Rho GTPases—Emerging Regulators of Glucose Homeostasis and Metabolic Health


Abstract
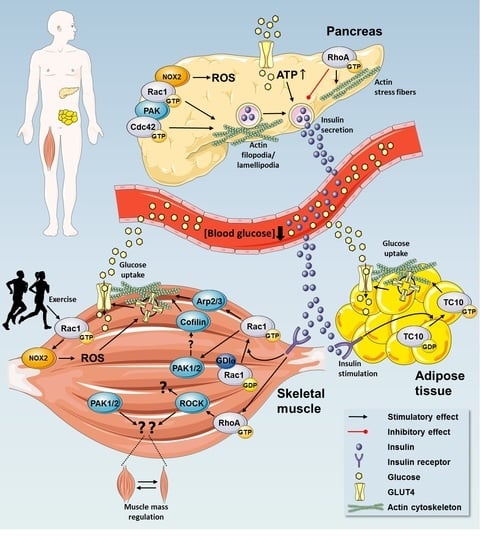
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
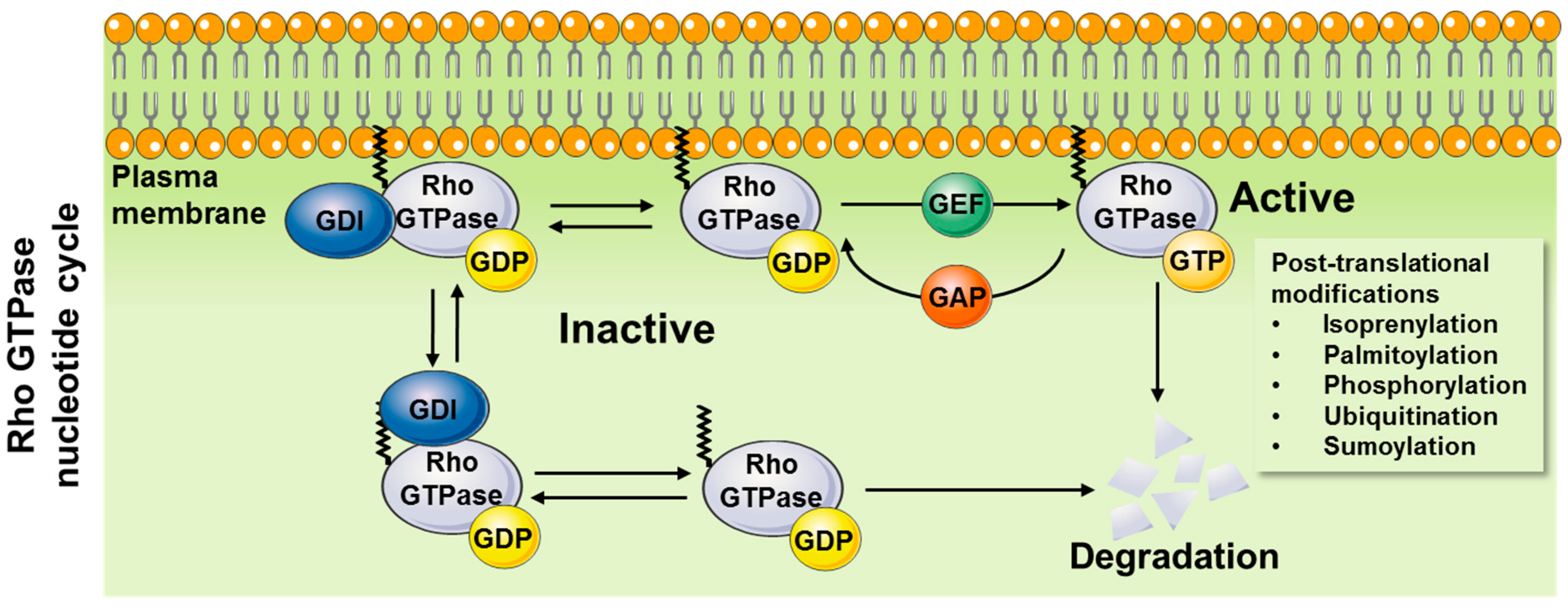
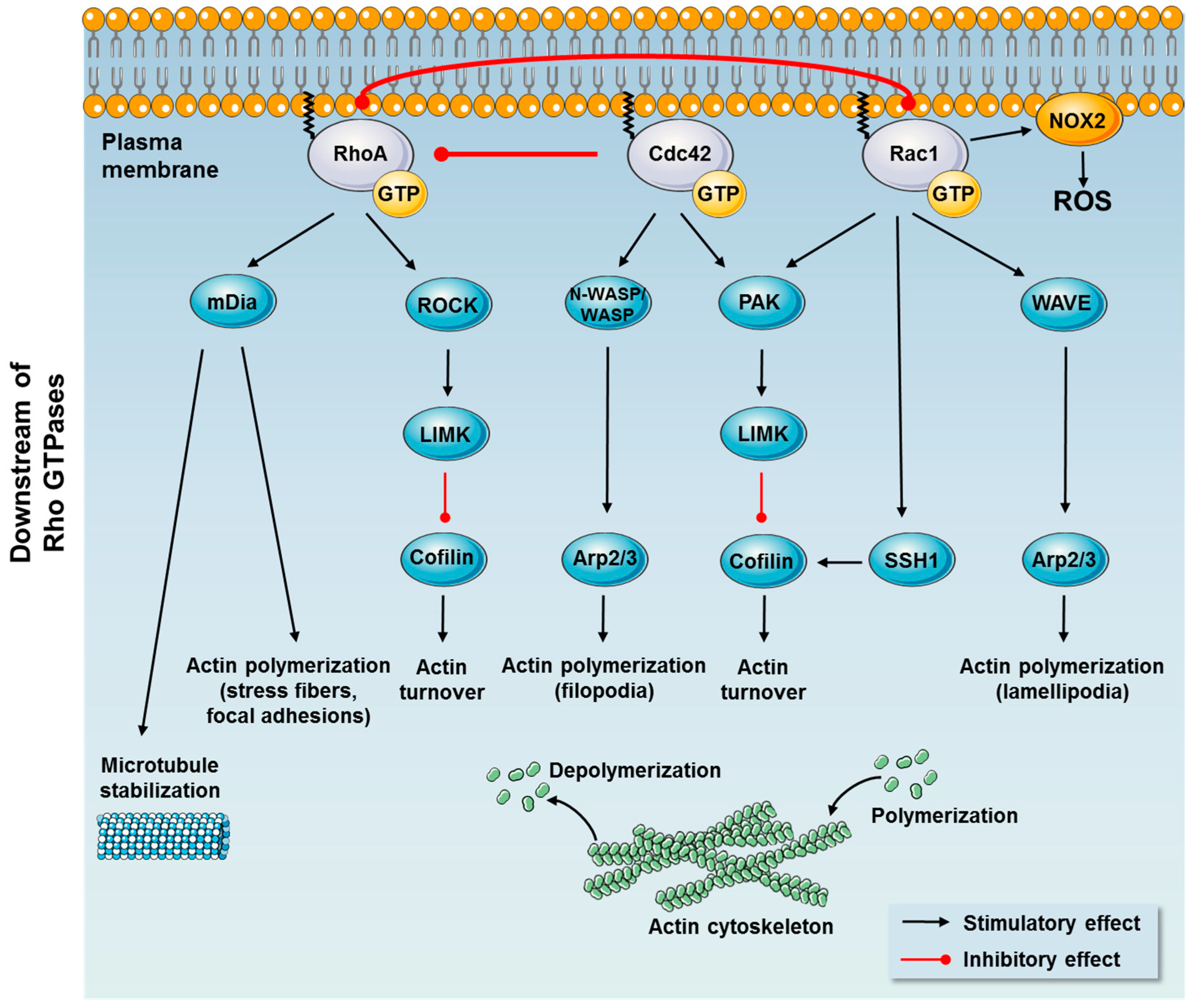
1.1. Mechanisms of Action
1.1.1. General Mechanisms of Activation
1.1.2. Downstream Effects
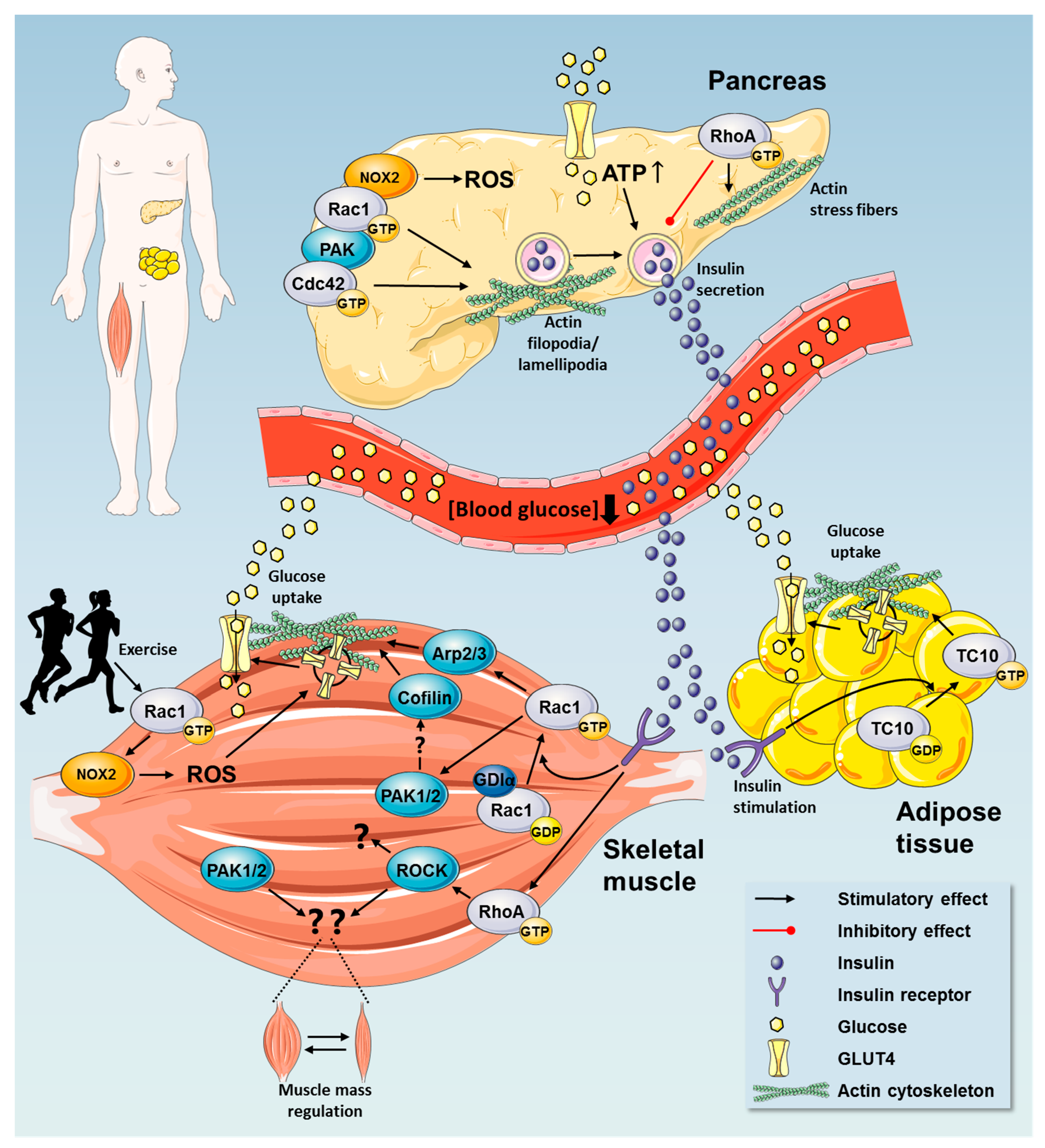
2. Rho GTPases in Regulation of Glucose Homeostasis
2.1. Insulin Secretion by Pancreatic β Cells
2.2. Rho GTPases are Implicated in β Cell Dysfunction in Metabolic Disease
2.3. Rho GTPases Regulate Glucose Uptake
2.3.1. Rho GTPases in Insulin Action in Skeletal Muscle
2.3.2. Cross-Talk Between Rho GTPases and Canonical Insulin Signaling
2.3.3. Exercise Elicits Metabolic Benefits via Regulation of Rho GTPases
2.4. A Possible Role for Rho GTPases in Adipose Tissue Glucose Uptake
2.5. Rho GTPases are Implicated in Insulin Resistance—A Key Contributor to Metabolic Disease
3. Rho GTPases as Hitherto Unrecognized Regulators of Muscle Mass
The Role of Rho GTPases in Muscle Wasting Conditions
Unresolved Issues
- Lack of in vivo experiments to support the in vitro literature on Rho GTPase regulation and in particular their role in metabolism.
- Evidence on the regulatory functions of Rho GTPases in humans is limited.
- Molecularly, the upstream activators and downstream effectors of Rho GTPases in different tissues and in response to different stimuli are poorly defined.
- Cross-talk between Rho GTPases is poorly defined but important to delineate, as they challenge all interpretation of data using knockdown or overexpression of a single Rho GTPase.
- High throughput methodological advances to directly measure in vivo GTP binding (fast hydrolysis) warranted.
- Whether Rho GTPases can be targeted pharmacologically to benefit metabolic diseases remains to be determined.
