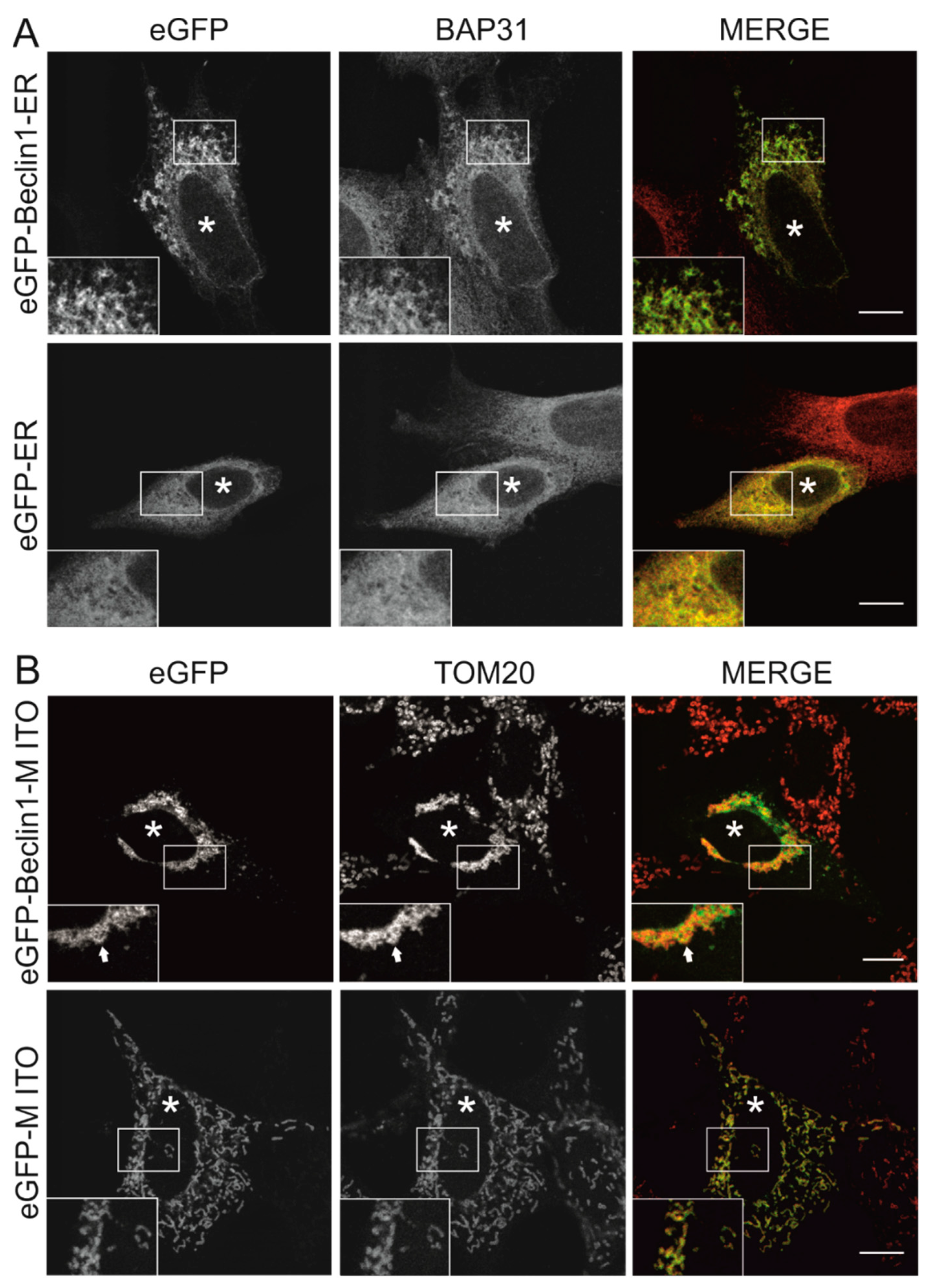
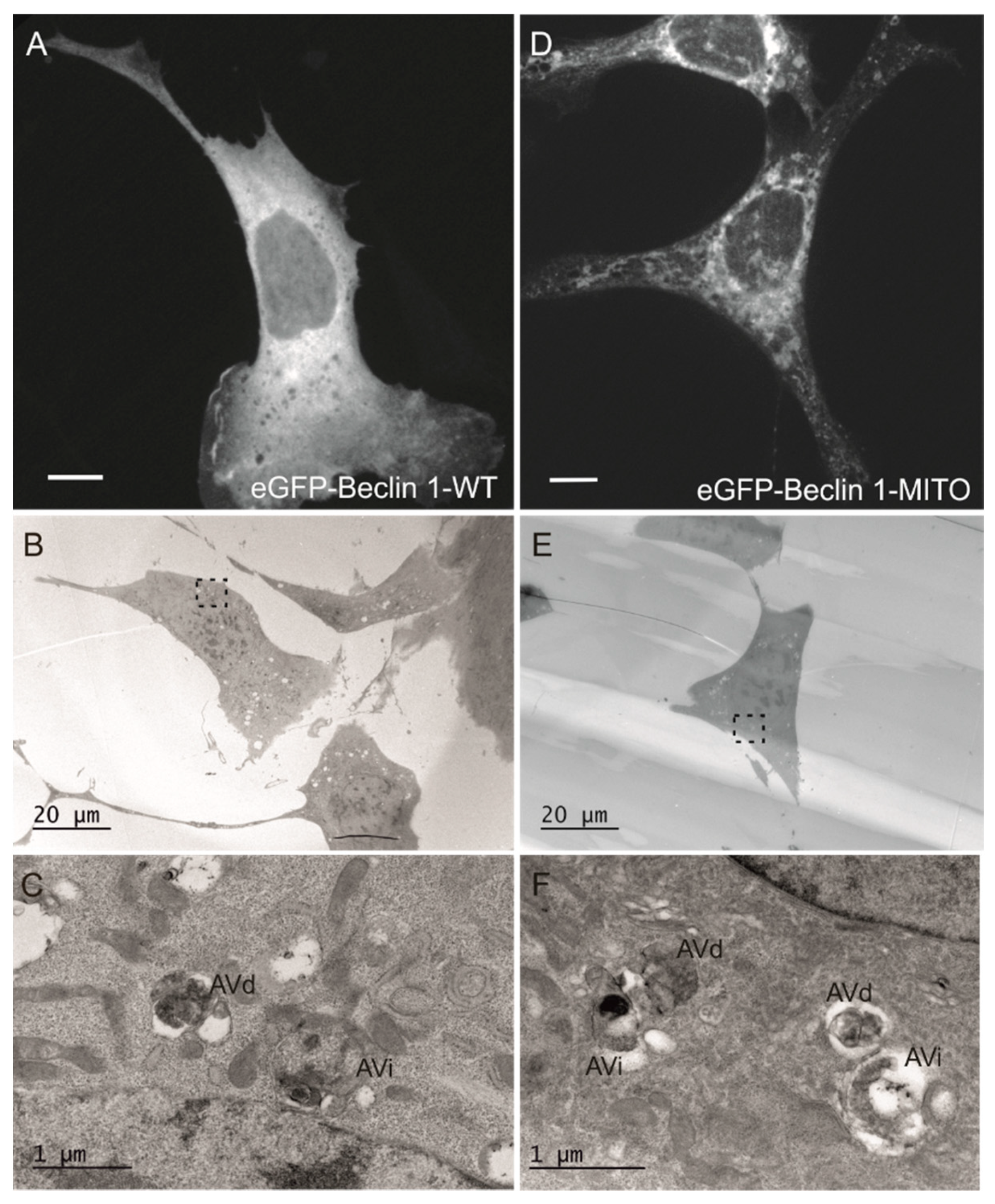
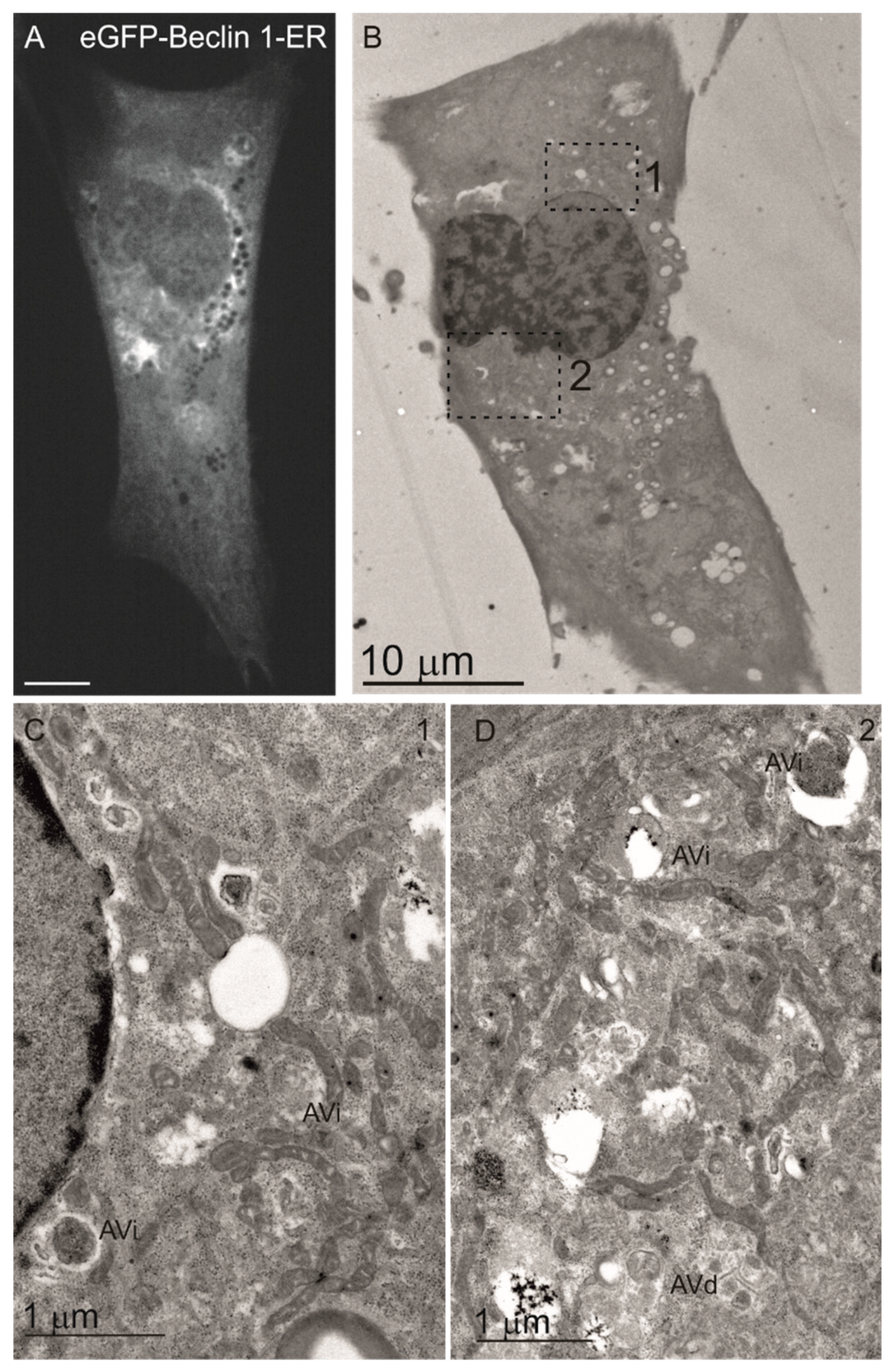
3.1. Beclin 1 Constructs Targeted to the Endoplasmic Reticulum and Mitochondria Localize to Their Expected Subcellular Compartments
3.2. Expression of the Targeted Beclin 1 Constructs Does Not Induce Organelle Stress
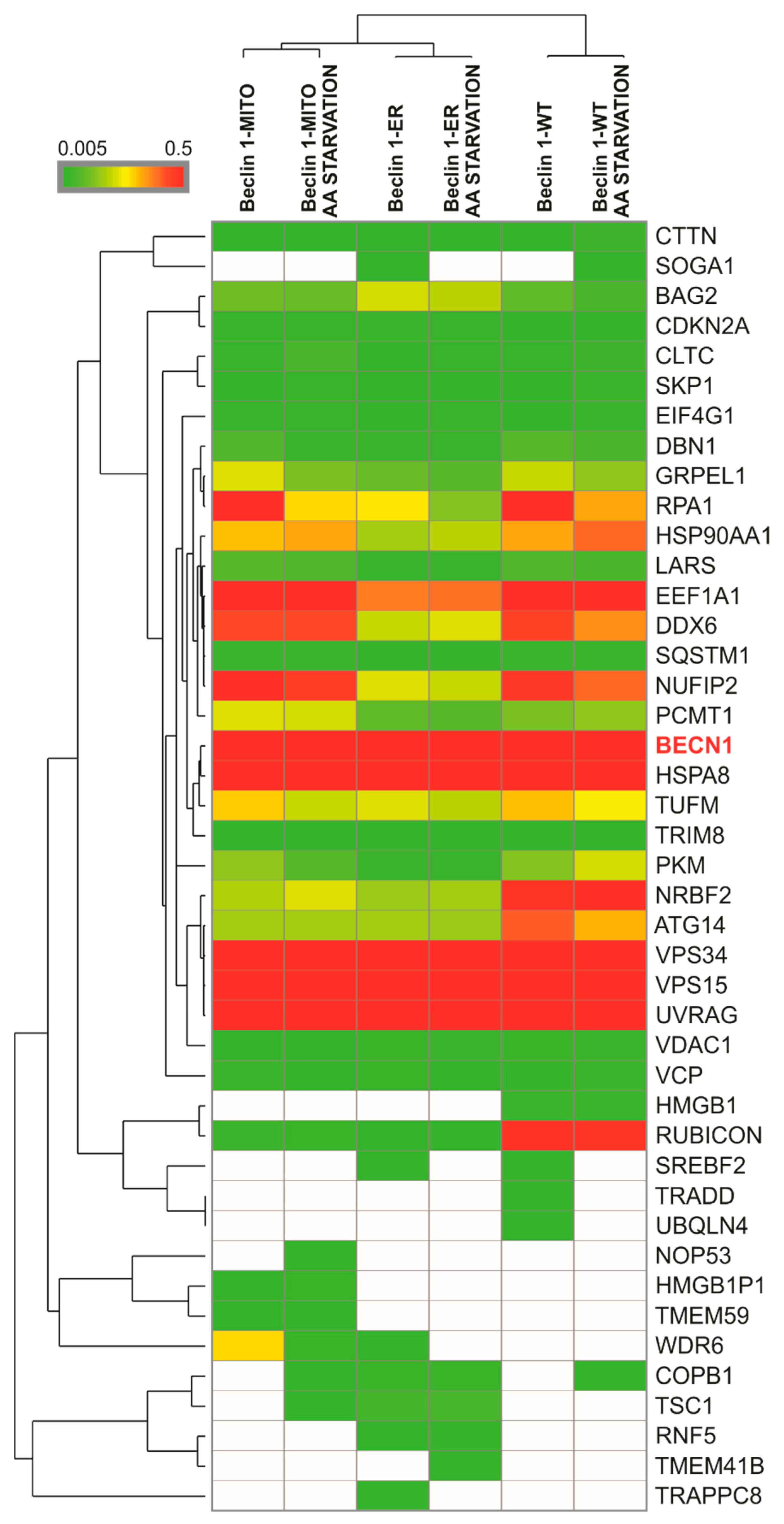
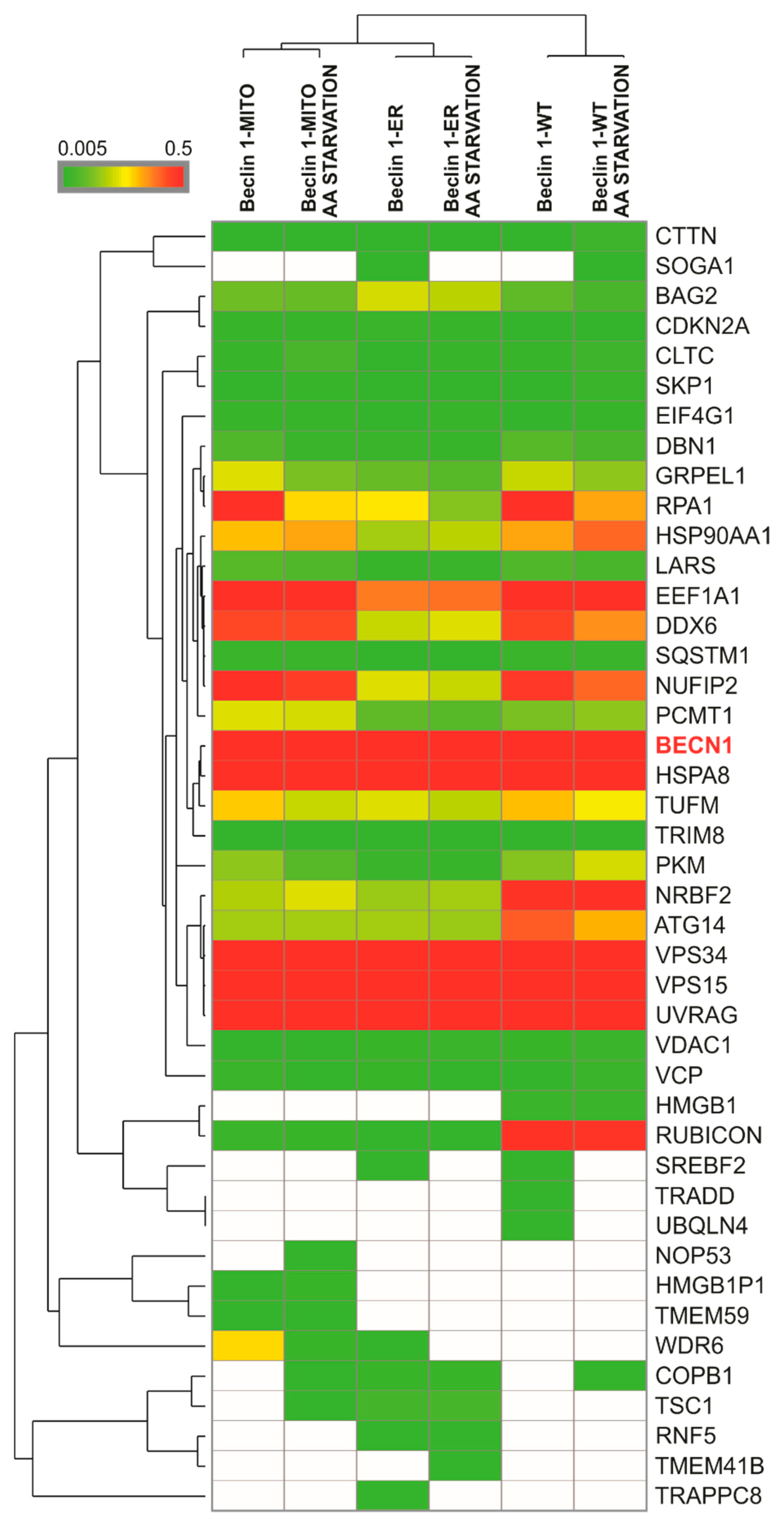
3.3. The Targeted Beclin 1 Constructs Bind to Known Autophagy-Related Interactors of Beclin 1
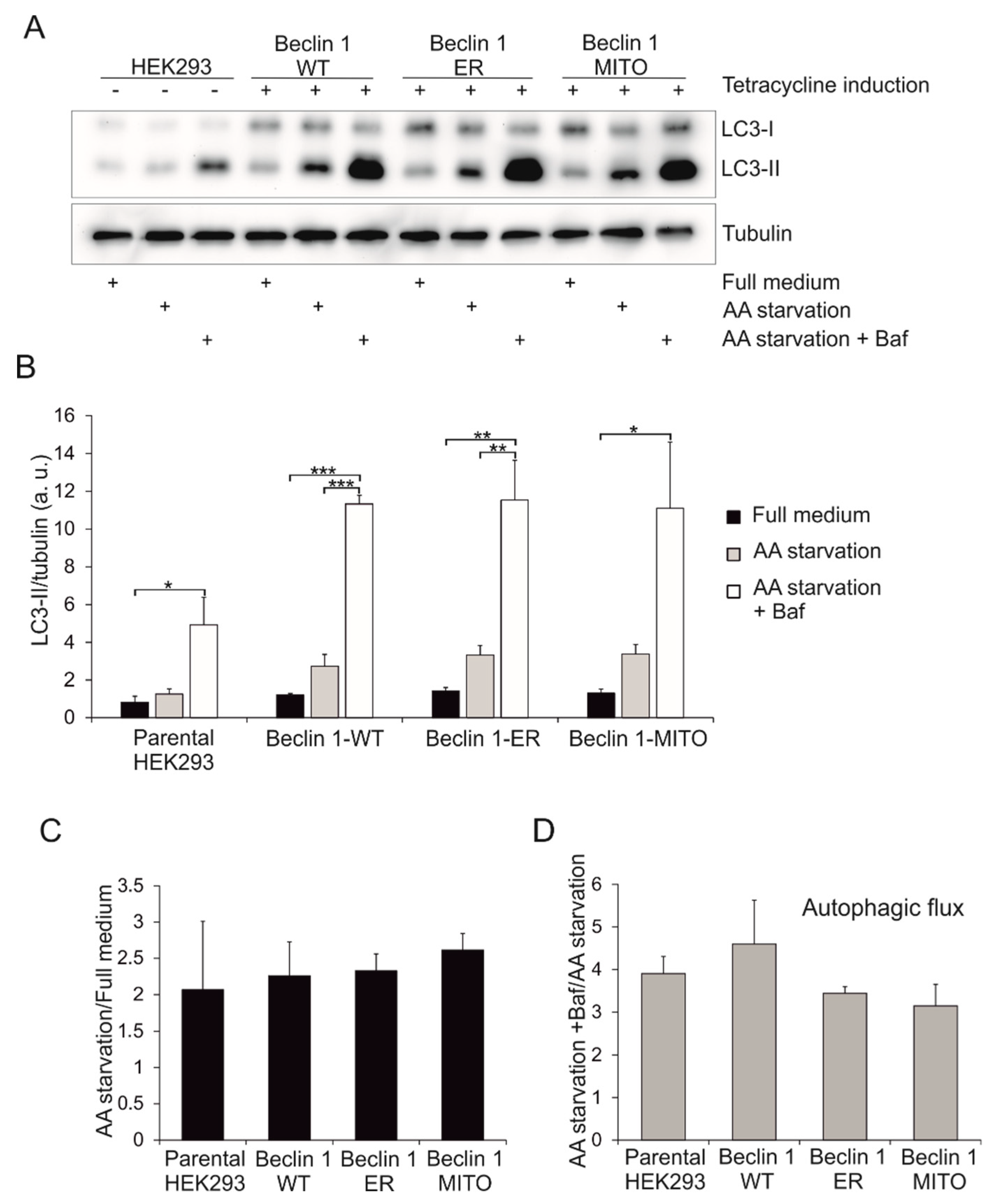
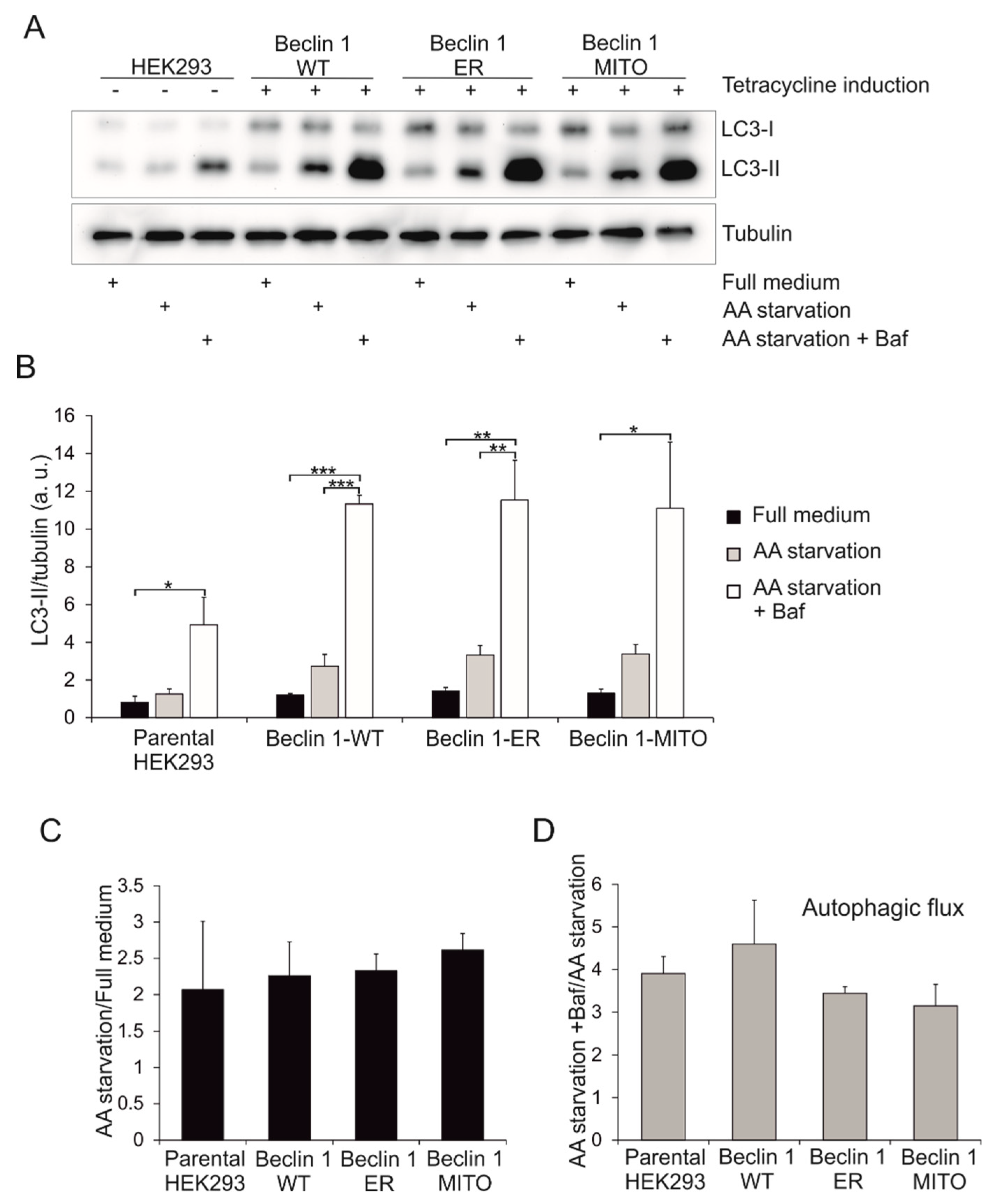
3.4. Effect of Beclin 1 Targeting on Autophagy in Human Embryonic Kidney (HEK293) Cells
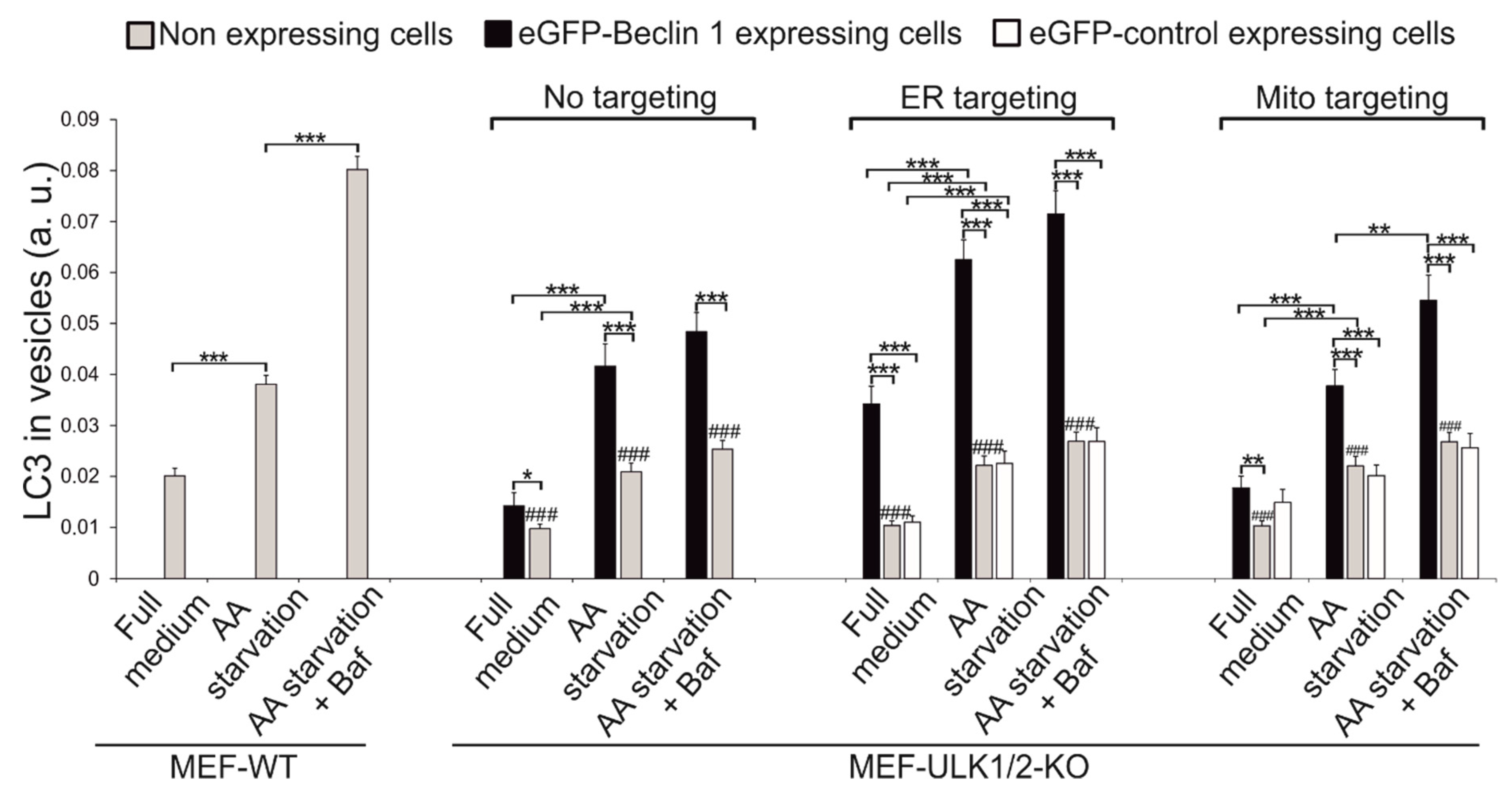
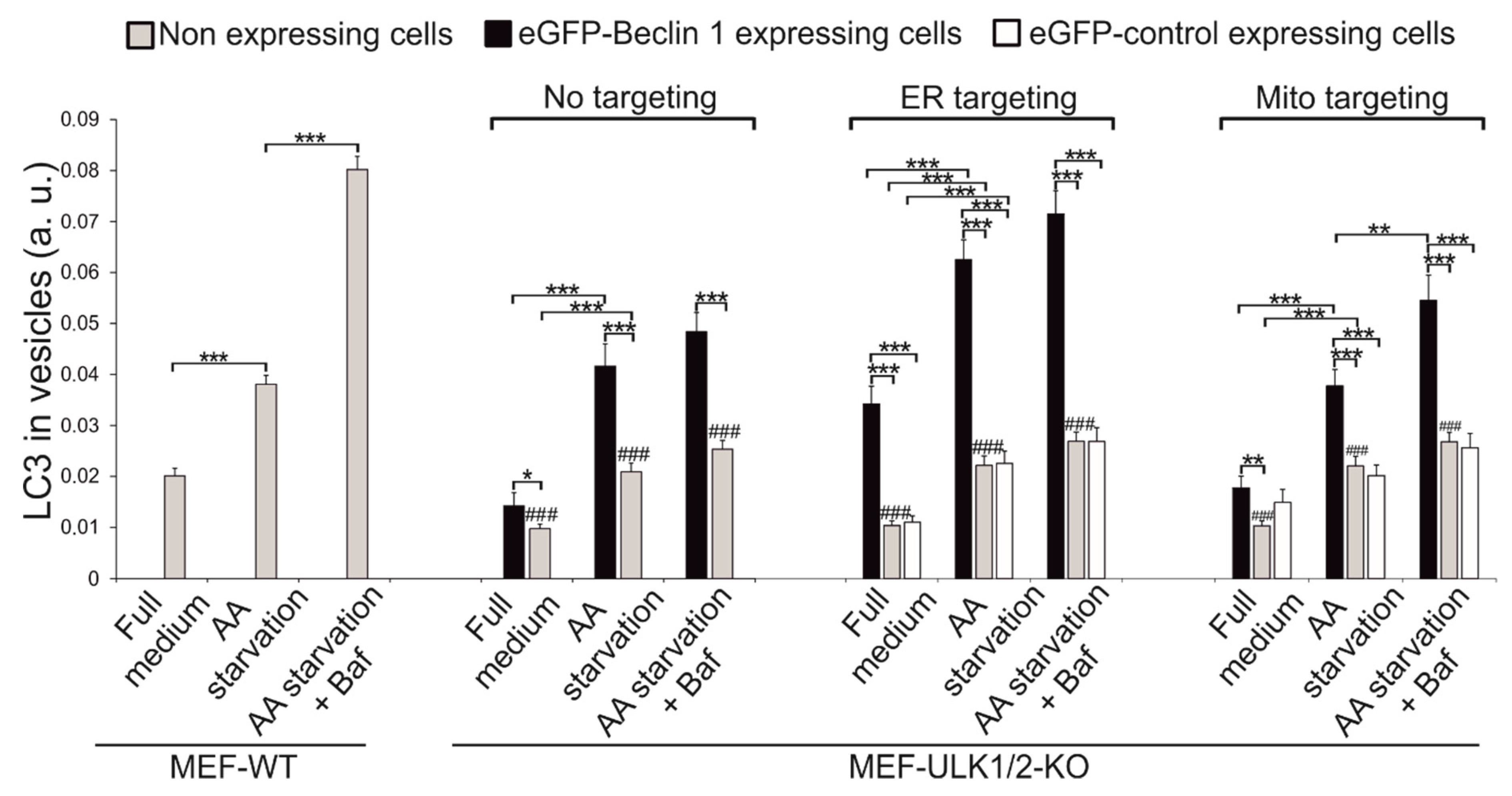
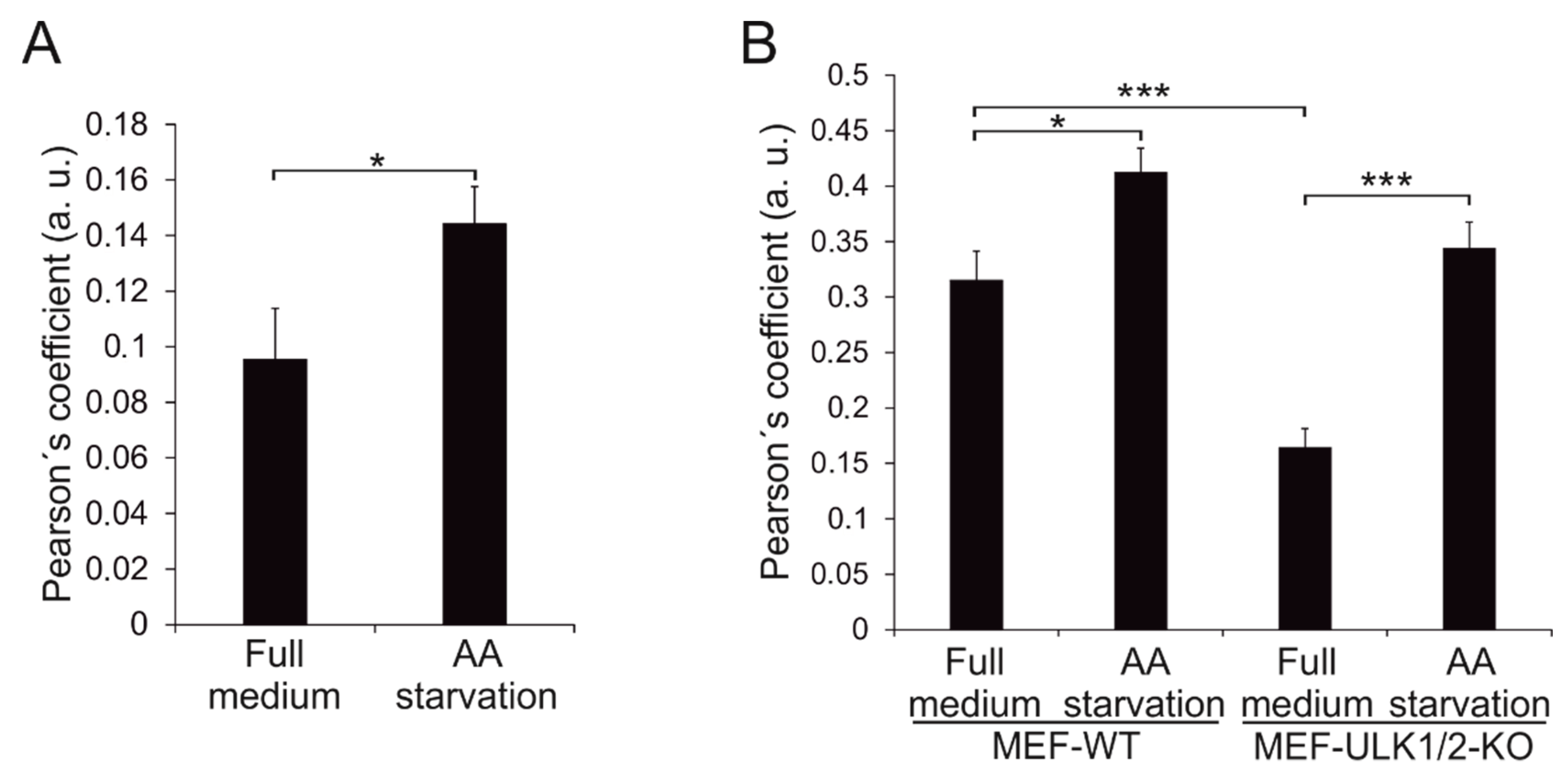
3.5. Effects of Beclin 1 Targeting on Autophagy in Wild-Type and ULK1/ULK2 Double Knockout Mouse Embryonic Fibroblasts (MEFs)
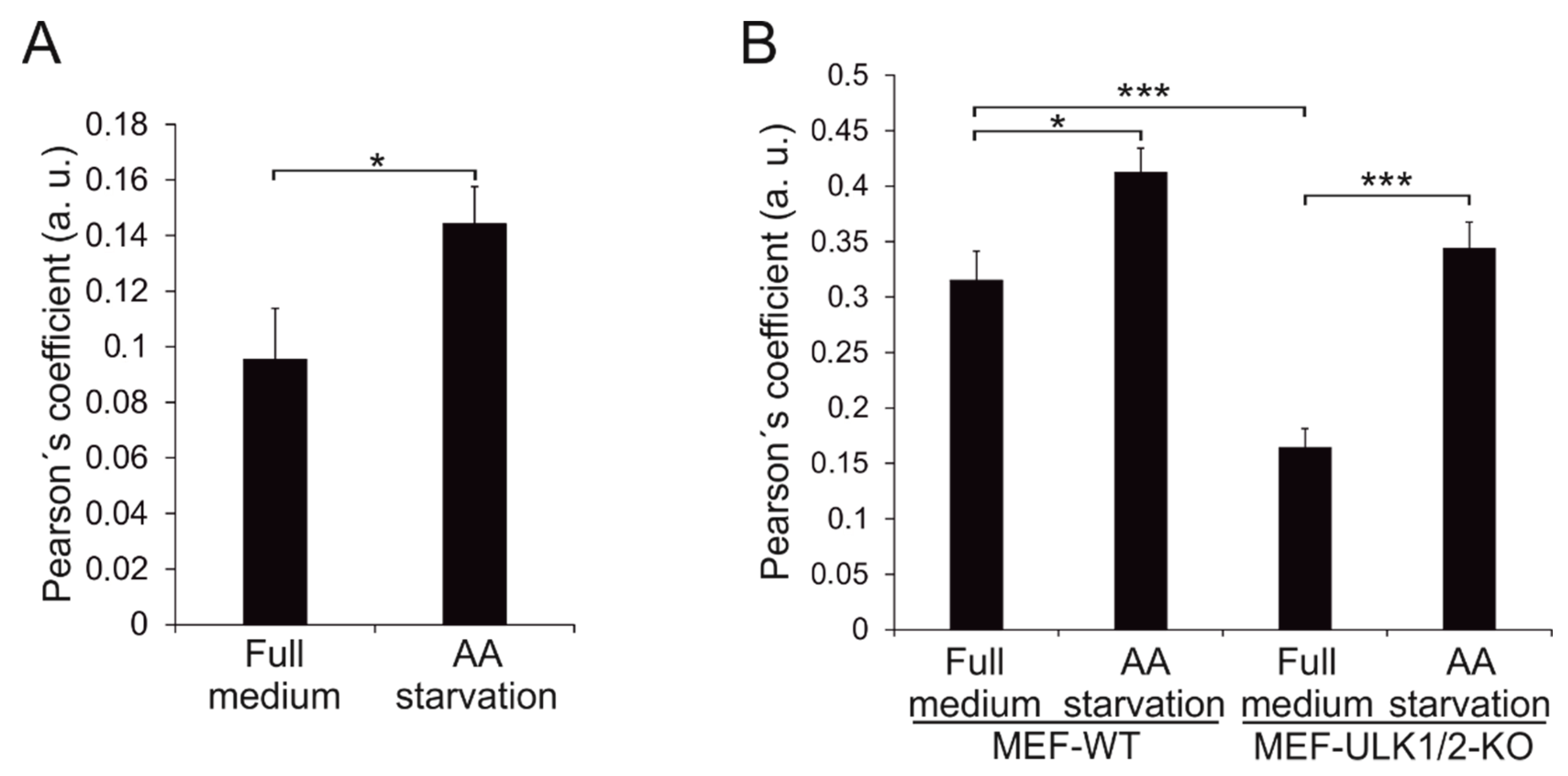
3.6. Wild-Type Beclin 1 Is Enriched in the Endoplasmic Reticulum (ER) during Amino Acid Starvation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- van Beek, N.; Klionsky, D.J.; Reggiori, F. Genetic aberrations in macroautophagy genes leading to diseases. Biochim. Biophys. Acta 2018, 1865, 803–816. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Boil. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Boil. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Levine, B.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Yu, J.; Brown, K.; Levine, B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res. 2001, 61, 3443–3449. [Google Scholar]
- Kihara, A.; Kabeya, Y.; Ohsumi, Y.; Yoshimori, T. Beclin–phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001, 2, 330–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskaran, S.; Carlson, L.-A.; Stjepanovic, G.; Young, L.N.; Kim, D.J.; Grob, P.; E Stanley, R.; Nogales, E.; Hurley, J.H.; Kühlbrandt, W. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. eLife 2014, 3, e05115. [Google Scholar] [CrossRef] [PubMed]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.C.; Meijer, A.J.; Codogno, P. Distinct Classes of Phosphatidylinositol 3’-Kinases Are Involved in Signaling Pathways That Control Macroautophagy in HT-29 Cells. J. Boil. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef]
- Stjepanovic, G.; Baskaran, S.; Lin, M.G.; Hurley, J.H. Vps34 kinase domain dynamics regulate the autophagic PI 3-kinase complex. Mol. Cell 2017, 67, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Fan, W.; Chen, K.; Ding, X.; Chen, S.; Zhong, Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 19211–19216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating Vps34 lipid kinase. Nat. Cell Boil. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.-C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.-M.; Starker, C.; Nho, R.S.; et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlpine, F.; Williamson, L.E.; Tooze, S.A.; Chan, E.Y. Regulation of nutrient-sensitive autophagy by uncoordinated 51-like kinases 1 and 2. Autophagy 2013, 9, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Mari, M.; Tooze, S.A.; Reggiori, F. The puzzling origin of the autophagosomal membrane. F1000 Boil. Rep. 2011, 3, 25. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Boil. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of Autophagosome Biogenesis. Curr. Boil. 2012, 22, R29–R34. [Google Scholar] [CrossRef] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Boil. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Boil. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanasios, E.; Stapleton, E.; Manifava, M.; Kaizuka, T.; Mizushima, N.; Walker, S.A.; Ktistakis, N.T. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J. Cell Sci. 2013, 126, 5224–5238. [Google Scholar] [CrossRef] [Green Version]
- Koyama-Honda, I.; Itakura, E.; Fujiwara, T.K.; Mizushima, N. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 2013, 9, 1491–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014, 127, 4089–4102. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Boil. 2010, 190, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Choi, W.; Hu, W.; Mi, N.; Guo, Q.; Ma, M.; Liu, M.; Tian, Y.; Lu, P.; Wang, F.