The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System


Abstract
1. Introduction
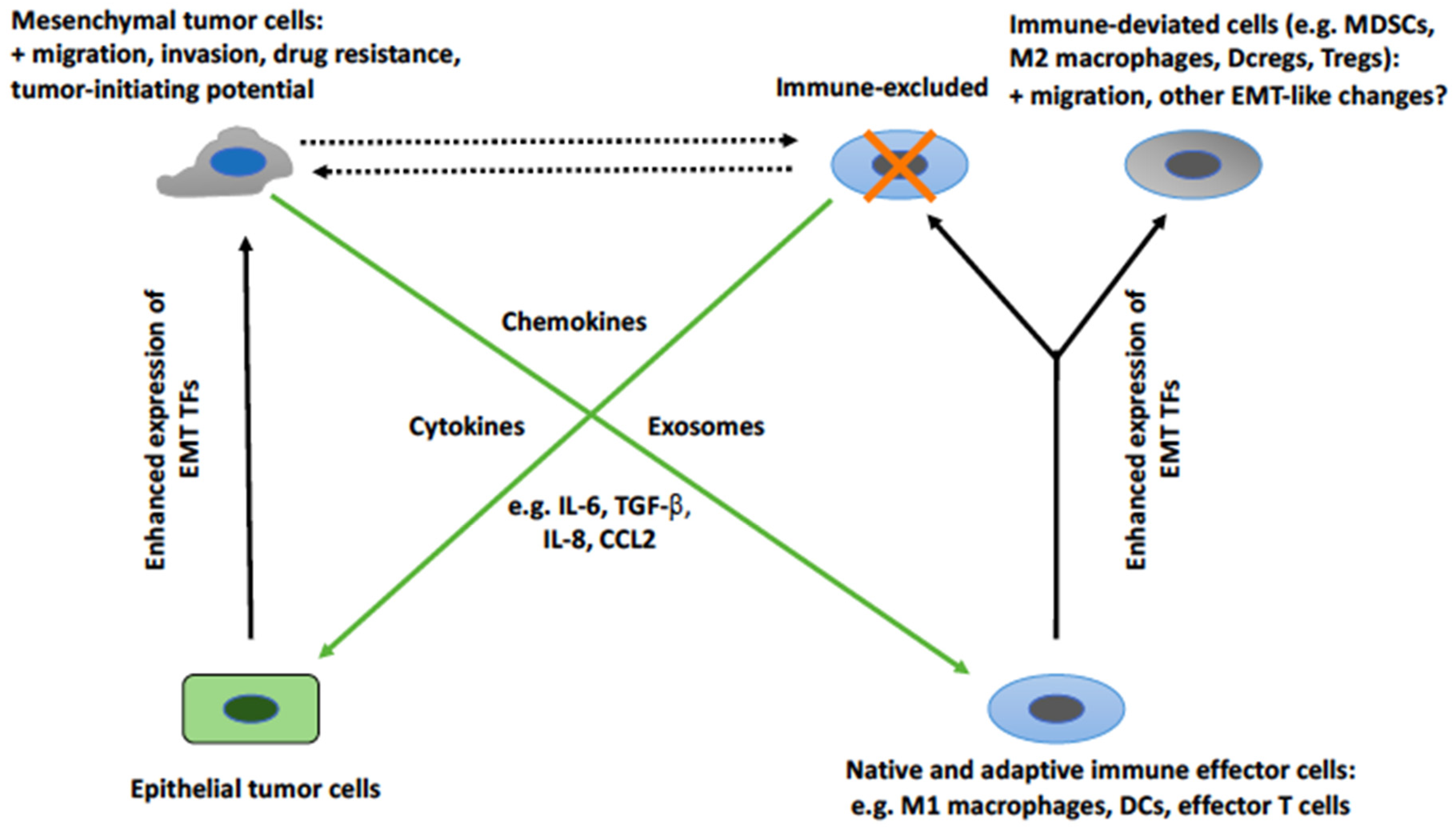
2. The Cross-Talk between EMT Tumor Cells and Cells of the Immune System
2.1. Association of EMT and an Immunosuppressive TME in Patients
2.2. EMT-Associated Changes in the Immunological Profile of Tumor Cells
2.3. EMT-Induced Effects on Cells of the Immune System
2.4. The Mediators of EMT-Induced Effects on Immune Cells
2.5. Induction of Tumor Cell EMT by Cells of the Immune System
2.6. The Mediators of the Induction of Tumor Cell EMT by Cells of the Immune System
3. Activation of Similar Programs in Response to Similar Stimuli in Tumor Cells and Immune Cells
4. Some Therapeutic Implications
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial–mesenchymal transition: A new target in anticancer drug discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Bellone, M.; Caserta, C.A.; Corti, A. Pushing tumor cells towards a malignant phenotype. Stimuli from the microenvironment, intercellular communications and alternative roads. Int. J. Cancer 2014, 135, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.; Wilson, M.B.; Crawford, Y.G.; Reynolds, P.A.; Sigaroudinia, M.; Tlsty, T.D. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 14867–14872. [Google Scholar] [CrossRef]
- Chung, S.S.; Giehl, N.; Wu, Y.Y.; Vadgama, J.V. STAT3 activation in HER2-overexpressing breast cancer promotes epithelial−mesenchymal transition and cancer stem cell traits. Int. J. Oncol. 2014, 44, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Adocque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Chui, M.H. Insights into cancer metastasis from a clinicopathologic perspective: Epithelial-Mesenchymal Transition is not a necessary step. Int. J. Cancer 2013, 132, 1487–1495. [Google Scholar] [CrossRef]
- Murakami, R.; Matsumura, N.; Mandai, M.; Yoshihara, K.; Tanabe, H.; Nakai, H.; Yamanoi, K.; Abiko, K.; Yoshioka, Y.; Hamanishi, J.; et al. Establishment of a novel histopathological classification of high-grade serous ovarian carcinoma correlated with prognostically distinct gene expression subtypes. Am. J. Pathol. 2016, 186, 1103–1113. [Google Scholar] [CrossRef]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, J.; Li, J.H.; Liu, X.; Wang, J.Z.; Qu, H.Y.; Wang, J.S.; Duan, X.Y. High tumor-associated macrophages infiltration is associated with poor prognosis and may contribute to the phenomenon of epithelial-mesenchymal transition in gastric cancer. Onco. Targets Ther. 2016, 9, 3975–3983. [Google Scholar] [CrossRef]
- Lou, Y.; Diao, L.; Cuentas, E.R.P.; Denning, W.L.; Chen, L.; Fan, Y.H.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.A.; Behrens, C.; et al. Epithelial–mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A patient-derived, pan-cancer EMT signature identifies global molecular alterations and immune target enrichment following epithelial-to-mesenchymal transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Koh, J.; Kim, M.Y.; Kwon, D.; Go, H.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H. PD-L1 expression is associated with epithelial-to-mesenchymal transition in adenocarcinoma of the lung. Hum. Pathol. 2016, 58, 7–14. [Google Scholar] [CrossRef]
- Alsuliman, A.; Colak, D.; Al-Harazi, O.; Fitwi, H.; Tulbah, A.; Al-Tweigeri, T.; Al-Alwan, M.; Ghebeh, H. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: Significance in claudin-low breast cancer cells. Mol. Cancer 2015, 14, 149. [Google Scholar] [CrossRef]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wölfel, T.; Hölzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–418. [Google Scholar] [CrossRef]
- Woods, K.; Pasam, A.; Jayachandran, A.; Andrews, M.C.; Cebon, J. Effects of epithelial to mesenchymal transition on T cell targeting of melanoma cells. Front. Oncol. 2014, 4, 367. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.L.; Lu, H.; Stone, B.; Reiman, J.M.; Behrens, M.D.; Prosperi, C.M.; Gad, E.A.; Smorlesi, A.; Disis, M.L. Immunoediting of cancers may lead to epithelial to mesenchymal transition. J. Immunol. 2006, 177, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl. Acad. Sci. USA 2016, 113, E1555–E1564. [Google Scholar] [CrossRef] [PubMed]
- López-Soto, A.; Huergo-Zapico, L.; Galvan, J.A.; Rodrigo, L.; de Herreros, A.G.; Astudillo, A.; Gonzalez, S. Epithelial-mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. J. Immunol. 2013, 190, 4408–4419. [Google Scholar] [CrossRef]
- Chen, X.H.; Liu, Z.C.; Zhang, G.; Wei, W.; Wang, X.X.; Wang, H.; Ke, H.P.; Zhang, F.; Wang, H.S.; Cai, S.H.; et al. TGF-β and EGF induced HLA-I downregulation is associated with epithelial-mesenchymal transition (EMT) through upregulation of snail in prostate cancer cells. Mol. Immunol. 2015, 65, 34–42. [Google Scholar] [CrossRef]