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-binding proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef]
- Boettner, B.; Van Aelst, L. The role of Rho GTPases in disease development. Gene 2002, 286, 155–174. [Google Scholar] [CrossRef]
- WHO Global status report on noncommunicable diseases 2014. Attaining the nine global noncommunicable diseases targets; a shared responsicility. World Heal. Organ. 2014, 176. [Google Scholar]
- Esposito, K.; Chiodini, P.; Colao, A.; Lenzi, A.; Giugliano, D. Metabolic Syndrome and Risk of Cancer: A systematic review and meta-analysis. Diabetes Care 2012, 35, 2402–2411. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Samson, S.L.; Garber, A.J. Metabolic Syndrome. Endocrinol. Metab. Clin. North Am. 2014, 43, 1–23. [Google Scholar] [CrossRef]
- Parada, L.F.; Tabin, C.J.; Shih, C.; Weinberg, R.A. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature 1982, 297, 474–478. [Google Scholar] [CrossRef]
- Santos, E.; Martin-Zanca, D.; Reddy, E.P.; Pierotti, M.A.; Della Porta, G.; Barbacid, M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science 1984, 223, 661–664. [Google Scholar] [CrossRef]
- Bos, J.L.; Fearon, E.R.; Hamilton, S.R.; Vries, M.V.; van Boom, J.H.; van der Eb, A.J.; Vogelstein, B. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327, 293–297. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Ras history. Small GTPases 2010, 1, 22–27. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef]
- Peurois, F.; Veyron, S.; Ferrandez, Y.; Ladid, I.; Benabdi, S.; Zeghouf, M.; Peyroche, G.; Cherfils, J. Characterization of the activation of small GTPases by their GEFs on membranes using artificial membrane tethering. Biochem. J. 2017, 474, 1259–1272. [Google Scholar] [CrossRef]
- Olofsson, B. Rho Guanine Dissociation Inhibitors. Cell. Signal. 1999, 11, 545–554. [Google Scholar] [CrossRef]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef]
- Wennerberg, K.; Der, C.J. Rho-family GTPases: It’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117, 1301–1312. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef]
- Nishimura, A.; Linder, M.E. Identification of a Novel Prenyl and Palmitoyl Modification at the CaaX Motif of Cdc42 That Regulates RhoGDI Binding. Mol. Cell. Biol. 2013, 33, 1417–1429. [Google Scholar] [CrossRef]
- Boulter, E.; Garcia-Mata, R.; Guilluy, C.; Dubash, A.; Rossi, G.; Brennwald, P.J.; Burridge, K. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat. Cell Biol. 2010, 12, 477–483. [Google Scholar] [CrossRef]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The “invisible hand”: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef]
- Byrne, K.M.; Monsefi, N.; Dawson, J.C.; Degasperi, A.; Bukowski-Wills, J.-C.; Volinsky, N.; Dobrzyński, M.; Birtwistle, M.R.; Tsyganov, M.A.; Kiyatkin, A.; et al. Bistability in the Rac1, PAK, and RhoA Signaling Network Drives Actin Cytoskeleton Dynamics and Cell Motility Switches. Cell Syst. 2016, 2, 38–48. [Google Scholar] [CrossRef]
- Sander, E.E.; ten Klooster, J.P.; van Delft, S.; van der Kammen, R.A.; Collard, J.G. Rac downregulates Rho activity: Reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 1999, 147, 1009–1022. [Google Scholar] [CrossRef]
- Nimnual, A.S.; Taylor, L.J.; Bar-Sagi, D. Redox-dependent downregulation of Rho by Rac. Nat. Cell Biol. 2003, 5, 236–241. [Google Scholar] [CrossRef]
- Rolli-Derkinderen, M.; Toumaniantz, G.; Pacaud, P.; Loirand, G. RhoA phosphorylation induces Rac1 release from guanine dissociation inhibitor alpha and stimulation of vascular smooth muscle cell migration. Mol. Cell. Biol. 2010, 30, 4786–4796. [Google Scholar] [CrossRef]
- Rolli-Derkinderen, M.; Sauzeau, V.; Boyer, L.; Lemichez, E.; Baron, C.; Henrion, D.; Loirand, G.; Pacaud, P. Phosphorylation of Serine 188 Protects RhoA from Ubiquitin/Proteasome-Mediated Degradation in Vascular Smooth Muscle Cells. Circ. Res. 2005, 96, 1152–1160. [Google Scholar] [CrossRef]
- Rosenfeldt, H.; Castellone, M.D.; Randazzo, P.A.; Gutkind, J.S. Rac inhibits thrombin-induced Rho activation: Evidence of a Pak-dependent GTPase crosstalk. J. Mol. Signal. 2006, 1, 8. [Google Scholar] [CrossRef]
- Barac, A.; Basile, J.; Vázquez-Prado, J.; Gao, Y.; Zheng, Y.; Gutkind, J.S. Direct Interaction of p21-Activated Kinase 4 with PDZ-RhoGEF, a G Protein-linked Rho Guanine Exchange Factor. J. Biol. Chem. 2004, 279, 6182–6189. [Google Scholar] [CrossRef]
- DerMardirossian, C.; Schnelzer, A.; Bokoch, G.M. Phosphorylation of RhoGDI by Pak1 Mediates Dissociation of Rac GTPase. Mol. Cell 2004, 15, 117–127. [Google Scholar] [CrossRef]
- Bustelo, X.R.; Sauzeau, V.; Berenjeno, I.M. GTP-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. BioEssays 2007, 29, 356–370. [Google Scholar] [CrossRef]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef]
- Hall, A. Ras-related GTPases and the cytoskeleton. Mol. Biol. Cell 1992, 3, 475–479. [Google Scholar] [CrossRef]
- Nobes, C.; Hall, A. Regulation and function of the Rho subfamily of small GTPases. Curr. Opin. Genet. Dev. 1994, 4, 77–81. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, rac and cdc42 GTPases: Regulators of actin structures, cell adhesion and motility. Biochem. Soc. Trans. 2015, 23, 456–459. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Spiering, D.; Hodgson, L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh. Migr. 2011, 5, 170–180. [Google Scholar] [CrossRef]
- Sit, S.-T.; Manser, E. Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci. 2011, 124, 679–683. [Google Scholar] [CrossRef]
- Ono, S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int. Rev. Cytol. 2007, 258, 1–82. [Google Scholar]
- Goley, E.D.; Welch, M.D. The ARP2/3 complex: An actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 2006, 7, 713–726. [Google Scholar] [CrossRef]
- Ghosh, M.; Song, X.; Mouneimne, G.; Sidani, M.; Lawrence, D.S.; Condeelis, J.S. Cofilin promotes actin polymerization and defines the direction of cell motility. Science 2004, 304, 743–746. [Google Scholar] [CrossRef]
- Kligys, K.; Claiborne, J.N.; DeBiase, P.J.; Hopkinson, S.B.; Wu, Y.; Mizuno, K.; Jones, J.C.R. The Slingshot Family of Phosphatases Mediates Rac1 Regulation of Cofilin Phosphorylation, Laminin-332 Organization, and Motility Behavior of Keratinocytes. J. Biol. Chem. 2007, 282, 32520–32528. [Google Scholar] [CrossRef]
