mTOR Signalling in Head and Neck Cancer: Heads Up



1
Division of Cancer Research, Peter MacCallum Cancer Centre, Grattan Street, Melbourne, Victoria 3000, Australia
2
Univ Lyon, Université Claude Bernard Lyon 1, INSERM 1052, CNRS 5286, Centre Léon Bérard, Centre de recherche en cancérologie de Lyon, 69008 Lyon, France
3
Department of Medical Oncology, Centre Léon Bérard, 69008 Lyon, France
4
Sir Peter MacCallum Department of Oncology, The University of Melbourne, Parkville, Victoria 3052, Australia
*
Author to whom correspondence should be addressed.
Cells 2019, 8(4), 333; https://doi.org/10.3390/cells8040333
Submission received: 9 March 2019
/
Revised: 8 April 2019
/
Accepted: 9 April 2019
/
Published: 9 April 2019
(This article belongs to the Special Issue mTOR Signaling in Metabolism and Cancer)
Abstract
:The mammalian target of rapamycin (mTOR) signalling pathway is a central regulator of metabolism in all cells. It senses intracellular and extracellular signals and nutrient levels, and coordinates the metabolic requirements for cell growth, survival, and proliferation. Genetic alterations that deregulate mTOR signalling lead to metabolic reprogramming, resulting in the development of several cancers including those of the head and neck. Gain-of-function mutations in EGFR, PIK3CA, and HRAS, or loss-of-function in p53 and PTEN are often associated with mTOR hyperactivation, whereas mutations identified from The Cancer Genome Atlas (TCGA) dataset that potentially lead to aberrant mTOR signalling are found in the EIF4G1, PLD1, RAC1, and SZT2 genes. In this review, we discuss how these mutant genes could affect mTOR signalling and highlight their impact on metabolic processes, as well as suggest potential targets for therapeutic intervention, primarily in head and neck cancer.
1. Background
Head and neck squamous cell carcinoma (HNSCC) is currently the sixth most frequently diagnosed malignancy worldwide [1]. It is the most common cancer of the head and neck, with anatomic subsites spanning the oral cavity, nasopharynx, larynx, oropharynx, and hypopharynx. HNSCC is a heterogeneous disease that harbours complex genetic defects. While the specific multiple risk factors for the development of HNSCC differ depending on the cancer site, chronic tobacco use and alcohol abuse are historically recognised as the main promoting factors associated with the overall occurrence of HNSCC [2,3,4]. Infection with high-risk human papillomaviruses (HPV) has also emerged as a risk factor for a subset of HNSCC (~25% of cases) but has more of a profound role in the development of oropharyngeal cancer [5,6,7]. Nonetheless, HPV positive HNSCCs are shown to have better prognosis compared to HPV negative patients (70–80% versus 25–40%) [8,9,10]. The standard of care for HNSCC patients involves surgery, radiation therapy, chemotherapy and most recently, targeted therapy and immunotherapy. However, these therapies are usually administered in the absence of accurate biomarkers of response, which often leads to treatment resistance, higher systemic toxicities and, in some cases, results in morbidity and mortality. Currently, the only Food and Drug Administration (FDA)-approved targeted therapy for recurrent or metastatic HNSCC patients is cetuximab, a monoclonal antibody that specifically binds and inhibits the epidermal growth factor receptor (EGFR). However, only ~10% of patients demonstrated a beneficial response to cetuximab therapy, while the remainder were at higher risk of relapse [11,12]. Pembrolizumab, an immune checkpoint inhibitor that targets tumour cells expressing high levels of PD-L1 has also been FDA-approved for the treatment of patients with recurrent or metastatic HNSCC. Unfortunately, the rate of pembrolizumab responders is also quite low (~20%) in the absence of patient stratification [13,14,15], and a significant proportion of patients may experience increased tumour growth kinetics (hyperprogressive disease) [16,17]. While significant advances in optimising therapeutic responses have been made, the five-year survival rate has remained between 25 and 60% [18]. Genetic alterations and complex signalling pathways have been shown to drive treatment resistance, allowing for continuous cancer cell survival and proliferation. These mechanisms render most HNSCC patients hard to cure, and therefore there is a need to identify biomarkers of treatment response that will serve to tailor treatment regimens in specific subsets of HNSCC patients.
2. Genomic Alterations in Head and Neck Cancer
The recent application of next generation sequencing to study patient cancer genomes has revolutionized medical oncology. In silico analyses provide great insights into the diverse genomic alterations within each cancer sample, allowing for a functional understanding of the drivers behind deregulated oncogenic pathways and biological mechanisms involved in cancer progression. Importantly, these approaches are being exploited to potentially personalise suitable treatment regimens, tailored towards targeting key oncogenic drivers based on the individual’s mutational profile. On this basis, The Cancer Genome Atlas (TCGA) has been extensively interrogated for a comprehensive genomic characterization of HNSCC, whereby several reports have identified hundreds of mutations in each cancer subtype [19,20,21,22]. This has resulted in common dysregulated pathways being identified across most HNSCC patients. Multiple genetic and epigenetic alterations, including point mutations, deletions, promoter methylation, and oncogene amplification, are strongly triggered by chronic exposure to the major risk factors associated with HNSCC development. Some of the mutated genes frequently associated with HNSCC are TP53, CDKN2A, FAT1, NOTCH1, EGFR, HRAS, and PI3KCA [23,24,25,26,27], and the mutations in these genes are recognised as drivers of tumour development and progression. In HPV+ subtype patients, high-risk HPV infection has been associated with the abnormal expression of proteins associated with cell cycle regulation, including p53 and p16 (CDKN2A) [21,28,29]. Functional TP53 inhibits the mammalian target of rapamycin (mTOR) pathway through AMPK in response to cellular stresses and DNA damage [21]. On the other hand, aberrant TP53 allows for persistent mTOR activity and has been associated with poor survival in HNSCC patients [30].
Furthermore, FAT1 is involved in the migration and invasion of HNSCC cells through the activation of the β-catenin pathway [31]. In addition, Notch1 has been reported to play a bimodal role as a tumour promoter and tumour suppressor [22,32,33]. Overall, genetic alterations in the above-mentioned genes and in EGFR and HRAS often lead to aberrant signalling and deregulation of important proto-oncogenic networks, such as the PI3K–mTOR pathway. For instance, PIK3CA mutations that directly activate the PI3K–mTOR signalling pathway have been reported in HNSCC at rates ranging from 2.6% to 19% [21]. Overall, genetic amplifications and overexpression of key proteins responsible for driving mTOR activation underlie the tumour progression that is often observed in cancers, including HNSCC. This review provides a comprehensive analysis of the driver mutations that lead to aberrant mTOR signalling in HNSCC and assesses a number of contemporary inhibitors.