-L.; et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012, 22, 473–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nat. Cell Boil. 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Mitoma, J.; Ito, A. The carboxy-terminal 10 amino acid residues of cytochrome b5 are necessary for its targeting to the endoplasmic reticulum. EMBO J. 1992, 11, 4197–4203. [Google Scholar] [CrossRef]
- Pistor, S.; Chakraborty, T.; Niebuhr, K.; Domann, E.; Wehland, J. The ActA protein of Listeria monocytogenes acts as a nucleator inducing reorganization of the actin cytoskeleton. EMBO J. 1994, 13, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Cowie, A.; Wasfy, G.W.; Penn, L.Z.; Leber, B.; Andrews, D.W. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 1996, 15, 4130–4141. [Google Scholar] [CrossRef]
- Varjosalo, M.; Sacco, R.; Stukalov, A.; Van Drogen, A.; Planyavsky, M.; Hauri, S.; Aebersold, R.; Bennett, K.L.; Colinge, J.; Gstaiger, M.; et al. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat. Methods 2013, 10, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Määttä, J.; Hallikas, O.; Welti, S.; Hilden, P.; Schröder, J.; Kuismanen, E. Limited caspase cleavage of human BAP31. FEBS Lett. 2000, 484, 202–206. [Google Scholar] [CrossRef] [Green Version]