- Cao, Z.; Li, X.; Li, J.; Kang, B.; Chen, J.; Luo, W.; Huang, C. SUMOylation of RhoGDIα is required for its repression of cyclin D1 expression and anchorage-independent growth of cancer cells. Mol. Oncol. 2014, 8, 285–296. [Google Scholar] [CrossRef]
- Chiu, T.T.; Patel, N.; Shaw, A.E.; Bamburg, J.R.; Klip, A. Arp2/3- and Cofilin-coordinated Actin Dynamics Is Required for Insulin-mediated GLUT4 Translocation to the Surface of Muscle Cells. Mol. Biol. Cell 2010, 21, 3529–3539. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.G.; Segal, A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef]
- Ohashi, K.; Nagata, K.; Maekawa, M.; Ishizaki, T.; Narumiya, S.; Mizuno, K. Rho-associated Kinase ROCK Activates LIM-kinase 1 by Phosphorylation at Threonine 508 within the Activation Loop. J. Biol. Chem. 2000, 275, 3577–3582. [Google Scholar] [CrossRef]
- Breitsprecher, D.; Goode, B.L. Formins at a glance. J. Cell Sci. 2013, 126, 1–7. [Google Scholar] [CrossRef]
- Copeland, J.W. The Diaphanous-related Formin mDia1 Controls Serum Response Factor Activity through its Effects on Actin Polymerization. Mol. Biol. Cell 2002, 13, 4088–4099. [Google Scholar] [CrossRef]
- Kühn, S.; Geyer, M. Formins as effector proteins of Rho GTPases. Small GTPases 2014, 5, e983876. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Hall, A. Rho family GTPases. Biochem. Soc. Trans. 2012, 40, 1378–1382. [Google Scholar] [CrossRef] [PubMed]
- Zegers, M.M.; Friedl, P. Rho GTPases in collective cell migration. Small GTPases 2014, 5, e983869. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef] [PubMed]
- Gandasi, N.R.; Yin, P.; Riz, M.; Chibalina, M.V.; Cortese, G.; Lund, P.-E.; Matveev, V.; Rorsman, P.; Sherman, A.; Pedersen, M.G.; et al. Ca2+ channel clustering with insulin-containing granules is disturbed in type 2 diabetes. J. Clin. Invest. 2017, 127, 2353–2364. [Google Scholar] [CrossRef]
- Zierath, J.R.; He, L.; Gumà, A.; Odegoard Wahlström, E.; Klip, A.; Wallberg-Henriksson, H. Insulin action on glucose transport and plasma membrane GLUT4 content in skeletal muscle from patients with NIDDM. Diabetologia 1996, 39, 1180–1189. [Google Scholar] [CrossRef]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Greiner, T.U.; Kesavan, G.; Ståhlberg, A.; Semb, H. Rac1 regulates pancreatic islet morphogenesis. BMC Dev. Biol. 2009, 9, 2. [Google Scholar] [CrossRef]
- Kesavan, G.; Lieven, O.; Mamidi, A.; Ohlin, Z.L.; Johansson, J.K.; Li, W.-C.; Lommel, S.; Greiner, T.U.; Semb, H. Cdc42/N-WASP signaling links actin dynamics to pancreatic cell delamination and differentiation. Development 2014, 141, 685–696. [Google Scholar] [CrossRef]
- Veluthakal, R.; Tunduguru, R.; Arora, D.K.; Sidarala, V.; Syeda, K.; Vlaar, C.P.; Thurmond, D.C.; Kowluru, A. VAV2, a guanine nucleotide exchange factor for Rac1, regulates glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia 2015, 58, 2573–2581. [Google Scholar] [CrossRef]
- Asahara, S.; Shibutani, Y.; Teruyama, K.; Inoue, H.Y.; Kawada, Y.; Etoh, H.; Matsuda, T.; Kimura-Koyanagi, M.; Hashimoto, N.; Sakahara, M.; et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 2013, 56, 1088–1097. [Google Scholar] [CrossRef]
- Rorsman, P.; Braun, M. Regulation of Insulin Secretion in Human Pancreatic Islets. Annu. Rev. Physiol. 2013, 75, 155–179. [Google Scholar] [CrossRef]
- Huang, Q.-Y.; Lai, X.-N.; Qian, X.-L.; Lv, L.-C.; Li, J.; Duan, J.; Xiao, X.-H.; Xiong, L.-X. Cdc42: A Novel Regulator of Insulin Secretion and Diabetes-Associated Diseases. Int. J. Mol. Sci. 2019, 20, 179. [Google Scholar] [CrossRef]
- Kalwat, M.A.; Thurmond, D.C. Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet β cells. Exp. Mol. Med. 2013, 45, e37. [Google Scholar] [CrossRef]
- Wang, Z.; Oh, E.; Clapp, D.W.; Chernoff, J.; Thurmond, D.C. Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J. Biol. Chem. 2011, 286, 41359–41367. [Google Scholar] [CrossRef]
- Prentki, M. Islet cell failure in type 2 diabetes. J. Clin. Invest. 2006, 116, 1802–1812. [Google Scholar] [CrossRef]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Invest. 2011, 121, 2118–2125. [Google Scholar] [CrossRef]
- Kowluru, A. Friendly, and not so friendly, roles of Rac1 in islet β-cell function: Lessons learnt from pharmacological and molecular biological approaches. Biochem. Pharmacol. 2011, 81, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Syed, I.; Kyathanahalli, C.N.; Jayaram, B.; Govind, S.; Rhodes, C.J.; Kowluru, R.A.; Kowluru, A. Increased Phagocyte-Like NADPH Oxidase and ROS Generation in Type 2 Diabetic ZDF Rat and Human Islets. Diabetes 2011, 60, 2843–2852. [Google Scholar] [CrossRef]
- Subasinghe, W.; Syed, I.; Kowluru, A. Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic β-cells: Evidence for regulation by Rac1. Am. J. Physiol. Integr. Comp. Physiol. 2011, 300, R12–R20. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, A.; Kowluru, R.A. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem. Pharmacol. 2014, 88, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Yu, D.; Ning, S.; Zhang, H.; Jiang, L.; He, L.; Li, M.; Sun, M. Augmented Rac1 Expression and Activity are Associated with Oxidative Stress and Decline of β Cell Function in Obesity. Cell. Physiol. Biochem. 2015, 35, 2135–2148. [Google Scholar] [CrossRef]
- Veluthakal, R.; Chepurny, O.G.; Leech, C.A.; Schwede, F.; Holz, G.G.; Thurmond, D.C. Restoration of Glucose-Stimulated Cdc42-Pak1 Activation and Insulin Secretion by a Selective Epac Activator in Type 2 Diabetic Human Islets. Diabetes 2018, 67, 1999–2011. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Yan, D.; Sun, J.; Wu, X.; Mulder, H.; Hua, X.; Ma, X. Glucagon-Like Peptide 1 Stimulates Insulin Secretion via Inhibiting RhoA/ROCK Signaling and Disassembling Glucotoxicity-Induced Stress Fibers. Endocrinology 2014, 155, 4676–4685. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Gunnarsson, R.; Björkman, O.; Olsson, M.; Wahren, J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J. Clin. Invest. 1985, 76, 149–155. [Google Scholar] [CrossRef]