3. The mTOR Complex and the Cellular Metabolism
The mammalian target of rapamycin (mTOR) is a serine-threonine kinase that senses growth factor cues, nutrient and oxygen status, and directs appropriate changes to maintain cellular and tissue homeostasis (Figure 1). The mTOR signalling pathway is recognised as a key driver and regulator of cell growth and proliferation, cell survival, metabolism, and protein synthesis. mTOR belongs to the phospho-inositide 3-kinase (PI3K)-related kinase family, and consists of two distinct complexes; mTOR complex 1 (mTORC1) and complex 2 (mTORC2). Whilst both complexes are tightly regulated in a normal context, they are often deregulated in multiple disease-associated metabolic alterations and in cancer development [34,35,36]. In normal conditions, activation of mTOR signalling occurs in response to the binding of specific growth factors to their cognate receptor tyrosine kinases (RTKs), including insulin-like growth factor (IGF), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and epidermal growth factor (EGF) [37]. The ligand-activated receptors recruit PI3K, which converts phosphatidylinositol bisphosphate (PIP2) to phosphatidylinositol triphosphate (PIP3), and provides binding sites for phosphoinositide-dependent protein kinase 1 (PDK1). PDK1 then phosphorylates and activates AKT, which in turn phosphorylates several downstream substrates that engage multiple pathways, including mTORC1. On the other hand, mTORC2 has been known to phosphorylate and activate members of the AGC kinase family, including AKT, serum and glucocorticoid-induced kinase (SGK1), and protein kinase C (PKC), whereby inhibition of these kinases results in tumour suppression [38,39,40].
Constitutive mTOR activation is known to promote metabolic changes, including dysregulation in glucose, fatty acid, amino acid, and lipid metabolism. For instance, cancer cells largely rely on glucose as the major source of cellular energy to sustain proliferation and survival. In the context of aberrant mTOR signalling, glucose metabolism is dysregulated as a result of increased synthesis of glucose transporter proteins and glycolytic enzyme activation, followed by lactic acid fermentation even when oxygen is available—a phenomenon known as the “Warburg effect” [41,42]. The Warburg effect links the rewiring of metabolism to sustained cancer cell survival and growth, in which increased glucose uptake and fermentation of glucose to lactate are key processes [43]. In several cancers, including HNSCC, the expression of glucose transporter 1 (GLUT1) is often elevated, and in conjunction with enhanced mTOR signalling (mTORC1 and C2), the pair activates key oncogenic drivers, including c-MYC and HIF-1α [44,45,46,47]. GLUT1 is a protein of the GLUT family, responsible for glucose uptake into the cytoplasm [48]. GLUT1 is negatively regulated by glycogen synthase kinase-3 (GSK-3) that, in turn, exerts its inhibitory effects through a tuberous sclerosis complex (TSC)- and mTOR-dependent pathway [44]. Glycolysis is also upregulated through mTOR signalling via elevated Hexokinase 2 (HK2) expression, which further promotes the activation of c-MYC and HIF-1α [49]. Furthermore, the mTOR signalling stimulates fatty acid synthesis in cancer, via the persistent activation of sterol regulatory element-binding protein-1c (SREBP1c) [50,51]. Although not widely described in HNSCC, Guri et al. observed that elevated lipogenesis correlated with enhanced mTOR activity in hepatocellular carcinoma patients, which in turn facilitated energy production and cancer growth [52]. Overall, deregulated or reprogrammed mTOR signalling is a key signature of cancer cellular metabolism, while the molecular manipulation of the internal and surrounding tumour environment both initiates and sustains cancer cell survival, growth, and proliferation.
4. Deregulated mTOR Signalling in HNSCC
The mTOR pathway is known to be hyperactivated in several cancers, including HNSCC, and both mTOR complexes play essential roles in HNSCC tumorigenesis. Interestingly, mTOR deregulation in HNSCC is the most commonly seen genomic alteration (~80–90% HNSCC) involved in aberrant mitogenic signalling, compared to other known pathways such as the JAK/STAT and MAPK, which harbour mutations in less than 10% of lesions [53,54].
In vivo analyses of mTOR signalling in HNSCC are commonly studied, and chemically-induced HNSCC mouse models have long been established. These include the widely used carcinogens DMBA-TPA and 4-nitroquinoline-1-oxide (4NQO), which have both been reported to result in persistent mTOR activation, leading to tumour development, and regression is observed after the administration of the mTOR inhibitor rapamycin [55,56,57]. Furthermore, several studies have analysed the effect of rapamycin and mTOR activity in other mouse models, including anal squamous cell carcinoma (SCC) and skin and breast cancers [58,59,60]. Aside from chemically-induced models, genetic mouse models have also been established for mTOR hyper activation. For instance, Sun et al. observed that conditional deletion of Tgfbr1 and Pten in an HNSCC mouse model was associated with the development of sporadic tongue tumours that were driven by mTOR activation [61]. Furthermore, tumour burden was significantly reduced following rapamycin treatment, confirming the role of mTOR in driving HNSCC [61]. Bozec et al. analysed the effect of temsirolimus, a potent mTOR inhibitor, in combination with cetuximab and conventional chemotherapeutic agents (cisplatin and 5-fluorouracil) on orthotopic CAL33 xenografts harbouring PIK3CA mutations [62]. The combination therapy was synergistic and resulted in almost complete tumour growth arrest, further associating a profound role between tumorigenesis and mTOR activity [62]. Furthermore, it has been reported that co-targeting mTOR and PD-L1 enhances tumour growth inhibition in a syngeneic oral cancer mouse model [63].
mTORC1 interacts with the Rag GTPases, which promote its translocation and activation at the lysosomal surface in response to amino acids [64,65]. Inhibition of mTOR reduced the lysosomal efflux of essential amino acids and converted the lysosome into a cellular depot for them [66]. This process can also be deregulated in cancers where inactivating mutations of the Rag GTPases regulators can lead to hyperactivation of mTORC1, even in the absence of amino acids [67]. mTOR not only has a major role in tumour progression but also plays a role as the central regulator of autophagy. Autophagy is an intracellular process mediated by lysosomes for the breakdown and recycling of damaged cellular components (e.g., organelles, proteins) [68]. In HNSCC, the oral cavity has been known to acquire mutations that are associated with impaired autophagy and correlate with reduced overall survival [69,70]. Whilst a number of inhibitors against the PI3K/AKT/mTOR signalling pathway have undergone extensive preclinical evaluation, the specific mechanism of the action remains elusive and successfully reversing defective autophagy has been variable [71,72].