- Kumar, S.; Davra, V.; Obr, A.E.; Geng, K.; Wood, T.L.; De Lorenzo, M.S.; Birge, R.B. Crk adaptor protein promotes PD-L1 expression, EMT and immune evasion in a murine model of triple-negative breast cancer. Oncoimmunology 2017, 7, e1376155. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Shan, B.; Man, H.; Liu, J.; Wang, L.; Zhu, T.; Ma, M.; Xv, Z.; Chen, X.; Yang, X.; Li, P. TIM-3 promotes the metastasis of esophageal squamous cell carcinoma by targeting epithelial-mesenchymal transition via the Akt/GSK-3β/Snail signaling pathway. Oncol. Rep. 2016, 36, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhang, T.; Liu, F.; Sun, Z.; Shi, H.; Hua, D.; Yang, C. The co-stimulatory molecule B7-H3 promotes the epithelial-mesenchymal transition in colorectal cancer. Oncotarget 2016, 7, 31755–31771. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Y.; Mou, Z.; Chen, J.; He, Y.; Dong, H.; Fu, X.; Wu, Y. B7H1 expression and epithelial-to-mesenchymal transition phenotypes on colorectal cancer stem-like cells. PLoS ONE 2015, 10, e0135528. [Google Scholar] [CrossRef]
- Noman, M.Z.; Van Moer, K.; Marani, V.; Gemmill, R.M.; Tranchevent, L.C.; Azuaje, F.; Muller, A.; Chouaib, S.; Thiery, J.P.; Berchem, G.; et al. CD47 is a direct target of SNAI1 and ZEB1 and its blockade activates the phagocytosis of breast cancer cells undergoing EMT. Oncoimmunology 2018, 7, e1345415. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Zhao, Q.; Xia, Y.; Hu, X.; Guo, J. PD-L1 induces epithelial-to-mesenchymal transition via activating SREBP-1c in renal cell carcinoma. Med. Oncol. 2015, 32, 212. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C.; Corti, A. Tumor cell-associated immune checkpoint molecules – Drivers of malignancy and stemness. Biochim. Biophys. Acta 2017, 1868, 571–583. [Google Scholar] [CrossRef]
- David, J.M.; Hamilton, D.H.; Palena, C. MUC1 upregulation promotes immune resistance in tumor cells undergoing brachyury-mediated epithelial-mesenchymal transition. Oncoimmunology 2016, 5, e1117738. [Google Scholar] [CrossRef]
- Hamilton, D.H.; Huang, B.; Fernando, R.I.; Tsang, K.-Y.; Palena, C. WEE1 inhibition alleviates resistance to immune attack of tumor cells undergoing epithelial–mesenchymal transition. Cancer Res. 2014, 74, 2510–2519. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Buart, S.; Tan, T.Z.; Gros, G.; Noman, M.Z.; Jorens, J.B.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: Consequences on susceptibility to cell-mediated cytotoxicity. Oncoimmunology 2017, 6, e1271858. [Google Scholar] [CrossRef] [PubMed]
- Akalay, I.; Tan, T.Z.; Kumar, P.; Janji, B.; Mami-Chouaib, F.; Charpy, C.; Vielh, P.; Larsen, A.K.; Thiery, J.P.; Sabbah, M.; et al. Targeting WNT1-inducible signaling pathway protein 2 alters human breast cancer cell susceptibility to specific lysis through regulation of KLF-4 and miR-7 expression. Oncogene 2015, 34, 2261–2271. [Google Scholar] [CrossRef]
- Noman, M.Z.; Buart, S.; Romero, P.; Ketari, S.; Janji, B.; Mari, B.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res. 2012, 72, 4629–4641. [Google Scholar] [CrossRef] [PubMed]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; André, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C. How tumor cells choose between epithelial-mesenchymal transition and autophagy to resist stress—Therapeutic implications. Front. Pharmacol. 2018, 9, 714. [Google Scholar] [CrossRef]
- Huergo-Zapico, L.; Acebes-Huerta, A.; López-Soto, A.; Villa-Álvarez, M.; Gonzalez-Rodriguez, P.; Gonzalez, S.L. Molecular bases for the regulation of NKG2D ligands in cancer. Front. Immunol. 2014, 5, 106. [Google Scholar] [CrossRef] [PubMed]
- Chockley, P.J.; Chen, J.; Chen, G.; Beer, D.G.; Standiford, T.J.; Keshamouni, V.G. Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer. J. Clin. Investig. 2018, 128, 1384–1396. [Google Scholar] [CrossRef]
- Huergo-Zapico, L.; Parodi, M.; Cantoni, C.; Lavarello, C.; Fernández-Martínez, J.L.; Petreto, A.; DeAndrés-Galiana, E.J.; Balsamo, M.; López-Soto, A.; Pietra, G.; et al. NK-cell editing mediates epithelial-to-mesenchymal transition via phenotypic and proteomic changes in melanoma cell lines. Cancer Res. 2018, 78, 3913–3925. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; McCampbell, K.K.; David, J.M.; Palena, C. Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight 2017, 2, 94296. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Bourcy, M.; Lesage, J.; Leroi, N.; Syne, L.; Blacher, S.; Hubert, P.; Erpicum, C.; Foidart, J.M.; Delvenne, P.; et al. Soluble factors regulated by epithelial-mesenchymal transition mediate tumour angiogenesis and myeloid cell recruitment. J. Pathol. 2015, 236, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Rao, Q.; Chen, Y.; Yeh, C.R.; Ding, J.; Li, L.; Chang, C.; Yeh, S. Recruited mast cells in the tumor microenvironment enhance bladder cancer metastasis via modulation of ERβ/CCL2/CCR2 EMT/MMP9 signals. Oncotarget 2016, 7, 7842–7855. [Google Scholar] [CrossRef]