- Lang, C.H. Rates and Tissue Sites of Noninsulin- and Insulin-Mediated Glucose Uptake in Diabetic Rats. Exp. Biol. Med. 2013, 199, 81–87. [Google Scholar] [CrossRef]
- Ploug, T.; van Deurs, B.; Ai, H.; Cushman, S.W.; Ralston, E. Analysis of GLUT4 Distribution in Whole Skeletal Muscle Fibers: Identification of Distinct Storage Compartments That Are Recruited by Insulin and Muscle Contractions. J. Cell Biol. 1998, 142, 1429–1446. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, H.P.M.M. Insulin- and Contraction-Induced Glucose Transporter 4 Traffic in Muscle. Exerc. Sport Sci. Rev. 2013, 41, 77–86. [Google Scholar] [CrossRef] [PubMed]
- JeBailey, L.; Rudich, A.; Huang, X.; Ciano-Oliveira, C.D.; Kapus, A.; Klip, A. Skeletal Muscle Cells and Adipocytes Differ in Their Reliance on TC10 and Rac for Insulin-Induced Actin Remodeling. Mol. Endocrinol. 2004, 18, 359–372. [Google Scholar] [CrossRef]
- Ishikura, S.; Koshkina, A.; Klip, A. Small G proteins in insulin action: Rab and Rho families at the crossroads of signal transduction and GLUT4 vesicle traffic. Acta Physiol. 2007, 192, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, N.; Ongusaha, P.; Jahng, W.J.; Araki, K.; Choi, C.S.; Kim, H.-J.; Lee, Y.H.; Kaibuchi, K.; Kahn, B.B.; Masuzaki, H.; et al. Role of Rho-kinase in regulation of insulin action and glucose homeostasis. Cell Metab. 2005, 2, 119–129. [Google Scholar] [CrossRef]
- Ueda, S.; Kitazawa, S.; Ishida, K.; Nishikawa, Y.; Matsui, M.; Matsumoto, H.; Aoki, T.; Nozaki, S.; Takeda, T.; Tamori, Y.; et al. Crucial role of the small GTPase Rac1 in insulin-stimulated translocation of glucose transporter 4 to the mouse skeletal muscle sarcolemma. FASEB J. 2010, 24, 2254–2261. [Google Scholar] [CrossRef]
- Sylow, L.; Jensen, T.E.; Kleinert, M.; Hojlund, K.; Kiens, B.; Wojtaszewski, J.; Prats, C.; Schjerling, P.; Richter, E.A. Rac1 Signaling Is Required for Insulin-Stimulated Glucose Uptake and Is Dysregulated in Insulin-Resistant Murine and Human Skeletal Muscle. Diabetes 2013, 62, 1865–1875. [Google Scholar] [CrossRef]
- Khayat, Z.A.; Tong, P.; Yaworsky, K.; Bloch, R.J.; Klip, A. Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J. Cell Sci. 2000, 113, 279–290. [Google Scholar]
- JeBailey, L.; Wanono, O.; Niu, W.; Roessler, J.; Rudich, A.; Klip, A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes 2007, 56, 394–403. [Google Scholar] [CrossRef]
- Ueda, S.; Kataoka, T.; Satoh, T. Activation of the small GTPase Rac1 by a specific guanine-nucleotide-exchange factor suffices to induce glucose uptake into skeletal-muscle cells. Biol. Cell 2008, 100, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Kleinert, M.; Pehmøller, C.; Prats, C.; Chiu, T.T.; Klip, A.; Richter, E.A.; Jensen, T.E. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell. Signal. 2014, 26, 323–331. [Google Scholar] [CrossRef]
- Chiu, T.T.; Jensen, T.E.; Sylow, L.; Richter, E.A.; Klip, A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell. Signal. 2011, 23, 1546–1554. [Google Scholar] [CrossRef]
- Madsen, A.B.; Knudsen, J.R.; Henriquez-Olguin, C.; Angin, Y.; Zaal, K.J.; Sylow, L.; Schjerling, P.; Ralston, E.; Jensen, T.E. β-Actin shows limited mobility and is required only for supraphysiological insulin-stimulated glucose transport in young adult soleus muscle. Am. J. Physiol. Metab. 2018, 315, E110–E125. [Google Scholar] [CrossRef]
- You, G.Y.; Lee, J.O.; Kim, J.H.; Kim, N.; Lee, S.K.; Moon, J.W.; Jie, S.; Lee, H.J.; Kim, S.J.; Park, S.H.; et al. Tiam-1, a GEF for Rac1, plays a critical role in metformin-mediated glucose uptake in C2C12 cells. Cell. Signal. 2013, 25, 2558–2565. [Google Scholar] [CrossRef]
- Charrasse, S.; Comunale, F.; Fortier, M.; Portales-Casamar, E.; Debant, A.; Gauthier-Rouviere, C. M-cadherin activates Rac1 GTPase through the Rho-GEF trio during myoblast fusion. Mol. Biol. Cell 2007, 18, 1734–1743. [Google Scholar] [CrossRef]
- Samson, T.; Will, C.; Knoblauch, A.; Sharek, L.; von der Mark, K.; Burridge, K.; Wixler, V. Def-6, a guanine nucleotide exchange factor for Rac1, interacts with the skeletal muscle integrin chain alpha7A and influences myoblast differentiation. J. Biol. Chem. 2007, 282, 15730–15742. [Google Scholar] [CrossRef]
- Kim, Y.B.; Inoue, T.; Nakajima, R.; Shirai-Morishita, Y.; Tokuyama, K.; Suzuki, M. Effect of long-term exercise on gene expression of insulin signaling pathway intermediates in skeletal muscle. Biochem. Biophys. Res. Commun. 1999, 254, 720–727. [Google Scholar] [CrossRef]
- Chen, E.H.; Pryce, B.A.; Tzeng, J.A.; Gonzalez, G.A.; Olson, E.N. Control of myoblast fusion by a guanine nucleotide exchange factor, loner, and its effector ARF6. Cell 2003, 114, 751–762. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, J.O.; Lee, Y.W.; Kim, S.A.; Park, S.H.; Kim, H.S. Kalirin, a GEF for Rac1, plays an important role in FSTL-1-mediated glucose uptake in skeletal muscle cells. Cell. Signal. 2017, 29, 150–157. [Google Scholar] [CrossRef]
- Kim, T.; Park, D. Molecular cloning and characterization of a novel mouse betaPix isoform. Mol. Cells 2001, 11, 89–94. [Google Scholar]
- Takenaka, N.; Nihata, Y.; Satoh, T. Rac1 Activation Caused by Membrane Translocation of a Guanine Nucleotide Exchange Factor in Akt2-Mediated Insulin Signaling in Mouse Skeletal Muscle. PLoS One 2016, 11, e0155292. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, G.; Closson, V.; Camonis, J.; Honoré, N.; Rousseau-Merck, M.F.; Tavitian, A.; Olofsson, B. RhoGDI-3 is a new GDP dissociation inhibitor (GDI). Identification of a non-cytosolic GDI protein interacting with the small GTP-binding proteins RhoB and RhoG. J. Biol. Chem. 1996, 271, 30366–30374. [Google Scholar] [CrossRef]
- Adra, C.N.; Manor, D.; Ko, J.L.; Zhu, S.; Horiuchi, T.; Van Aelst, L.; Cerione, R.A.; Lim, B. RhoGDI: A GDP-dissociation inhibitor for Rho proteins with preferential expression in brain and pancreas. Proc. Natl. Acad. Sci. 1997, 94, 4279–4284. [Google Scholar] [CrossRef]
- Møller, L.L.V.; Davey, J.; Jedrychowski, M.; Qian, H.; Nielsen, J.; Ørtenblad, N.; Han, X.; Jensen, T.E.; Højlund, K.; Wojtaszewski, J.F.P.; et al. RhoGDIα is a negative regulator of Rac1 activity and glucose metabolism in skeletal muscle (Unpublished work).