5. HPV Status, mTOR Activation and Metabolism in HNSCC
HPV infection is known to activate mTOR signalling in HNSCC and is further sustained through deregulation of metabolic pathways. For instance, HPV-positive cells utilise mitochondrial respiration, as evidenced by increased oxygen consumption in comparison to HPV-negative HNSCC cells, which exhibit increased glucose metabolism, as evidenced by the over production of lactate [73]. HPV-negative cells express HIF-1α, which is responsible for upregulating downstream mediators involved in glucose metabolism, including hexokinase II (HKII) and carbonic anhydrase IX (CAIX), while HPV-negative cells show greater expression of cytochrome c oxidase (COX) [74,75]. Moreover, as a result of increased lactate and pyruvate production, Jung et al. found that HPV-negative HNSCC cells exhibit advantageous growth, survival and radioresistance [73]. Inhibition of pyruvate dehydrogenase kinase (PDK) sensitises HPV-negative HNSCC to irradiation, which could potentially explain why those tumours are more inclined to have an unfavourable prognosis compared to HPV-positive tumours [9,10]. Therefore, both HPV-positive and HPV-negative HNSCC cells are characterised by deregulated mTOR signalling, which impairs their metabolism and thus sustains the survival and growth of cancer cells in a vicious cycle.
mTOR inhibitors have shown promising anti-cancer effects in HPV-positive HNSCC mouse models. The mTOR inhibitors Rapamycin and RAD001 reduced tumour burden in HPV-positive HNSCC xenografts through the inhibition of mTOR activity [76]. Moreover, HPV E6/E7 mouse models develop SCC lesions with high mTOR activation, and, unsurprisingly, tumour development was abolished using the mTOR inhibitor Rapamycin [56]. Despite the link between HPV infection and mTOR signalling activation with altered metabolic processes, the potential inhibition of both mTOR and HPV-deregulated pathways in HNSCC is still not well explored.
Furthermore, the E6 and E7 HPV oncoproteins are known to correlate with PIK3CA mutations or amplifications in over half of HPV-positive HNSCC, leading to drug resistance. Brand et al. showed that PI3K inhibition resulted in increased expression of the HER3 receptor and, in turn, elevated the abundance of E6 and E7 oncoproteins to promote resistance to PI3K inhibition [77]. This study also assessed the targeting of HER3 with the monoclonal antibody CDX-3379, which resulted in reduced E6 and E7 expression and enhanced the treatment efficacy of PI3K-targeted inhibition. As concluded by the authors, this suggests that co-targeting HER3 and PI3K may be an effective treatment strategy for HPV+ tumours where HER3 and HPV oncoproteins promote resistance to PI3K inhibitors. In addition, Madera et al. inhibited mTOR signalling using metformin, a ubiquitous anti-diabetic drug. This resulted in reduced tumour growth that was driven by the PIK3CA and HPV oncogenes in oral SCC (OSCC) [78].
6. Validated Mutant Genes Known to Drive Activation of mTOR Signalling in HNSCC
Mutations in EGFR, PIK3CA, and HRAS, as well as others found in potential genes such as EIF4G1, RAC1, SZT2, and PLD1, can result in aberrant mTOR signalling (Figure 2). Deregulation of mTOR signalling can equally be induced by a loss of tumour suppressors such as PTEN, APC, and NF1 [79,80,81]. A hyperactive mTORC1 engages downstream effectors through phosphorylation of the eukaryotic translation initiation factor 4E-binding proteins (4EBP-1), p70 and ribosomal protein S6 kinase (S6K1), promoting tumour development and progression [82,83,84]. In a similar manner, hyperactivation of mTORC2 drives cancer cell survival, proliferation and migration mainly through the oncogenic activation of AKT [34,85,86]. We next discuss the role of these validated and proposed genes (those that have not been well studied in HNSCC) that directly or indirectly activate mTOR signalling in HNSCC.
6.1. EGFR-PI3K-AKT-mTOR Pathway
The hyperactivation of EGFR through various epigenetic and genetic mechanisms is known to activate the PI3K-AKT-mTOR pathway. This is evident in HNSCC patient samples that show high EGFR-mTOR signalling, often associated with poor clinical outcomes [87]. A study conducted by Li et al. demonstrated that a positive feedback loop involving EGFR-mTOR and the inhibitor of nuclear factor kappa-B kinase (IKK)-NF-κB signalling regulates HNSCC cell growth [88]. One regulator of the EGFR-mTOR pathway is the AXL protein, which was shown to dimerize with and phosphorylate EGFR, and to activate phospholipase Cy (PLCy)-protein kinase C (PKC), resulting in the hyper activation of mTOR [89]. Furthermore, genetic alterations of the EGFR gene result in a common cancer-associated variant III (EGFRvIII), in several cancers. Widely studied in gliomas, the EGFRvIII is characterized by the absence of exons 2–7, leading to disruption of the ligand-binding region and is therefore constitutively active in a ligand-independent manner [90]. However, in HNSCC cases the role of EGFRvIII has remained controversial. For instance, although the sample number analysed was low, Sok et al. reported EGFRvIII to be hyperactive in >42% of HNSCC samples (14/33) [91]. However, in a recent study conducted by Khattri et al., it was established that EGFRvIII is rarely seen in HNSCC samples (2/540, 0.37%) and the clinical significance remains unclear [92]. Moreover, EGFR amplification (chromosome 7) has been reported in 11% of HNSCC cases [93,94]. Hashmi et al. observed that EGFR copy number gain occurs in oral leukoplakia and is tightly linked with an increased risk of oral cancer development [94].