- Knab, L.M.; Ebine, K.; Chow, C.R.; Raza, S.S.; Sahai, V.; Patel, A.P.; Kumar, K.; Bentrem, D.J.; Grippo, P.J.; Munshi, H.G. Snail cooperates with Kras G12D in vivo to increase stem cell factor and enhance mast cell infiltration. Mol. Cancer Res. 2014, 12, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Yao, W.; Yang, T.; Yang, Y.; Liu, Y.; Shen, Q.; Zhang, J.; Qi, W.; Wang, J. aPKC-ι/P-Sp1/Snail signaling induces epithelial-mesenchymal transition and immunosuppression in cholangiocarcinoma. Hepatology 2017, 66, 1165–1182. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Moccia, T.; Zhu, C.; Paradiso, G.; Budillon, A.; Chiao, P.J.; Abbruzzese, J.L.; Melisi, D. Anti-VEGF treatment–resistant pancreatic cancers secrete proinflammatory factors that contribute to malignant progression by inducing an EMT cell phenotype. Clin. Cancer Res. 2011, 17, 5822–5832. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Abiko, K.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Murakami, R.; Yamanoi, K.; Horikawa, N.; Hosoe, Y.; Nakamura, E.; et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat. Commun. 2018, 9, 1685. [Google Scholar] [CrossRef] [PubMed]
- Martínez, V.G.; Rubio, C.; Martínez-Fernández, M.; Segovia, C.; López-Calderón, F.; Garín, M.I.; Teijeira, A.; Munera-Maravilla, E.; Varas, A.; Sacedón, R.; et al. BMP4 induces M2 macrophage polarization and favors tumor progression in bladder cancer. Clin. Cancer Res. 2017, 23, 7388–7399. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.Y.; Chen, W.; Bai, X.L.; Xu, X.Y.; Zhang, Q.; Xia, X.F.; Sun, X.; Li, G.G.; Hu, Q.D.; Fu, Q.H.; et al. Hypoxia-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res. 2016, 76, 818–830. [Google Scholar] [CrossRef]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Shirako, H.; Ohike, M.; Tsukamoto, N.; Kawakami, Y. CCL2 is critical for immunosuppression to promote cancer metastasis. Clin. Exp. Metast. 2013, 30, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075. [Google Scholar] [CrossRef]
- Sangaletti, S.; Tripodo, C.; Santangelo, A.; Castioni, N.; Portararo, P.; Gulino, A.; Botti, L.; Parenza, M.; Cappetti, B.; Orlandi, R.; et al. Mesenchymal transition of high-grade breast carcinomas depends on extracellular matrix control of myeloid suppressor cell activity. Cell Rep. 2016, 17, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Faget, J.; Groeneveld, S.; Boivin, G.; Sankar, M.; Zangger, N.; Garcia, M.; Guex, N.; Zlobec, I.; Steiner, L.; Piersigilli, A.; et al. Neutrophils and Snail orchestrate the establishment of a pro-tumor microenvironment in lung cancer. Cell Rep. 2017, 21, 3190–3204. [Google Scholar] [CrossRef] [PubMed]
- Visciano, C.; Liotti, F.; Prevete, N.; Calì, G.; Franco, R.; Collina, F.; de Paulis, A.; Marone, G.; Santoro, M.; Melillo, R.M. Mast cells induce epithelial-to-mesenchymal transition and stem cell features in human thyroid cancer cells through an IL-8-Akt-Slug pathway. Oncogene 2015, 34, 5175–5186. [Google Scholar] [CrossRef]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Tsang, K.Y.; Palena, C. Short-term EGFR blockade enhances immune-mediated cytotoxicity of EGFR mutant lung cancer cells: Rationale for combination therapies. Cell Death Dis. 2016, 7, e2380. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.L.; Duan, W.; Su, C.Y.; Mao, F.Y.; Lv, Y.P.; Teng, Y.S.; Yu, P.W.; Zhuang, Y.; Zhao, Y.L. Interleukin 6 induces M2 macrophage differentiation by STAT3 activation that correlates with gastric cancer progression. Cancer Imunol. Immunother. 2017, 66, 1597–1608. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Tai, S.K.; Yang, M.H. Snail-overexpressing cancer cells promote M2-like polarization of tumor-associated macrophages by delivering miR-21-abundant exosomes. Neoplasia 2018, 20, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hao, X.; Sun, R.; Wei, H.; Tian, Z. Natural killer cell-derived interferon-gamma promotes hepatocellular carcinoma through the epithelial cell adhesion molecule-epithelial-to-mesenchymal transition axis in hepatitis B virus transgenic mice. Hepatology 2018, 69, 1735–1750. [Google Scholar] [CrossRef]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.P.; Abastado, J.P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef]