- E Carpenter, A.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; A Guertin, D.; Chang, J.H.; A Lindquist, R.; Moffat, J.; et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Boil. 2006, 7, R100. [Google Scholar] [CrossRef]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.-A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpenter, A.E. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef] [Green Version]
- Sage, D.; Neumann, F.; Hediger, F.; Gasser, S.; Unser, M. Automatic tracking of individual fluorescence particles: application to the study of chromosome dynamics. IEEE Trans. Image Process. 2005, 14, 1372–1383. [Google Scholar] [CrossRef] [Green Version]
- Yadav, L.; Tamene, F.; Göös, H.; Van Drogen, A.; Katainen, R.; Aebersold, R.; Gstaiger, M.; Varjosalo, M. Systematic Analysis of Human Protein Phosphatase Interactions and Dynamics. Cell Syst. 2017, 4, 430–444.e5. [Google Scholar] [CrossRef]
- Teo, G.; Liu, G.; Zhang, J.; Nesvizhskii, A.I.; Gingras, A.C.; Choi, H. SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J. Proteom. 2014, 100, 37–43, Epub 2014/02/12. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219, Epub 2012/01/03. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Christensen, M.E.; Waterhouse, N.J. Measuring Mitochondrial Transmembrane Potential by TMRE Staining. Cold Spring Harbor Protoc. 2016, 2016. Epub 2016/12/10. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease - the long and short of it. J. Cell Sci. 2016, 129, 2297–2306, Epub 2016/05/18. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Boil. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, Y.; Saab, W.F.A.; Yang, F.; Pessin, J.E.; Backer, J.M. NRBF2 regulates macroautophagy as a component of Vps34 Complex I. Biochem. J. 2014, 461, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Lee, J.-S.; Inn, K.-S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Boil. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, M.; Joachim, J.; Tooze, S.A. Autophagosome formation—The role of ULK1 and Beclin1–PI3KC3 complexes in setting the stage. Semin. Cancer Boil. 2013, 23, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Jhonson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Intracellular Compartments and Protein Sorting. In Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2015; pp. 641–694. ISBN1 978-0-8153-4464-3. ISBN2 978-0-81534432-2. [Google Scholar]
- Sahani, M.H.; Itakura, E.; Mizushima, N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 2014, 10, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.-L. Fine Structure of the Autophagosome. Methods Mol. Biol. 2008, 445, 11–28. [Google Scholar]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.-H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Boil. 2006, 8, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-L. Ambra1 in autophagy and apoptosis: Implications for cell survival and chemotherapy resistance. Oncol. Lett. 2016, 12, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alers, S.; Löffler, A.S.; Paasch, F.; Dieterle, A.M.; Keppeler, H.; Lauber, K.; Campbell, D.G.; Fehrenbacher, B.; Schaller, M.; Wesselborg, S.; et al. Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy 2011, 7, 1424–1433. [Google Scholar] [CrossRef] [Green Version]
- Manzoni, C.; Mamais, A.; Roosen, D.A.; Dihanich, S.; Soutar, M.P.M.; Plun-Favreau, H.; Bandopadhyay, R.; Hardy, J.; Tooze, S.A.; Cookson, M.R.; et al. mTOR independent regulation of macroautophagy by Leucine Rich Repeat Kinase 2 via Beclin-1. Sci. Rep. 2016, 6, 35106. [Google Scholar] [CrossRef] [Green Version]
- Cheong, H.; Lindsten, T.; Wu, J.; Lu, C.; Thompson, C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA 2011, 108, 11121–11126. [Google Scholar] [CrossRef] [Green Version]
- Gammoh, N.; Florey, O.; Overholtzer, M.; Jiang, X. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat. Struct. Mol. Biol. 2013, 20, 144. [Google Scholar] [CrossRef]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A Non-canonical MEK/ERK Signaling Pathway Regulates Autophagy via Regulating Beclin 1*. J. Boil. Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molejon, M.I.; Ropolo, A.; Re, A.L.; Boggio, V.; Vaccaro, M.I. The VMP1-Beclin 1 interaction regulates autophagy induction. Sci. Rep. 2013, 3, 1055. [Google