Aberrant EGFR signalling was also shown to mediate aerobic glycolysis and to upregulate GLUT1 expression. In EGFR mutated lung adenocarcinoma, Makinoshima et al. found that mTOR signalling plays a crucial role in regulating glycolysis and in upregulating GLUT1 localisation [95]. In a panel of EGFR-mutated lung cancer cell lines, mTOR inhibitors significantly suppressed glycolysis and down regulation of GLUT1 by RNAi reduced cell proliferation [95]. Conversely, Chiang et al. found that mTOR signalling contributes to metabolic reprogramming in erlotinib (EGFR inhibitor) resistant lung cancer cells and strongly correlates with poor clinical outcomes of EGFR-mutated lung cancer patients [96].
The frequent activation of the EGFR pathway led to the development of EGFR inhibitors targeting receptor function to prevent downstream signalling, including mTOR activation. To date, cetuximab is the only FDA-approved EGFR inhibitor in combination with chemotherapy or with radiation therapy. Tyrosine kinase inhibitors with reversible-binding activity, such as erlotinib and gefitinib, have been disappointing in the head and neck setting, while irreversible-binding Tyrosine Kinase Inhibitors (TKI), including afatinib, appear clinically promising [97,98,99]. The combination of inhibitors targeting both mTOR and EGFR has also emerged as beneficial. For instance, combinatorial treatment targeting mTOR and EGFR has been successful in other cancers, including small cell lung cancers [100,101]. Furthermore, Bozec et al. investigated combined mTOR (temsirolimus) and EGFR (cetuximab) targeting in an orthotopic xenograft model of HNSCC, which culminated in synergistic effects against tumour growth [62]. In agreement, Lattanzio et al. observed a similar result in HNSCC cell lines [100] and Wang et al. also demonstrated reduced tumour burden in both PIK3CA- and RAS-expressing HNSCC xenografts, particularly in cetuximab resistant HNSCC cell lines [53]. Overall, targeting both EGFR and mTOR related pathways could be a promising personalised targeted therapy for HNSCC patients.
6.2. PIK3CA Mutation and PTEN Loss
Mutations that activate the catalytic unit of phosphoinositide-3-kinase (PI3K) have been implicated in several cancers, including HNSCC. Gain of function mutations of PIK3CA, the most common activator of the PI3K pathway, is detected in approximately 6–20% of HNSCC cases [21,22]. Lui et al. analysed whole-exome sequencing data from 151 tumours and revealed frequent oncogenic mutations in 30.5% (46/151) of the cases affecting the PI3K-mTOR pathway, whereas only 9.3% (14/151) and 8% (46/151) of tumours harboured mutations in the JAK/STAT or the MAPK pathways, respectively [21]. Furthermore, all tumours exhibiting PI3K pathway mutations were advanced (stage IV) cancers, implying a strong role in cancer progression.
Aberrant PI3K-mTOR signalling was shown to also regulate the properties of key cancer stem cell (CSC) factors, including the sex determining region Y box 2 (SOX2) [102,103]. SOX2 is involved in cancer stem cell (CSC) maintenance and is also associated with increased levels of CSC markers, including aldehyde dehydrogenase (ALDH1) [104]. Keysar et al. characterised the CSC from patient-derived xenografts and defined the molecular features of tumours caused by tobacco smoking and HPV infection [103]. This work unraveled the consequences of deregulated PI3K signalling, such as increased SOX2 translation and expression of ALDH, resulting in enhanced spheroid and tumour formation. This study also observed reduced SOX2 levels after silencing AKT1 (downstream of PI3K) or EIF4E (downstream of mTORC1), suggesting a direct link between SOX2 regulation and PI3K. Additionally, SOX2 knocks down suppressed ALDH transcripts and protein levels. Moreover, Suda et al. revealed that copy-number amplification of PIK3CA, within 3q (found in up to 30% of HNSCC) is associated with a poor prognosis of HNSCC patients [105] and partially overlaps with PIK3CA driving mutations. In addition, it has been shown that PIK3CA mutations are associated with an elevated uptake of glucose and glutamine in colorectal cancer [106], and a similar effect is observed in PIK3CA mutant breast cancer cells [107]. The elevated glucose and glutamine uptakes fuel the growth and progression of tumourigenicity.
Conversely, inactivation of phosphatase and tensin homologue (PTEN), a potent tumour suppressor and negative regulator of PI3K, also leads to hyperactivation of PI3K-driven mTOR signalling [108]. Although the penetrance of PTEN mutations in HNSCC ranges between 5 and 16%, loss of PTEN expression is observed in 29% of tongue cancers, and loss of heterozygosity of the PTEN locus occurs in 40% of HNSCC tumours [109,110]. Genetic alterations were even lower in SCC of the skin, in which loss of PTEN was mainly due to loss of gene transcription [111,112]. Deletion of the developmental transcription factor Grainyhead-like 3 (Grhl3) induces HNSCC in both humans and mice [111,113,114,115], and GRHL3 functions as a tumour suppressor against SCC of the skin through the direct transcriptional regulation of Pten [111,116,117]. Loss of Grhl3 leads to PTEN downregulation and the development of aggressive cutaneous SCC via the activation of the PI3K–mTOR signalling pathway [111]. Inhibition of PI3K/mTOR using BEZ235 was able to prevent the initiation as well as the promotion to malignancy of carcinogen-induced SCC, but was not efficient against the established cancer [118]. Interestingly, mutations in the PTEN gene are rare in human skin SCC and common in HNSCC, which could be a prognostic marker for patients with tongue cancer [111,114,119,120]. Moreover, suppression of PTEN in concert with other tumour suppressors, like transforming growth factor beta-receptor 1 (TGFBR1), can also contribute to deregulated PI3K-mTOR signalling. Bian et al. unraveled the relationship between TGF-β signalling and the PI3K-mTOR pathway by conditionally deleting both TGFBR1 and PTEN in HNSCC mouse models using the Cre-LoxP system. Enhanced cell proliferation and decreased apoptosis occurred, which promoted HNSCC tumour development [121].