- Caiado, F.; Carvalho, T.; Rosa, I.; Remédio, L.; Costa, A.; Matos, J.; Heissig, B.; Yagita, H.; Hattori, K.; da Silva, J.P.; et al. Bone marrow-derived CD11b+Jagged2+ cells promote epithelial-to-mesenchymal transition and metastasization in colorectal cancer. Cancer Res. 2013, 73, 4233–4246. [Google Scholar] [CrossRef]
- De Cock, J.M.; Shibue, T.; Dongre, A.; Keckesova, Z.; Reinhardt, F.; Weinberg, R.A. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res. 2016, 76, 6778–6784. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Dan, Z.; Hu, X.; Gesang, L.; Ze, Y.; Bianba, Z. CD14 regulates gastric cancer cell epithelial-mesenchymal transition and invasion in vitro. Oncol. Rep. 2013, 30, 2725–2732. [Google Scholar] [CrossRef]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.T.; Dai, Z.; Song, K.; Zhang, Z.J.; Zhou, Z.J.; Zhou, S.L.; Zhao, Y.M.; Xiao, Y.S.; Sun, Q.M.; Ding, Z.B.; et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int. J. Oncol. 2015, 46, 587–596. [Google Scholar] [CrossRef]
- Ravi, J.; Elbaz, M.; Wani, N.A.; Nasser, M.W.; Ganju, R.K. Cannabinoid receptor-2 agonist inhibits macrophage induced EMT in non-small cell lung cancer by downregulation of EGFR pathway. Mol. Carcinog. 2016, 55, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef]
- Deng, Y.R.; Liu, W.B.; Lian, Z.X.; Li, X.; Hou, X. Sorafenib inhibits macrophage-mediated epithelial-mesenchymal transition in hepatocellular carcinoma. Oncotarget 2016, 7, 38292–38305. [Google Scholar] [CrossRef]
- Hu, Y.; He, M.Y.; Zhu, L.F.; Yang, C.C.; Zhou, M.L.; Wang, Q.; Zhang, W.; Zheng, Y.Y.; Wang, D.M.; Xu, Z.Q.; et al. Tumor-associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 12. [Google Scholar] [CrossRef]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Shen, M.; Zhang, P.; Zheng, C.; Pang, Z.; Zhu, L.; Du, J. Intratumoral neutrophil granulocytes contribute to epithelial-mesenchymal transition in lung adenocarcinoma cells. Tumour Biol. 2015, 36, 7789–7796. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Sun, X.; Yang, X.; Zhu, H.; Liu, J.; Wang, Y.; Chen, G.; Sun, X. Exosomes derived from irradiated esophageal carcinoma-infiltrating T cells promote metastasis by inducing the epithelial-mesenchymal transition in esophageal cancer cells. Pathol. Oncol. Res. 2018, 24, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.N.; Gao, H.; Anfossi, S.; Mego, M.; Reddy, N.G.; Debeb, B.; Giordano, A.; Tin, S.; Wu, Q.; Garza, R.J.; et al. Inflammation mediated metastasis: Immune induced epithelial-to-mesenchymal transition in inflammatory breast cancer cells. PLoS ONE 2015, 10, e0132710. [Google Scholar] [CrossRef]
- Goebel, L.; Grage-Griebenow, E.; Gorys, A.; Helm, O.; Genrich, G.; Lenk, L.; Wesch, D.; Ungefroren, H.; Freitag-Wolf, S.; Sipos, B.; et al. CD4+ T cells potently induce epithelial-mesenchymal-transition in premalignant and malignant pancreatic ductal epithelial cells-novel implications of CD4+ T cells in pancreatic cancer development. Oncoimmunology 2015, 4, e1000083. [Google Scholar] [CrossRef]
- Santisteban, M.; Reiman, J.M.; Asiedu, M.K.; Behrens, M.D.; Nassar, A.; Kalli, K.R.; Haluska, P.; Ingle, J.N.; Hartmann, L.C.; Manjili, M.H.; et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009, 69, 2887–2895. [Google Scholar] [CrossRef]
- Koizumi, M.; Hiasa, Y.; Kumagi, T.; Yamanishi, H.; Azemoto, N.; Kobata, T.; Matsuura, B.; Abe, M.; Onji, M. Increased B cell-activating factor promotes tumor invasion and metastasis in human pancreatic cancer. PLoS ONE 2013, 8, e71367. [Google Scholar] [CrossRef] [PubMed]
- Panni, R.Z.; Sanford, D.E.; Belt, B.A.; Mitchem, J.B.; Worley, L.A.; Goetz, B.D.; Mukherjee, P.; Wang-Gillam, A.; Link, D.C.; Denardo, D.G.; et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol. Immunother. 2014, 63, 513–528. [Google Scholar] [CrossRef]
- Lo, U.G.; Pong, R.C.; Yang, D.; Gandee, L.; Hernandez, E.; Dang, A.; Lin, C.J.; Santoyo, J.; Ma, S.; Sonavane, R.; et al. IFN-γ-induced IFIT5 promotes epithelial-to-mesenchymal transition in prostate cancer via microRNA processing. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lv, N.; Gao, Y.; Guan, H.; Wu, D.; Ding, S.; Teng, W.; Shan, Z. Inflammatory mediators, tumor necrosis factor-α and interferon-γ, induce EMT in human PTC cell lines. Oncol. Lett. 2015, 10, 2591–2597. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Wang, J.R.; Shen, T.; Gao, S.S.; He, X.S.; Li, J.N.; Yang, T.Y.; Zhang, S.; Gan, W.J.; Li, J.M.; et al. Nur77 deficiency in mice accelerates tumor invasion and metastasis by facilitating TNFα secretion and lowering CSF-1R expression. PLoS ONE 2017, 12, e0171347. [Google Scholar] [CrossRef]