- Wang, Z.; Thurmond, D.C. Differential phosphorylation of RhoGDI mediates the distinct cycling of Cdc42 and Rac1 to regulate second-phase insulin secretion. J. Biol. Chem. 2010, 285, 6186–6197. [Google Scholar] [CrossRef] [PubMed]
- DerMardirossian, C.; Rocklin, G.; Seo, J.-Y.; Bokoch, G.M. Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol. Biol. Cell 2006, 17, 4760–4768. [Google Scholar] [CrossRef] [PubMed]
- Tsakiridis, T.; Taha, C.; Grinsteinl, S.; Klip, A. Insulin activates a p21-activated kinase in muscle cells via phosphatidylinositol 3-kinase. J. Biol. Chem. 1996, 271, 19664–19667. [Google Scholar] [CrossRef]
- Tunduguru, R.; Chiu, T.T.; Ramalingam, L.; Elmendorf, J.S.; Klip, A.; Thurmond, D.C. Signaling of the p21-activated kinase (PAK1) coordinates insulin-stimulated actin remodeling and glucose uptake in skeletal muscle cells. Biochem. Pharmacol. 2014, 92, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Moller, L.L.V.; Jaurji, M.; Nielsen, I.L.; Joseph, G.A.; Kjobsted, R.; Knudsen, J.R.; Madsen, A.B.; Schjerling, P.; Jensen, T.E.; Krauss, R.S.; et al. The role of p-21 activated kinases (PAKs) in glucose homeostasis and skeletal muscle glucose uptake. bioRxiv 2019, 543736. [Google Scholar]
- Joseph, G.A.; Hung, M.; Goel, A.J.; Hong, M.; Rieder, M.-K.; Beckmann, N.D.; Serasinghe, M.N.; Chipuk, J.E.; Devarakonda, P.M.; Goldhamer, D.J.; et al. Late-onset megaconial myopathy in mice lacking group I Paks. Skelet. Muscle 2019, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lee, D.-H.; Zabolotny, J.M.; Kim, Y.-B. Metabolic actions of Rho-kinase in periphery and brain. Trends Endocrinol. Metab. 2013, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Lee, D.H.; Shi, J.; Jeoung, N.H.; Kim, M.S.; Zabolotny, J.M.; Lee, S.W.; White, M.F.; Wei, L.; Kim, Y.-B. Targeted Disruption of ROCK1 Causes Insulin Resistance in Vivo. J. Biol. Chem. 2009, 284, 11776–11780. [Google Scholar] [CrossRef]
- Chun, K.-H.; Araki, K.; Jee, Y.; Lee, D.-H.; Oh, B.-C.; Huang, H.; Park, K.S.; Lee, S.W.; Zabolotny, J.M.; Kim, Y.-B. Regulation of Glucose Transport by ROCK1 Differs from That of ROCK2 and Is Controlled by Actin Polymerization. Endocrinology 2012, 153, 1649–1662. [Google Scholar] [CrossRef]
- Kanda, T.; Wakino, S.; Homma, K.; Yoshioka, K.; Tatematsu, S.; Hasegawa, K.; Takamatsu, I.; Sugano, N.; Hayashi, K.; Saruta, T. Rho-kinase as a molecular target for insulin resistance and hypertension. FASEB J. 2006, 20, 169–171. [Google Scholar] [CrossRef]
- Noda, K.; Nakajima, S.; Godo, S.; Saito, H.; Ikeda, S.; Shimizu, T.; Enkhjargal, B.; Fukumoto, Y.; Tsukita, S.; Yamada, T.; et al. Rho-Kinase Inhibition Ameliorates Metabolic Disorders through Activation of AMPK Pathway in Mice. PLoS One 2014, 9, e110446. [Google Scholar] [CrossRef]
- Tao, W.; Wu, J.; Xie, B.X.; Zhao, Y.Y.; Shen, N.; Jiang, S.; Wang, X.X.; Xu, N.; Jiang, C.; Chen, S.; et al. Lipid-induced muscle insulin resistance is mediated by GGPPS via modulation of the RhoA/Rho kinase signaling pathway. J. Biol. Chem. 2015, 290, 20086–20097. [Google Scholar] [CrossRef]
- Kramer, H.F.; Witczak, C.A.; Taylor, E.B.; Fujii, N.; Hirshman, M.F.; Goodyear, L.J. AS160 Regulates Insulin- and Contraction-stimulated Glucose Uptake in Mouse Skeletal Muscle. J. Biol. Chem. 2006, 281, 31478–31485. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef]
- Delaney, M.K.; Liu, J.; Zheng, Y.; Berndt, M.C.; Du, X. The role of Rac1 in glycoprotein Ib-IX-mediated signal transduction and integrin activation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2761–2768. [Google Scholar] [CrossRef]
- Ijuin, T.; Takenawa, T. Regulation of Insulin Signaling by the Phosphatidylinositol 3,4,5-Triphosphate Phosphatase SKIP through the Scaffolding Function of Pak1. Mol. Cell. Biol. 2012, 32, 3570–3584. [Google Scholar] [CrossRef]
- Takenaka, N.; Yasuda, N.; Nihata, Y.; Hosooka, T.; Noguchi, T.; Aiba, A.; Satoh, T. Role of the guanine nucleotide exchange factor in Akt2-mediated plasma membrane translocation of GLUT4 in insulin-stimulated skeletal muscle. Cell. Signal. 2014, 26, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, N.; Izawa, R.; Wu, J.; Kitagawa, K.; Nihata, Y.; Hosooka, T.; Noguchi, T.; Ogawa, W.; Aiba, A.; Satoh, T. A critical role of the small GTPase Rac1 in Akt2-mediated GLUT4 translocation in mouse skeletal muscle. FEBS J. 2014, 281, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, N.; Araki, N.; Satoh, T. Involvement of the protein kinase Akt2 in insulin-stimulated Rac1 activation leading to glucose uptake in mouse skeletal muscle. PLoS One 2019, 14, e0212219. [Google Scholar] [CrossRef]
- Chiu, T.T.; Sun, Y.; Koshkina, A.; Klip, A. Rac-1 superactivation triggers insulin-independent glucose transporter 4 (GLUT4) translocation that bypasses signaling defects exerted by c-Jun N-terminal kinase (JNK)- and ceramide-induced insulin resistance. J. Biol. Chem. 2013, 288, 17520–17531. [Google Scholar] [CrossRef]
- Sylow, L.; Richter, E.S. Current advances in our understanding of exercise as medicine in metabolic disease. Curr. Opin. Physiol. 2019. [Google Scholar] [CrossRef]
- Boulé, N.G.; Haddad, E.; Kenny, G.P.; Wells, G.A.; Sigal, R.J. Effects of Exercise on Glycemic Control and Body Mass in Type 2 Diabetes Mellitus. JAMA 2001, 286, 1218. [Google Scholar] [CrossRef] [PubMed]
- Dela, F.; Larsen, J.J.; Mikines, K.J.; Ploug, T.; Petersen, L.N.; Galbo, H. Insulin-Stimulated Muscle Glucose Clearance in Patients With NIDDM: Effects of One-Legged Physical Training. Diabetes 1995, 44, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Fatone, C.; Guescini, M.; Balducci, S.; Battistoni, S.; Settequattrini, A.; Pippi, R.; Stocchi, L.; Mantuano, M.; Stocchi, V.; De Feo, P. Two weekly sessions of combined aerobic and resistance exercise are sufficient to provide beneficial effects in subjects with Type 2 diabetes mellitus and metabolic syndrome. J. Endocrinol. Invest. 2010, 33, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Diabetes Prevention Program Research Group Reduction in the Incidence of Type 2 Diabetes with Lifestyle Intervention or Metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [PubMed]