PTEN loss also promotes cancer progression by enhancing glucose metabolism and reducing DNA repair and checkpoint pathways. Martin et al. observed PTEN loss in prostate cancer cell lines and increased pAKT expression and enhanced glucose metabolism, resulting in the survival of tumour cells [122]. Mathur et al. also observed enhanced glutamine metabolism in PTEN mutant breast cancer cells [123]. Conversely, Garcia-Cao et al. showed that transgenic overexpression of PTEN in mice decreased the levels of PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3) and glutaminase, key rate-limiting enzymes responsible for glycolysis and glutaminolysis respectively, and two important metabolic features of tumour cell growth [124]. Together, these data predict that tumours with loss of PTEN function will respond to treatment with inhibitors of glycolysis and glutaminolysis, therefore providing a potential targeted therapy for these tumours.
6.3. HRAS
HRAS belongs to the Ras oncogene family, and HRAS mutants are known to aberrantly activate mTOR signalling in HNSCC tumours [125]. The Cancer Genome Atlas analysed 279 HNSCC samples and reported that a subgroup of oral cavity tumours had favourable clinical outcomes displaying infrequent copy number alterations in conjunction with activating mutations of HRAS [20]. Nakagaki et al. utilised next-generation sequencing on a cohort of 80 Japanese OSCC patients, and identified HRAS mutations in 5% of samples [126]. Su et al. analysed whole-exome sequencing of 120 Taiwanese OSCC patients and identified 11.7% of the samples were positive for HRAS mutations [127]. Furthermore, Koumaki et al. identified HRAS mutations in 86 OSCC patients (8.6%) of Greek descent, and defined a role for HRAS in driving PI3K-mTOR signalling in OSCC [128]. Adding to the evaluations of several ethnicities, Murugan et al. identified mutant HRAS in 10 out of 56 Vietnamese OSCC patients (18%) and associated these events with an advanced tumour stage [129]. In addition to promoting mTOR activation, HRAS mutations have been shown to play a role in altering metabolic processes. Zheng et al. suggest that the HRAS transformed breast cancer cell line MCF10a, derived from an early stage cancer, exhibits a profound alteration in glucose metabolism and is strongly regulated by the oncogenic proteins HIF-1α and c-Myc [130].
Additional investigations have implicated the HRAS protein in driving resistance to therapies in HNSCC. In a recent study conducted by Ruicci et al., HRAS mutant HNSCC cell lines did not respond to PI3K inhibition (BYL719). This inhibitor induced constitutive MAPK signalling suggests feedbacks between MAPK and PI3K, resulting in persistent mTOR activity [131]. Hah et al. also outlined an association between HRAS mutations and resistance to the EGFR tyrosine kinase inhibitor erlotinib in a panel of HNSCC cell lines [132]. Likewise, Rampias et al. demonstrated that oncogenic HRAS leads to the activation of MAPK signalling, which results in resistance to cetuximab in HNSCC cells [133]. Furthermore, the authors evaluated a cohort of 55 HNSCC patients, and identified HRAS mutations in 7 out of 55 samples (12.7%) that were associated with a poorer response to Cetuximab treatment.
7. Potential Mutant Genes Activating the mTOR Signalling Pathway
7.1. EIF4G1
Eukaryotic translation initiation factor (EIF) 4 gamma 1 (EIF4G1) plays a crucial role downstream of mTOR signalling. EIF4G1 functions as a modular scaffold in the translational initiation complex, interacting with EIF3, EIF4A, EIF4E, poly (A)-binding proteins, and MNK1 [134,135]. When mTOR phosphorylates EIF4E-binding proteins (4E-BPs), it releases 4E (EIF4E) to bind to 4G1 and initiate cap-dependent translation [136]. Moreover, the downstream target of mTOR, programmed cell death 4 (PDCD4), disrupts the interaction between 4A and 4G1, leading to translational inhibition [137]. In this study, PDCD4 was shown to be downregulated by miR-21 in HNSCC, suggesting the initiation of mTOR-regulated translation by 4G1.
Aberrant over-expression of EIF4G1 is tightly linked with the prognosis of several cancers, such as lung squamous cell carcinoma, inflammatory breast cancer, cervical cancers, and nasopharyngeal carcinoma [138,139,140,141]. Although not well studied in HNSCC, the 2015 TCGA analysis of 279 HNSCC patients has provided evidence that EIF4G1 has a high alteration frequency in HNSCC, with 19% of patients harbouring EIF4G1 amplifications and 3.9% somatic mutations [20]. This is supported by studies in breast and nasopharyngeal cancers, indicating that the overexpression of 4G1 facilitates tumourigenesis, malignant transformation, and invasion, while the depletion of 4G1 remarkably leads to the inhibition of cell cycle progression, invasion, and colony formation in vitro and in vivo [139,140,142]. Although there are limited studies testing EIF4G1 inhibitors in HNSCC, small molecule inhibitors have been investigated in other cancer types. For instance, 4EGI1 has been designed to block the interaction of EIF4G1/EIF4E, resulting in decreased translation of oncogenic proteins and abrogating lung tumour growth in vivo [143,144,145,146]. Moreover, SBI-0640756 and RNA aptamers have been designed to directly target 4G1 in melanoma models [147,148]. Overall, despite limited available evidence, inhibitors of EIF4G or EIF4G1/EIF4E interactions could be emerging as novel strategies to indirectly target mTOR signalling in HNSCC.
7.2. RAC1
The Rac family of small GTPase 1 (RAC1) functions downstream of the mTOR signalling to regulate the reorganization of F-actin, lamellipodia formation, and cell motility [149]. mTORC1-mediated activation is essential to increase RAC1 expression, while mTORC2 directly facilitates RAC1 activation via the inhibition of Rho GDP dissociation inhibitor beta ARHGDIB. This results in the initiation of phosphatidylinositol-3,4,5-trisphosphate dependent rac exchange factor 1/2 (PREX1 and PREX2) and T cell lymphoma invasion and metastasis 1 (TIAM-1) expression, which all contribute to tumour growth [150,151]. Increasing evidence suggests that RAC1 modulates mTOR activity, whereby the binding of RAC1 to mTOR regulates the plasma membrane localization of the mTORC1/2 complex. This in turn promotes the phosphorylation of mTOR downstream substrates [152,153]. Collectively, RAC1 is therefore considered to be both an upstream and downstream effector of mTOR activity.