- Zhu, Y.; Cheng, Y.; Guo, Y.; Chen, J.; Chen, F.; Luo, R.; Li, A. Protein kinase D2 contributes to TNF-α-induced epithelial mesenchymal transition and invasion via the PI3K/GSK-3β/β-catenin pathway in hepatocellular carcinoma. Oncotarget 2016, 7, 5327–5341. [Google Scholar] [CrossRef]
- Chuang, M.J.; Sun, K.H.; Tang, S.J.; Deng, M.W.; Wu, Y.H.; Sung, J.S.; Cha, T.L.; Sun, G.H. Tumor-derived tumor necrosis factor-alpha promotes progression and epithelial-mesenchymal transition in renal cell carcinoma cells. Cancer Sci. 2008, 99, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Öner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gao, Q.; Han, S.; Pan, F.; Fan, W. The CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal transition by cooperatively activating STAT3-Twist signaling. Tumour Biol. 2015, 36, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.W.; Liu, L.J.; Huang, J. Interleukin-6-induced epithelial-mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int. J. Oncol. 2014, 45, 165–176. [Google Scholar] [CrossRef]
- Colomiere, M.; Ward, A.C.; Riley, C.; Trenerry, M.K.; Cameron-Smith, D.; Findlay, J.; Ackland, L.; Ahmed, N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br. J. Cancer 2009, 100, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Asgeirsson, K.S.; Olafsdóttir, K.; Jónasson, J.G.; Ogmundsdóttir, H.M. The effects of IL-6 on cell adhesion and E-cadherin expression in breast cancer. Cytokine 1998, 10, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, S.; Parajuli, K.R.; Zhang, W.; Zhang, K.; Mo, Z.; Liu, J.; Chen, Z.; Yang, S.; Wang, A.R.; et al. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene 2017, 36, 687–699. [Google Scholar] [CrossRef]
- Wang, L.; Ma, R.; Kang, Z.; Zhang, Y.; Ding, H.; Guo, W.; Gao, Q.; Xu, M. Effect of IL-17A on the migration and invasion of NPC cells and related mechanisms. PLoS ONE 2014, 9, e108060. [Google Scholar] [CrossRef]
- Zhang, L.M.; Zhang, Y.; Fei, C.; Zhang, J.; Wang, L.; Yi, Z.W.; Gao, G. Neutralization of IL-18 by IL-18 binding protein ameliorates bleomycin-induced pulmonary fibrosis via inhibition of epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2019, 508, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Kawata, M.; Koinuma, D.; Ogami, T.; Umezawa, K.; Iwata, C.; Watabe, T.; Miyazono, K. TGF-β-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW 264.7 macrophage cells. J. Biochem. 2012, 151, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, X.; Chen, Y.; Fang, J.; Ge, Z. High-mobility group box 1: A novel inducer of the epithelial-mesenchymal transition in colorectal carcinoma. Cancer Lett. 2015, 357, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Funamizu, N.; Hu, C.; Lacy, C.; Schetter, A.; Zhang, G.; He, P.; Gaedcke, J.; Ghadimi, M.B.; Ried, T.; Yfantis, H.G.; et al. Macrophage migration inhibitory factor induces epithelial to mesenchymal transition, enhances tumor aggressiveness and predicts clinical outcome in resected pancreatic ductal adenocarcinoma. Int. J. Cancer 2013, 132, 785–794. [Google Scholar] [CrossRef]
- Yin, J.; Zeng, F.; Wu, N.; Kang, K.; Yang, Z.; Yang, H. Interleukin-8 promotes human ovarian cancer cell migration by epithelial-mesenchymal transition induction in vitro. Clin. Transl. Oncol. 2015, 17, 365–370. [Google Scholar] [CrossRef]
- Long, X.; Ye, Y.; Zhang, L.; Liu, P.; Yu, W.; Wei, F.; Ren, X.; Yu, J. IL-8, a novel messenger to cross-link inflammation and tumor EMT via autocrine and paracrine pathways (Review). Int. J. Oncol. 2016, 48, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Li, W.; Li, C.; Gao, Z.; Guo, K.; Song, S. CCL18 promotes epithelial-mesenchymal transition, invasion and migration of pancreatic cancer cells in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2015, 46, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, D.; Li, Y.; Liu, Y.; Xie, X.; Wu, Y.; Zhou, Y.; Ren, J.; Zhang, J.; Zhu, H.; et al. CCL21/CCR7 axis contributed to CD133+ pancreatic cancer stem-like cell metastasis via EMT and Erk/NF-κB pathway. PLoS ONE 2016, 11, e0158529. [Google Scholar] [CrossRef]