Scholar] [CrossRef]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation—At the Center of Autophagy Regulation. Front. Cell Dev. Boil. 2018, 6, 137. [Google Scholar] [CrossRef]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Boil. 2010, 12, 831–835. [Google Scholar] [CrossRef]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anwar, T.; Liu, X.; Suntio, T.; Marjamäki, A.; Biazik, J.; Chan, E.Y.W.; Varjosalo, M.; Eskelinen, E.-L. ER-Targeted Beclin 1 Supports Autophagosome Biogenesis in the Absence of ULK1 and ULK2 Kinases. Cells 2019, 8, 475. https://doi.org/10.3390/cells8050475
Anwar T, Liu X, Suntio T, Marjamäki A, Biazik J, Chan EYW, Varjosalo M, Eskelinen E-L. ER-Targeted Beclin 1 Supports Autophagosome Biogenesis in the Absence of ULK1 and ULK2 Kinases. Cells. 2019; 8(5):475. https://doi.org/10.3390/cells8050475
Chicago/Turabian StyleAnwar, Tahira, Xiaonan Liu, Taina Suntio, Annika Marjamäki, Joanna Biazik, Edmond Y. W. Chan, Markku Varjosalo, and Eeva-Liisa Eskelinen. 2019. "ER-Targeted Beclin 1 Supports Autophagosome Biogenesis in the Absence of ULK1 and ULK2 Kinases" Cells 8, no. 5: 475. https://doi.org/10.3390/cells8050475
APA StyleAnwar, T., Liu, X., Suntio, T., Marjamäki, A., Biazik, J., Chan, E. Y. W., Varjosalo, M., & Eskelinen, E.-L. (2019). ER-Targeted Beclin 1 Supports Autophagosome Biogenesis in the Absence of ULK1 and ULK2 Kinases. Cells, 8(5), 475. https://doi.org/10.3390/cells8050475