Overexpression of RAC1 is frequently observed in oral, breast, gastric, testicular, and prostate cancers and increased RAC1 expression is positively associated with cancer progression [154,155,156,157]. Aberrant RAC1 activity was shown to facilitate metastasis of colorectal and lung cancer cells by multiple mechanisms, including epithelial-mesenchymal transition (EMT), migration, and invasion [158,159]. Although not well studied in HNSCC, the TCGA database reports that 3.2% of HNSCC patients harbour RAC1 somatic mutation [20]. Moreover, persistent RAC1 overexpression has been shown to drive resistance to radio/chemotherapy [20,160,161]. In recent years, limited inhibitors targeting RAC1 have been developed. These have included EHop-016 [162] and EHT 1864, which was designed to prevent RAC1-GTP interactions and the RAC1 downstream effectors in order to block RAC1-mediated metastasis [163]. Alongside monotherapy, recent reports suggest that the combined inhibition of RAC1 and mTOR could dramatically increase treatment efficacy against renal cell carcinoma by dephosphorylating the retinoblastoma transcriptional corepressor 1 (RB1) [164]. Although the exact mechanism of the action of RAC1 inhibitors has not been thoroughly explored, their use as single or combination agents seems to have a synergistic effect with mTOR inhibition that could be considered for the treatment of HNSCC.
7.3. SZT2
Deficiency of seizure threshold 2 protein homolog (SZT2) is commonly detected in patients with intellectual disability, epilepsy, and autism [165,166,167,168]. Only recently however, SZT2 was identified as a component of KICSTOR, which negatively regulates the mTOR signalling pathway [168,169]. Interestingly, the SZT2 gene shows a relatively high somatic mutation frequency (3.6%) in HNSCC in the TCGA database. In addition, low expression of SZT2 is correlated with a low five-year survival rate of HNSCC patients [20]. Future investigation of the SZT2 function is therefore warranted to determine whether it could act as a prognostic factor for HNSCC patients and/or a possible biomarker of response to mTOR inhibitors.
7.4. PLD1
Phospholipase D1 (PLD1) is an established upstream regulator of mTOR signalling [140]. Once activated, PLD1 leads to the accumulation of phosphatidic acid (PA), resulting in mTOR activation via the ERK signaling pathway, an acquired resistance to mTORC1 inhibitors, and a feed-forward loop, resulting in constitutive PLD1 activity [170,171,172]. High PLD1 expression is frequently detected in various cancers, including glioma, pancreatic ductal adenocarcinoma, colorectal cancer, hepatocellular carcinoma, breast cancer, and melanoma [173,174,175,176,177,178]. Although PLD1 has not been extensively investigated in HNSCC, data from the TCGA show that 20% of HNSCC patients harbour copy number amplification, while 2.9% of patients harbour mutant PLD1. Based on the high percentage of its genetic alteration, we anticipate that PLD1 could function as a driver or a prognostic marker for HNSCC [20].
Multiple inhibitors targeting PLD1 have been developed, such as VU-0155069 and VU-0359595, which directly bind to the N-terminus, allosterically suppressing the catalytic activity of PLD1 [179]. In addition, inhibitors such as Fifi, ML-299, VU-0155056, and VU-0285655-1 show less selectivity by targeting both PLD1 and PLD2 [180]. Kang et al. found that inhibition of PLD1 suppresses the PI3K–mTOR pathway and results in reduced cell proliferation, migration, and invasion in vitro, as well as reduced tumour growth and EMT of patient-derived xenografts in colorectal and hepatocellular carcinoma [175,178]. Since the published literature is establishing a clear relationship between PLD1 and mTOR, further investigations are required to explore the inhibition of PLD1 for mTOR-driven malignancies, as well as the inclusion of PLD1 inhibitors in HNSCC clinical trials.
8. Current Clinical Trials Targeting mTOR in HNSCC
Because multiple mutant genes are directly associated with the oncogenic activation of the mTOR pathway, it is not surprising that multiple clinical trials are currently targeting aberrant mTOR signalling in cancer (Table 1). For instance, the multicentre Phase II trial recruited platinum/cetuximab-refractory HNSCC patients for treatment with the mTOR inhibitor temsirolimus (NCT01172769). Results from this trial indicate that in a total of 40 patients, the treatment was well tolerated, and tumour shrinkage was observed in 13/40 (39.4%) patients [181]. This study indicated that mTOR inhibition alleviates tumour burden, although further molecular analysis is required to identify predictive parameters for temsirolimus guided treatment response. Patients included in this study showed no mutations of KRAS or BRAF. Following from this trial, a Phase II study of temsirolimus in combination with carboplatin and paclitaxel has been conducted on recurrent and/or metastatic HNSCC patients. This resulted in an objective response in 15/36 (41.7%) patients and stable disease progression in 19/36 (52.3%) patients (NCT01016769) [182]. This trial confirmed that a relatively high response can be observed with combination treatment and suggests that genetic alterations associated with aberrant mTOR signalling necessitate further exploration.
Several clinical trials are currently recruiting HNSCC patients for the assessment of other mTOR pathway inhibitors. Following promising results obtained in vitro and in vivo with BYL719, a potent PI3Kα inhibitor, this drug has progressed to Phase II trials for the treatment of recurrent or metastatic HNSCC patients who have previously failed to respond to platinum-based therapy (NCT02145312) [183]. Moreover, in a large multi-centre clinical trial, the Phase II molecular analysis for therapy choice (MATCH) trial tailors personalised inhibitors to each patient’s individual mutational status (NCT02465060). This study includes HNSCC patients with mutations that activate mTOR signalling, who received the inhibitor sapanisertib, which binds to and inhibits both mTOR complexes. Of the targeted therapies related to the mTOR pathway, patients with PIK3CA mutations received the PI3K inhibitor taselisib, patients harbouring EGFR mutations received the EGFR inhibitor afatinib, while patients with loss or mutated PTEN received the PI3K-beta inhibitor GSK2636771. In addition to monotherapies, combination treatments are scheduled with the PI3K/mTOR inhibitor gedatolisib and the cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor palbociclib, and HNSCC patients are currently being recruited for this Phase I trial (NCT03065062).