- Kai, K.; Iwamoto, T.; Zhang, D.; Shen, L.; Takahashi, Y.; Rao, A.; Thompson, A.; Sen, S.; Ueno, N.T. CSF-1/CSF-1R axis is associated with epithelial/mesenchymal hybrid phenotype in epithelial-like inflammatory breast cancer. Sci. Rep. 2018, 8, 9427. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 secreted from cancer-associated fibroblasts mediates chemoresistance in NSCLC by increasing epithelial-mesenchymal transition signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.S.; Li, Y.F.; Tan, J.; Sun, B.; Xiao, Y.C.; Fang, X.B.; Zhang, X.F.; Li, Q.; Dong, J.H.; Li, M.; et al. CCL20 and CXCL8 synergize to promote progression and poor survival outcome in patients with colorectal cancer by collaborative induction of the epithelial-mesenchymal transition. Cancer Lett. 2014, 348, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.H.; Sun, G.H.; Wu, Y.C.; Ko, B.J.; Hsu, H.T.; Wu, S.T. TNF-α augments CXCR2 and CXCR3 to promote progression of renal cell carcinoma. J. Cell. Mol. Med. 2016, 20, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Tang, S.J.; Sun, G.H.; Sun, K.H. CXCR7 mediates TGFβ1-promoted EMT and tumor-initiating features in lung cancer. Oncogene 2016, 35, 2123–2132. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell. Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4 + CD25- naive T cells to CD4 + CD25 + regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Gorelik, L.; Constant, S.; Flavell, R.A. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J. Exp. Med. 2002, 195, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Yamashita, M.; Shinoda, K.; Tofukuji, S.; Onodera, A.; Shinnakasu, R.; Motohashi, S.; Hosokawa, H.; Tumes, D.; Iwamura, C.; et al. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-beta and suppresses T(H)2 differentiation. Nat. Immunol. 2012, 13, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef]
- Johansson, J.; Tabor, V.; Wikell, A.; Jalkanen, S.; Fuxe, J. TGF-β1-induced epithelial-mesenchymal transition promotes monocyte/macrophage properties in breast cancer cells. Front. Oncol. 2015, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Ikeda, K.; Sato, W.; Horie-Inoue, K.; Inoue, S. Extracellular vesicle-mediated EBAG9 transfer from cancer cells to tumor microenvironment promotes immune escape and tumor progression. Oncogenesis 2018, 7, 7. [Google Scholar] [CrossRef]
- Hübel, J.; Hieronymus, T. HGF/Met-signaling contributes to immune regulation by modulating tolerogenic and motogenic properties of dendritic cells. Biomedicines 2015, 3, 138–148. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Hlubek, F.; Löhberg, C.; Meiler, J.; Suchy, U.; Kirchner, T. Negative regulation of CD4 expression in T cells by the transcriptional repressor ZEB. Int. Immunol. 1999, 11, 1701–1708. [Google Scholar] [CrossRef]
- Wang, J.; Lee, S.; The, C.E.; Binting, K.; Ma, L.; Shannon, M.F. The transcription repressor, ZEB1, cooperates with CtBP2 and HDAC1 to suppress IL-2 gene activation in T cells. Int. Immunol. 2009, 21, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Genetta, T.; Ruezinsky, D.; Kadesch, T. Displacement of an E-box-binding repressor by basic helix-loop-helix proteins: Implications for B-cell specificity of the immunoglobulin heavy-chain enhancer. Mol. Cell Biol. 1994, 14, 6153–6163. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lyons, J.G.; Patel, V.; Poue, N.C.; Fok, S.Y.; Soon, L.L.; Halliday, G.M.; Gutkind, J.S. Snail up-regulates proinflammatory mediators and inhibits differentiation in oral keratinocytes. Cancer Res. 2008, 68, 4525–4530. [Google Scholar] [CrossRef] [PubMed]
- Black, A.P.; Ardern-Jones, M.R.; Kasprowicz, V.; Bowness, P.; Jones, L.; Bailey, A.S.; Ogg, G.S. Human keratinocyte induction of rapid effector function in antigen-specific memory CD4+ and CD8+ T cells. Eur. J. Immunol. 2007, 37, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Konradi, S.; Yasmin, N.; Haslwanter, D.; Weber, M.; Gesslbauer, B.; Sixt, M.; Strobl, H. Langerhans cell maturation is accompanied by induction of N-cadherin and the transcriptional regulators of epithelial-mesenchymal transition ZEB1/2. Eur. J. Immunol. 2014, 44, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Dean, D.C. Independent repressor domains in ZEB regulate muscle and T-cell differentiation. Mol. Cell Biol. 1999, 19, 7961–7971. [Google Scholar] [CrossRef] [PubMed]
- Omilusik, K.D.; Best, J.A.; Yu, B.; Goossens, S.; Weidemann, A.; Nguyen, J.V.; Seuntjens, E.; Stryjewska, A.; Zweier, C.; Roychoudhuri, R.; et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med. 2015, 212, 2027–2039. [Google Scholar] [CrossRef] [PubMed]
- van Helden, M.J.; Goossens, S.; Daussy, C.; Mathieu, A.L.; Faure, F.; Marçais, A.; Vandamme, N.; Farla, N.; Mayol, K.; Viel, S.; et al. Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J. Exp. Med. 2015, 212, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-β signaling in cancer. Trends Cancer 2017, 3, 56–61. [Google Scholar] [CrossRef]