- Snowling, N.J.; Hopkins, W.G. Effects of Different Modes of Exercise Training on Glucose Control and Risk Factors for Complications in Type 2 Diabetic Patients: A meta-analysis. Diabetes Care 2006, 29, 2518–2527. [Google Scholar] [CrossRef]
- Kiens, B. Skeletal Muscle Lipid Metabolism in Exercise and Insulin Resistance. Physiol. Rev. 2006, 86, 205–243. [Google Scholar] [CrossRef]
- Sylow, L.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Exercise-stimulated glucose uptake — regulation and implications for glycaemic control. Nat. Rev. Endocrinol. 2017, 13, 133–148. [Google Scholar] [CrossRef]
- Ploug, T.; Galbo, H.; Richter, E.A. Increased muscle glucose uptake during contractions: No need for insulin. Am. J. Physiol. Metab. 1984, 247, E726–E731. [Google Scholar] [CrossRef]
- Wallberg-Henriksson, H.; Holloszy, J.O. Contractile activity increases glucose uptake by muscle in severely diabetic rats. J. Appl. Physiol. 1984, 57, 1045–1049. [Google Scholar] [CrossRef]
- Martin, I.K.; Katz, A.; Wahren, J. Splanchnic and muscle metabolism during exercise in NIDDM patients. Am. J. Physiol. Metab. 1995, 269, E583–E590. [Google Scholar] [CrossRef]
- Sylow, L.; Jensen, T.E.; Kleinert, M.; Mouatt, J.R.; Maarbjerg, S.J.; Jeppesen, J.; Prats, C.; Chiu, T.T.; Boguslavsky, S.; Klip, A.; et al. Rac1 Is a Novel Regulator of Contraction-Stimulated Glucose Uptake in Skeletal Muscle. Diabetes 2013, 62, 1139–1151. [Google Scholar] [CrossRef]
- Sylow, L.; Nielsen, I.L.; Kleinert, M.; Møller, L.L.V.; Ploug, T.; Schjerling, P.; Bilan, P.J.; Klip, A.; Jensen, T.E.; Richter, E.A. Rac1 governs exercise-stimulated glucose uptake in skeletal muscle through regulation of GLUT4 translocation in mice. J. Physiol. 2016, 594, 4997–5008. [Google Scholar] [CrossRef]
- Sylow, L.; Møller, L.L.V.; Kleinert, M.; D’Hulst, G.; De Groote, E.; Schjerling, P.; Steinberg, G.R.; Jensen, T.E.; Richter, E.A. Rac1 and AMPK Account for the Majority of Muscle Glucose Uptake Stimulated by Ex Vivo Contraction but Not In Vivo Exercise. Diabetes 2017, 66, 1548–1559. [Google Scholar] [CrossRef]
- Hu, F.; Li, N.; Li, Z.; Zhang, C.; Yue, Y.; Liu, Q.; Chen, L.; Bilan, P.J.; Niu, W. Electrical pulse stimulation induces GLUT4 translocation in a Rac-Akt-dependent manner in C2C12 myotubes. FEBS Lett. 2018, 592, 644–654. [Google Scholar] [CrossRef]
- Henriquez-Olguin, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; Jensen, T. Exercise-stimulated muscle ROS production and glucose uptake requires NADPH oxidase 2. bioRxiv 2019. [Google Scholar] [CrossRef]
- Muñoz, V.R.; Gaspar, R.C.; Kuga, G.K.; da Rocha, A.L.; Crisol, B.M.; Botezelli, J.D.; Baptista, I.L.; Mekary, R.A.; da Silva, A.S.R.; Cintra, D.E.; et al. Exercise increases Rho-kinase activity and insulin signaling in skeletal muscle. J. Cell. Physiol. 2018, 233, 4791–4800. [Google Scholar] [CrossRef]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef]
- Chiang, S.-H.; Baumann, C.A.; Kanzaki, M.; Thurmond, D.C.; Watson, R.T.; Neudauer, C.L.; Macara, I.G.; Pessin, J.E.; Saltiel, A.R. Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature 2001, 410, 944–948. [Google Scholar] [CrossRef]
- Watson, R.T.; Shigematsu, S.; Chiang, S.H.; Mora, S.; Kanzaki, M.; Macara, I.G.; Saltiel, A.R.; Pessin, J.E. Lipid raft microdomain compartmentalization of TC10 is required for insulin signaling and GLUT4 translocation. J. Cell Biol. 2001, 154, 829–840. [Google Scholar] [CrossRef]
- Usui, I.; Imamura, T.; Huang, J.; Satoh, H.; Olefsky, J.M. Cdc42 Is a Rho GTPase Family Member That Can Mediate Insulin Signaling to Glucose Transport in 3T3-L1 Adipocytes. J. Biol. Chem. 2003, 278, 13765–13774. [Google Scholar] [CrossRef]
- Takenaka, N.; Nihata, Y.; Ueda, S.; Satoh, T. In situ detection of the activation of Rac1 and RalA small GTPases in mouse adipocytes by immunofluorescent microscopy following in vivo and ex vivo insulin stimulation. Cell. Signal. 2017, 39, 108–117. [Google Scholar] [CrossRef]
- Karnam, P.; Standaert, M.L.; Galloway, L.; Farese, R. V Activation and translocation of Rho (and ADP ribosylation factor) by insulin in rat adipocytes: Apparent involvement of phosphatidylinositol 3- kinase. J. Biol. Chem. 1997, 272, 6136–6140. [Google Scholar] [CrossRef]
- Standaert, M.; Bandyopadhyay, G.; Galloway, L.; Ono, Y.; Mukai, H.; Farese, R. Comparative effects of GTPgammaS and insulin on the activation of Rho, phosphatidylinositol 3-kinase, and protein kinase N in rat adipocytes. Relationship to glucose transport. J. Biol. Chem. 1998, 273, 7470–7477. [Google Scholar] [CrossRef]
- Marcusohn, J.; Isakoff, S.J.; Rose, E.; Symons, M.; Skolnik, E.Y. The GTP-binding protein Rac does not couple PI 3-kinase to insulin-stimulated glucose transport in adipocytes. Curr. Biol. 1995, 5, 1296–1302. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Chawla, A.; Bose, A.; Way, M.; Czech, M.P. A Phosphatidylinositol 3-Kinase-independent Insulin Signaling Pathway to N-WASP/Arp2/3/F-actin Required for GLUT4 Glucose Transporter Recycling. J. Biol. Chem. 2002, 277, 509–515. [Google Scholar] [CrossRef]
- Kanzaki, M.; Watson, R.T.; Hou, J.C.; Stamnes, M.; Saltiel, A.R.; Pessin, J.E. Small GTP-binding Protein TC10 Differentially Regulates Two Distinct Populations of Filamentous Actin in 3T3L1 Adipocytes. Mol. Biol. Cell 2002, 13, 2334–2346. [Google Scholar] [CrossRef]
- Lopez, J.A.; Burchfield, J.G.; Blair, D.H.; Mele, K.; Ng, Y.; Vallotton, P.; James, D.E.; Hughes, W.E. Identification of a Distal GLUT4 Trafficking Event Controlled by Actin Polymerization. Mol. Biol. Cell 2009, 20, 3918–3929. [Google Scholar] [CrossRef]
- Stierwalt, H.D.; Ehrlicher, S.E.; Bergman, B.C.; Robinson, M.M.; Newsom, S.A. Insulin-stimulated Rac1-GTP binding is not impaired by palmitate treatment in L6 myotubes. Physiol. Rep. 2018, 6, e13956. [Google Scholar] [CrossRef]
- Raun, S.H.; Ali, M.; Kjøbsted, R.; Møller, L.L.V.; Federspiel, M.A.; Richter, E.A.; Jensen, T.E.; Sylow, L. Rac1 muscle knockout exacerbates the detrimental effect of high-fat diet on insulin-stimulated muscle glucose uptake independently of Akt. J. Physiol. 2018, 596, 2283–2299. [Google Scholar] [CrossRef]
- Noda, K.; Godo, S.; Saito, H.; Tsutsui, M. Opposing Roles of Nitric Oxide and Rho-Kinase in Lipid Metabolism in Mice. Tohoku J. Exp. Med. 2015, 235, 171–183. [Google Scholar] [CrossRef][Green Version]