Despite mTOR signalling driving aberrant metabolic processes, there is currently no clinical trial investigating combinational treatment against both mTOR and dysregulated metabolism in HNSCC patients. Nonetheless, there are several studies targeting key transporters involved in metabolism and HNSCC progression. In a recent study conducted by Mehibel et al., the authors investigated the use of simvastatin (which specifically inhibits lipid and cholesterol biosynthesis) and AZD3965 (which inhibits monocarboxylate transporter 1 and results in enhanced glycolysis) in HNSCC cell lines [184]. They found that prophylactic simvastatin lead to the upregulation of xenograft tumour MCT1 expression that effectively primed these cells for MCT1 inhibition using AZD3965. The combination of these inhibitors led to a delay in tumour growth in HNSCC xenograft models and showed no signs of toxicity. Moreover, AZD3965 has been independently assessed in pre-clinical xenograft studies for other cancer types, such as small cell lung cancer, where it was shown to reduce tumour growth via the inhibition of lactate release and glycolysis [185,186].
9. Conclusions and Perspectives
The mTOR pathway integrates multiple intrinsic genetic alterations and extrinsic cues, leading to aberrant signalling and metabolic alterations. Since the validated and potential mutant genes, as identified from the TCGA dataset, directly affect mTOR activation status in cancer, they could be used as biomarkers for response and mTOR targeted inhibition in a tissue-specific manner. In fact, multiple biomarkers for predicting drug sensitivity have been proposed, including those related to PTEN loss, PTEN mutations, NOTCH1 mutation, and EGFR expression in other cancers, but could be further established in HNSCC. Furthermore, the functional characterisation of these mutant genes and the molecular dissection of their associated oncogenic networks could provide targets for combinatorial therapies to alleviate resistance to mTOR inhibition.
Funding
This research was funded by the Australian National Health and Medical Research (NHMRC, grant numbers APP1049870, APP1106697) and The Association for International Cancer Research (AICR, 11-0060). The authors were funded by Fellowships from the Victorian Cancer Agency (Clare Oliver Memorial, COF11_04 and Mid-Career, MCRF16017) to C.D. and by the French LYriCAN grant (INCa-DGOS-Inserm_12563) to P.S.
Acknowledgments
The authors acknowledge Bryce van Denderen for his careful corrections of the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Wang, X.; Xu, J.; Wang, L.; Liu, C.; Wang, H. The role of cigarette smoking and alcohol consumption in the differentiation of oral squamous cell carcinoma for the males in China. J. Cancer Res. Ther. 2015, 11, 141–145. [Google Scholar] [CrossRef]
- Lubin, J.H.; Purdue, M.; Kelsey, K.; Zhang, Z.F.; Winn, D.; Wei, Q.; Talamini, R.; Szeszenia-Dabrowska, N.; Sturgis, E.M.; Smith, E.; et al. Total exposure and exposure rate effects for alcohol and smoking and risk of head and neck cancer: A pooled analysis of case-control studies. Am. J. Epidemiol. 2009, 170, 937–947. [Google Scholar] [CrossRef]
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol consumption and site-specific cancer risk: A comprehensive dose-response meta-analysis. Br. J. Cancer 2015, 112, 580–593. [Google Scholar] [CrossRef]
- Dalla Torre, D.; Burtscher, D.; Soelder, E.; Offermanns, V.; Rasse, M.; Puelacher, W. HPV prevalence in a Mid-European oral squamous cell cancer population: A cohort study. Oral Dis. 2018, 24, 948–956. [Google Scholar] [CrossRef]
- Young, D.; Xiao, C.C.; Murphy, B.; Moore, M.; Fakhry, C.; Day, T.A. Increase in head and neck cancer in younger patients due to human papillomavirus (HPV). Oral Oncol. 2015, 51, 727–730. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: A systematic review. Cancer Epidemiol. Biomark. Prev. 2005, 14, 467–475. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Engels, E.A.; Anderson, W.F.; Gillison, M.L. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J. Clin. Oncol. 2008, 26, 612–619. [Google Scholar] [CrossRef]
- Marur, S.; D’Souza, G.; Westra, W.H.; Forastiere, A.A. HPV-associated head and neck cancer: A virus-related cancer epidemic. Lancet Oncol. 2010, 11, 781–789. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef]
- Griffin, S.; Walker, S.; Sculpher, M.; White, S.; Erhorn, S.; Brent, S.; Dyker, A.; Ferrie, L.; Gilfillan, C.; Horsley, W.; et al. Cetuximab plus radiotherapy for the treatment of locally advanced squamous cell carcinoma of the head and neck. Health Technol. Assess. 2009, 13 Suppl. 1, 49–54. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Haddad, R.; Gupta, S.; Mahipal, A.; Mehra, R.; Tahara, M.; Berger, R.; Eder, J.P.; Burtness, B.; Lee, S.H.; et al. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results From the Phase Ib KEYNOTE-012 Expansion Cohort. J. Clin. Oncol. 2016, 34, 3838–3845. [Google Scholar] [CrossRef]
- Champiat, S.; Ferrara, R.; Massard, C.; Besse, B.; Marabelle, A.; Soria, J.C.; Ferte, C. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat. Rev. Clin. Oncol. 2018, 15, 748–762. [Google Scholar] [CrossRef]
- Saada-Bouzid, E.; Defaucheux, C.; Karabajakian, A.; Coloma, V.P.; Servois, V.; Paoletti, X.; Even, C.; Fayette, J.; Guigay, J.; Loirat, D.; et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2017, 28, 1605–1611. [Google Scholar] [CrossRef]
- Pulte, D.; Brenner, H. Changes in survival in head and neck cancers in the late 20th and early 21st century: A period analysis. Oncologist 2010, 15, 994–1001. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhang, J.; Yoo, S.Y.; Bengtsson, L.; Moorthy, S.; Neskey, D.M.; Zhao, M.; Ortega Alves, M.V.; Chang, K.; Drummond, J.; et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013, 3, 770–781. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, J.; Fu, X.; Yang, A. Identification of Key Genes and Pathways in Tongue Squamous Cell Carcinoma Using Bioinformatics Analysis. Med. Sci. Monit. 2017, 23, 5924–5932. [Google Scholar] [CrossRef]