- Hamilton, D.H.; Litzinger, M.T.; Jales, A.; Huang, B.; Fernando, R.I.; Hodge, J.W.; Ardiani, A.; Apelian, D.; Schlom, J.; Palena, C. Immunological targeting of tumor cells undergoing an epithelial-mesenchymal transition via a recombinant brachyury-yeast vaccine. Oncotarget 2013, 4, 1777–1790. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C.; Lefoulon, F. Anti-cancer stem-like cell compounds in clinical development – An overview and critical appraisal. Front. Oncol. 2016, 6, 115. [Google Scholar] [CrossRef]
- Marcucci, F.; Caserta, C.A.; Romeo, E.; Rumio, C. Antibody-drug conjugates (ADC) against cancer stem-like cells (CSC)—Is there still room for optimism? Front. Oncol. 2019, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Corti, A. Improving drug penetration to curb tumor drug resistance. Drug Discov. Today 2012, 17, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type of Alteration | Consequences | References |
|---|---|---|
| Reduced Expression of Tumor Antigens | ||
| Reduced expression of tumor antigens in melanoma cells | Escape from antigen-specific killing by CTLs | [16] |
| Emergence of antigen-loss variants in EMT tumor cells from neu-transgenic mice | Escape from antigen-specific killing by CTLs | [18] |
| Reduced Expression of Antigen-Presenting Molecules | ||
| Downregulation of HLA class I molecules | Reduced antigen presentation and escape from antigen-specific killing by CTLs. | [20,21] |
| Enhanced Expression of Inhibitory Immune Checkpoint Molecules | ||
| Upregulation of PD-L1, TIM-3, B7-H1, B7-H3, CD47 | Downregulation of antitumor immune responses, resistance to killing by CTLs, amplification of tumor cell EMT | [15,20,22,23,24,25,26,27,28,29,30] |
| Enhanced Resistance of EMT Tumor Cells to Killing by T Cells | ||
| Overexpression of MUC-1 | Reduced susceptibility to killing by TRAIL and CTLs | [31] |
| Expression of the EMT TF brachyury, leading to inefficient apoptosis, with normal levels of HLA class I, antigenic peptides, and components of antigen presentation machinery | Reduced susceptibility to killing by CTLs and NK cells | [32] |
| EMT tumor cells showing defective immune synapse signaling | Resistance to killing by CTLs and NK cells | [33] |
| Upregulation of KLF-4 and downregulation of miR-7 | Reduced susceptibility to killing by CTLs | [34] |
| Expression of hypoxia-inducible miR-210 | Reduced susceptibility to killing by CTLs | [35] |
| Actin cytoskeleton remodeling, autophagy, and attenuation of an immunological synapse | Autophagy-dependent reduced susceptibility to killing by CTLs | [36] |
| Downregulation of HL -I and upregulation of ligands activating NK cells | Reduced recognition by CTLs and enhanced recognition by NK cells | [20,38] |
| Modulation of E-cadherin and CADM1 | Increased susceptibility to killing by NK cells and reduced metastasis formation | [39] |
| EMT-like changes in melanoma cells accompanied by upregulation of HLA class I and downregulation of activating receptors on NK cells | Escape from killing by NK cells | [40] |
| Mediator | Summary of Experimental Observations. | References |
|---|---|---|
| Cytokines | ||
| IL-2 | IL-2 from cholangiocarcinoma cells with EMT-like features induced generation of CD4+ CD25+ natural Tregs. | [46] |
| IL-6 | IL-6 induced macrophages to differentiate into M2-polarized macrophages. | [60] |
| TGF-β | TGF-β from cholangiocarcinoma cells with EMT-like features induced generation of CD4+ CD25+ natural Tregs. | [46] |
| BMP-4 | Recombinant BMP4 and BMP4-containing conditioned media from bladder cancer cell lines promoted monocyte/macrophage polarization toward an M2 phenotype. | [49] |
| GM-CSF | GM-CSF from mesenchymal-like breast cancer cells (BT-549, MDA-MB-436, and MDA-MB-231) promoted the acquisition of a TAM-like phenotype by macrophages. | [51] |
| Chemokines | ||
| IL-8/CXCL8 | IL-8 from claudin-low TNBC cells induced recruitment of PMN-MDSCs in vitro and in vivo, as determined through neutralization experiments with mAb HuMax-IL8. | [42] |
| CXCL1, CXCL2 | CXCL1 and CXCL2 from mouse ovarian cancer cells promoted tumor infiltration of MDSCs, as determined by knockdown of EMT transcription factor Snail. | [48] |
| CCL2 | CCL2 derived from various tumor cell lines induced, in cooperation with Lipocalin-2, DCregs, which in turn induced Tregs, and finally impaired the induction of tumor-specific CTLs. | [53] |
| CCL20 | CCL20 derived from EMT hepatoma cells induced IDO in monocyte-derived macrophages, which in turn suppressed T-cell proliferations and promoted the expansion of Tregs. | [50] |
| Other Soluble Mediators | ||
| Thrombospondin-1 | Snail-transduced melanoma cells with EMT features produced thrombospondin-1, which induced Tregs and impaired DCs in vitro and in vivo. | [52] |