- Tang, S.; Wu, W.; Tang, W.; Ge, Z.; Wang, H.; Hong, T.; Zhu, D.; Bi, Y. Suppression of Rho-kinase 1 is responsible for insulin regulation of the AMPK/SREBP-1c pathway in skeletal muscle cells exposed to palmitate. Acta Diabetol. 2017, 54, 635–644. [Google Scholar] [CrossRef]
- Chun, K.-H.; Choi, K.-D.; Lee, D.-H.; Jung, Y.; Henry, R.R.; Ciaraldi, T.P.; Kim, Y.-B. In vivo activation of ROCK1 by insulin is impaired in skeletal muscle of humans with type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E536–E542. [Google Scholar] [CrossRef]
- Gordon, S.E.; Flück, M.; Booth, F.W. Selected Contribution: Skeletal muscle focal adhesion kinase, paxillin, and serum response factor are loading dependent. J. Appl. Physiol. 2001, 90, 1174–1183. [Google Scholar] [CrossRef]
- Sakuma, K.; Nakao, R.; Inashima, S.; Hirata, M.; Kubo, T.; Yasuhara, M. Marked reduction of focal adhesion kinase, serum response factor and myocyte enhancer factor 2C, but increase in RhoA and myostatin in the hindlimb dy mouse muscles. Acta Neuropathol. 2004, 108, 241–249. [Google Scholar] [CrossRef]
- McClung, J.M.; Thompson, R.W.; Lowe, L.L.; Carson, J.A. RhoA expression during recovery from skeletal muscle disuse. J. Appl. Physiol. 2004, 96, 1341–1348. [Google Scholar] [CrossRef]
- Tsai, F.C.; Pai, M.H.; Chiu, C.C.; Chou, C.M.; Hsieh, M.S. Denervation dynamically regulates integrin α7 signaling pathways and microscopic structures in rats. J. Trauma 2011, 70, 220–227. [Google Scholar] [CrossRef]
- Sakuma, K.; Watanabe, K.; Hotta, N.; Koike, T.; Ishida, K.; Katayama, K.; Akima, H. The adaptive responses in several mediators linked with hypertrophy and atrophy of skeletal muscle after lower limb unloading in humans. Acta Physiol. 2009, 197, 151–159. [Google Scholar] [CrossRef]
- McClung, J.M.; Lee, W.J.; Thompson, R.W.; Lowe, L.L.; Carson, J.A. RhoA induction by functional overload and nandrolone decanoate administration in rat skeletal muscle. Pflugers Arch. Eur. J. Physiol. 2003, 447, 345–355. [Google Scholar] [CrossRef]
- Lamon, S.; Wallace, M.A.; Léger, B.; Russell, A.P. Regulation of STARS and its downstream targets suggest a novel pathway involved in human skeletal muscle hypertrophy and atrophy. J. Physiol. 2009, 587, 1795–1803. [Google Scholar] [CrossRef]
- Lang, F.; Aravamudhan, S.; Nolte, H.; Türk, C.; Hölper, S.; Müller, S.; Günther, S.; Blaauw, B.; Braun, T.; Krüger, M. Dynamic changes in the mouse skeletal muscle proteome during denervation-induced atrophy. Dis. Model. Mech. 2017, 10, 881–896. [Google Scholar] [CrossRef]
- Chockalingam, P.S.; Cholera, R.; Oak, S.A.; Zheng, Y.; Jarrett, H.W.; Thomason, D.B. Dystrophin-glycoprotein complex and Ras and Rho GTPase signaling are altered in muscle atrophy. Am. J. Physiol. 2002, 283, C500–C511. [Google Scholar] [CrossRef]
- Fernandes, J.J.; Atreya, K.B.; Desai, K.M.; Hall, R.E.; Patel, M.D.; Desai, A.A.; Benham, A.E.; Mable, J.L.; Straessle, J.L. A dominant negative form of Rac1 affects myogenesis of adult thoracic muscles in Drosophila. Dev. Biol. 2005, 285, 11–27. [Google Scholar] [CrossRef]
- Joseph, G.A.; Lu, M.; Radu, M.; Lee, J.K.; Burden, S.J.; Chernoff, J.; Krauss, R.S. Group I Paks Promote Skeletal Myoblast Differentiation In Vivo and In Vitro. Mol. Cell. Biol. 2017, 37, e00222-16. [Google Scholar] [CrossRef]
- Cerquone Perpetuini, A.; Re Cecconi, A.D.; Chiappa, M.; Martinelli, G.B.; Fuoco, C.; Desiderio, G.; Castagnoli, L.; Gargioli, C.; Piccirillo, R.; Cesareni, G. Group I Paks support muscle regeneration and counteract cancer-associated muscle atrophy. J. Cachexia Sarcopenia Muscle 2018. [Google Scholar] [CrossRef]
- Han, X.; Møller, L.L.V.; Groote, E.D.; Bojsen-Møller, K.N.; Davey, J.; Henríquez-Olguin, C.; Li, Z.; Knudsen, J.R.; Jensen, T.E.; Madsbad, S.; et al. Mechanisms involved in follistatin-induced increased insulin action in skeletal muscle. bioRxiv 2019, 568097. [Google Scholar] [CrossRef]
- Barbé, C.; Bray, F.; Gueugneau, M.; Devassine, S.; Lause, P.; Tokarski, C.; Rolando, C.; Thissen, J.-P. Comparative Proteomic and Transcriptomic Analysis of Follistatin-Induced Skeletal Muscle Hypertrophy. J. Proteome Res. 2017, 16, 3477–3490. [Google Scholar] [CrossRef]
- Leong, D.P.; Teo, K.K.; Rangarajan, S.; Lopez-Jaramillo, P.; Avezum, A.; Orlandini, A.; Seron, P.; Ahmed, S.H.; Rosengren, A.; Kelishadi, R.; et al. Prognostic value of grip strength: Findings from the Prospective Urban Rural Epidemiology (PURE) study. Lancet 2015, 386, 266–273. [Google Scholar] [CrossRef]
- Sakuma, K.; Akiho, M.; Nakashima, H.; Akima, H.; Yasuhara, M. Age-related reductions in expression of serum response factor and myocardin-related transcription factor A in mouse skeletal muscles. Biochim. Biophys. Acta 2008, 1782, 453–461. [Google Scholar] [CrossRef]
- Murgia, M.; Toniolo, L.; Nagaraj, N.; Ciciliot, S.; Vindigni, V.; Schiaffino, S.; Reggiani, C.; Mann, M. Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017, 19, 2396–2409. [Google Scholar] [CrossRef]
- Muñoz, V.R.; Gaspar, R.C.; Kuga, G.K.; Pavan, I.C.B.; Simabuco, F.M.; da Silva, A.S.R.; de Moura, L.P.; Cintra, D.E.; Ropelle, E.R.; Pauli, J.R. The Effects of Aging on Rho-Kinase and Insulin Signaling in Skeletal Muscle and White Adipose Tissue of Rats. J. Gerontol. A. Biol. Sci. Med. Sci 2018. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 2018, 4, 17105. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Møller, L.L.V.; Klip, A.; Sylow, L. Rho GTPases—Emerging Regulators of Glucose Homeostasis and Metabolic Health. Cells 2019, 8, 434. https://doi.org/10.3390/cells8050434
Møller LLV, Klip A, Sylow L. Rho GTPases—Emerging Regulators of Glucose Homeostasis and Metabolic Health. Cells. 2019; 8(5):434. https://doi.org/10.3390/cells8050434
Chicago/Turabian StyleMøller, Lisbeth Liliendal Valbjørn, Amira Klip, and Lykke Sylow. 2019. "Rho GTPases—Emerging Regulators of Glucose Homeostasis and Metabolic Health" Cells 8, no. 5: 434. https://doi.org/10.3390/cells8050434
APA StyleMøller, L. L. V., Klip, A., & Sylow, L. (2019). Rho GTPases—Emerging Regulators of Glucose Homeostasis and Metabolic Health. Cells, 8(5), 434. https://doi.org/10.3390/cells8050434