| Lipocalin-2 | CCL2 derived from various tumor cell lines induced, in cooperation with Lipocalin-2, DCregs, which in turn induced Tregs and finally impaired the induction of tumor-specific CTLs. | [53] |
| Exosomes | ||
| Snail-expressing EMT human head and neck cancer cells activated the transcription of miR-21 to produce tumor-derived exosomes, which were engulfed by CD14+ human monocytes, suppressing the expression of M1 markers and increasing that of M2 markers. | [61] | |
| Mediator | Summary of Experimental Observations. | References |
|---|---|---|
| Cytokines | ||
| IFN-γ | IFN-γ and other cytokines mediated NK cell-induced increased malignancy of melanoma cells; | [40,61,80,81] |
| IFN-γ mediated NK cell-induced EMT and HCC formation in hepatocytes from HBsAg transgenic mice; | ||
| IFN-γ induced EMT in prostate and papillary thyroid cancer cells. | ||
| TNF-α | TNF-α and other cytokines mediated NK cell-induced increased malignancy of melanoma cells; | [40,75,81,82,83,84] |
| TNF-α, with other cytokines, induced EMT in IBC cells in a manner similar to soluble factors from activated T cells; | ||
| TNF-α induced EMT in papillary thyroid cancer and HCC cells; | ||
| TNF-α induced EMT in HCC cells, with synergistic induction in combination with IL-1β and IL-6; | ||
| TNF-α derived from macrophages induced tumor cell EMT. | ||
| TGF-β | TGF-β induced HLA class I downregulation and EMT in prostate cancer cells; | [21,62,66,70,73,75,95] |
| TGF-β mediated, with other cytokines, EMT-like changes induced by MDSCs in melanoma cells; | ||
| TGF-β mediated EMT of HCCs induced by TAMs; | ||
| TGF-β was the main mediator of EMT induced in teratocarcinoma cells by macrophage-conditioned medium; | ||
| TGF-β mediated EMT in lung adenocarcinoma cells induced by polymorphonuclear neutrophils; | ||
| TGF-β, with other cytokines, induced EMT in IBC cells in a manner similar to soluble factors from activated T cells; | ||
| TGF-β induced EMT in lung adenocarcinoma cells, with synergistic effects with other cytokines from a macrophage cell line. | ||
| EGF | EGF induced HLA-I downregulation and EMT in prostate cancer cells; | [21,62,89] |
| EGF mediated, with other cytokines, EMT-like changes induced by MDSCs in melanoma cells; | ||
| EGF induced EMT and enhanced the migration of ovarian carcinoma cells. | ||
| HGF | HGF mediated, with other cytokines, EMT-like changes induced by MDSCs in melanoma cells. | [62] |
| IL-6 | IL-6, with other cytokines, induced EMT in IBC cells in a manner similar to soluble factors from activated T cells; | [75,76,85,86,87,88,89,90] |
| IL-6 from activated T cells induced EMT in premalignant pancreatic cells; | ||
| IL-6 induced EMT in breast cancer, CRC, NSCLC, cervical carcinoma, and ovarian carcinoma cells; | ||
| IL-17 | IL-6 induced EMT in prostate cancer cell lines. | [92,93] |
| IL-17 induced EMT and enhanced migration and invasion in nasopharyngeal carcinoma cell lines. | ||
| IL-18 | IL-18 induced EMT in lung adenocarcinoma cells. | [94] |
| HMGB1 | HMGB1 induced EMT in CRC cells. | [96] |
| MIF | MIF induced EMT in pancreatic cancer cells. | [97] |
| BAF | BAF induced EMT in pancreatic cancer cells. | [78] |
| Chemokines | ||
| IL-8 | IL-8 mediated mast cell-induced EMT in human thyroid cancer cells; | [57,67,91,98] |
| IL-8 mediated macrophage-induced EMT in HCC cells; | ||
| IL-8 induced EMT and migration in human ovarian cancer cells; | ||
| review of IL-8 as a mediator between inflammation and tumor cell EMT. | ||
| CCL2 | CCL2 enhanced IL-6 induced EMT and invasion of NSCLC cells. | [87] |
| CCL18 | CCL18 from TAM induced EMT in breast cancer cells; | [51,99] |
| CCL18 induced EMT and the invasion and migration of pancreatic cancer cells. | ||
| CCL21 | CCL21 induced EMT and metastasis in CD133+ pancreatic cancer CSCs. | [100] |
| Exosomes | ||
| Exosomes from TILs induced EMT in human esophageal squamous cell carcinoma cells. | [74] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romeo, E.; Caserta, C.A.; Rumio, C.; Marcucci, F. The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System. Cells 2019, 8, 460. https://doi.org/10.3390/cells8050460
Romeo E, Caserta CA, Rumio C, Marcucci F. The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System. Cells. 2019; 8(5):460. https://doi.org/10.3390/cells8050460
Chicago/Turabian StyleRomeo, Elisabetta, Carmelo Antonio Caserta, Cristiano Rumio, and Fabrizio Marcucci. 2019. "The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System" Cells 8, no. 5: 460. https://doi.org/10.3390/cells8050460
APA StyleRomeo, E., Caserta, C. A., Rumio, C., & Marcucci, F. (2019). The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System. Cells, 8(5), 460. https://doi.org/10.3390/cells8050460