- Cuevas Gonzalez, J.C.; Gaitan Cepeda, L.A.; Borges Yanez, S.A.; Cornejo, A.D.; Mori Estevez, A.D.; Huerta, E.R. p53 and p16 in oral epithelial dysplasia and oral squamous cell carcinoma: A study of 208 cases. Indian J. Pathol. Microbiol. 2016, 59, 153–158. [Google Scholar]
- Karpathiou, G.; Monaya, A.; Forest, F.; Froudarakis, M.; Casteillo, F.; Marc Dumollard, J.; Prades, J.M.; Peoc’h, M. p16 and p53 expression status in head and neck squamous cell carcinoma: A correlation with histological, histoprognostic and clinical parameters. Pathology 2016, 48, 341–348. [Google Scholar] [CrossRef]
- Lin, S.C.; Lin, L.H.; Yu, S.Y.; Kao, S.Y.; Chang, K.W.; Cheng, H.W.; Liu, C.J. FAT1 somatic mutations in head and neck carcinoma are associated with tumor progression and survival. Carcinogenesis 2018, 39, 1320–1330. [Google Scholar] [CrossRef]
- Lee, S.H.; Do, S.I.; Lee, H.J.; Kang, H.J.; Koo, B.S.; Lim, Y.C. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Lab. Investig. 2016, 96, 508–516. [Google Scholar] [CrossRef]
- Plath, M.; Broglie, M.A.; Forbs, D.; Stoeckli, S.J.; Jochum, W. Prognostic significance of cell cycle-associated proteins p16, pRB, cyclin D1 and p53 in resected oropharyngeal carcinoma. J. Otolaryngol. Head Neck Surg. 2018, 47, 53. [Google Scholar] [CrossRef]
- Belobrov, S.; Cornall, A.M.; Young, R.J.; Koo, K.; Angel, C.; Wiesenfeld, D.; Rischin, D.; Garland, S.M.; McCullough, M. The role of human papillomavirus in p16-positive oral cancers. J. Oral Pathol. Med.: Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2018, 47, 18–24. [Google Scholar] [CrossRef]
- Cheng, H.; Yang, X.; Si, H.; Saleh, A.D.; Xiao, W.; Coupar, J.; Gollin, S.M.; Ferris, R.L.; Issaeva, N.; Yarbrough, W.G.; et al. Genomic and Transcriptomic Characterization Links Cell Lines with Aggressive Head and Neck Cancers. Cell Rep. 2018, 25, 1332–1345.e5. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Miyazaki, T.; Nakashiro, K.; Yamagata, H.; Isokane, M.; Goda, H.; Tanaka, H.; Oka, R.; Hamakawa, H. Human FAT1 cadherin controls cell migration and invasion of oral squamous cell carcinoma through the localization of beta-catenin. Oncol. Rep. 2011, 26, 587–592. [Google Scholar]
- Yoshida, R.; Nagata, M.; Nakayama, H.; Niimori-Kita, K.; Hassan, W.; Tanaka, T.; Shinohara, M.; Ito, T. The pathological significance of Notch1 in oral squamous cell carcinoma. Lab. Investig. 2013, 93, 1068–1081. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef]
- Li, S.; Wang, Z.; Huang, J.; Cheng, S.; Du, H.; Che, G.; Peng, Y. Clinicopathological and prognostic significance of mTOR and phosphorylated mTOR expression in patients with esophageal squamous cell carcinoma: A systematic review and meta-analysis. BMC Cancer 2016, 16, 877. [Google Scholar] [CrossRef]
- Driscoll, D.R.; Karim, S.A.; Sano, M.; Gay, D.M.; Jacob, W.; Yu, J.; Mizukami, Y.; Gopinathan, A.; Jodrell, D.I.; Evans, T.R.; et al. mTORC2 Signaling Drives the Development and Progression of Pancreatic Cancer. Cancer Res. 2016, 76, 6911–6923. [Google Scholar] [CrossRef]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef]
- Tenkerian, C.; Krishnamoorthy, J.; Mounir, Z.; Kazimierczak, U.; Khoutorsky, A.; Staschke, K.A.; Kristof, A.S.; Wang, S.; Hatzoglou, M.; Koromilas, A.E. mTORC2 Balances AKT Activation and eIF2alpha Serine 51 Phosphorylation to Promote Survival under Stress. Mol. Cancer Res. 2015, 13, 1377–1388. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Buller, C.L.; Loberg, R.D.; Fan, M.H.; Zhu, Q.; Park, J.L.; Vesely, E.; Inoki, K.; Guan, K.L.; Brosius, F.C., 3rd. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am. J. Physiol. Cell Physiol. 2008, 295, C836–C843. [Google Scholar]
- Li, S.; Yang, X.; Wang, P.; Ran, X. The effects of GLUT1 on the survival of head and neck squamous cell carcinoma. Cell Physiol. Biochem. 2013, 32, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Simopoulos, C.; Turley, H.; Talks, K.; Gatter, K.C.; Harris, A.L. Hypoxia-inducible factor (HIF1A and HIF2A), angiogenesis, and chemoradiotherapy outcome of squamous cell head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2002, 53, 1192–1202. [Google Scholar] [CrossRef]
- Kleszcz, R.; Paluszczak, J.; Krajka-Kuzniak, V.; Baer-Dubowska, W. The inhibition of c-MYC transcription factor modulates the expression of glycolytic and glutaminolytic enzymes in FaDu hypopharyngeal carcinoma cells. Adv. Clin. Exp. Med. 2018, 27, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta 2013, 1835, 164–169. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, S.; Li, Y.; Tang, Z.; Kong, W. Hexokinase 2 overexpression promotes the proliferation and survival of laryngeal squamous cell carcinoma. Tumour Biol. 2014, 35, 3743–3753. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pilo, G.M.; Li, X.; Cigliano, A.; Latte, G.; Che, L.; Joseph, C.; Mela, M.; Wang, C.; Jiang, L.; et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016, 64, 333–341. [Google Scholar] [CrossRef]
- Ricoult, S.J.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823 e12. [Google Scholar] [CrossRef]
- Wang, Z.; Martin, D.; Molinolo, A.A.; Patel, V.; Iglesias-Bartolome, R.; Degese, M.S.; Vitale-Cross, L.; Chen, Q.; Gutkind, J.S. mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Amornphimoltham, P.; Leelahavanichkul, K.; Molinolo, A.; Patel, V.; Gutkind, J.S. Inhibition of Mammalian target of rapamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clin. Cancer Res. 2008, 14, 8094–8101. [Google Scholar] [CrossRef]
- Callejas-Valera, J.L.; Iglesias-Bartolome, R.; Amornphimoltham, P.; Palacios-Garcia, J.; Martin, D.; Califano, J.A.; Molinolo, A.A.; Gutkind, J.S. mTOR inhibition prevents rapid-onset of carcinogen-induced malignancies in a novel inducible HPV-16 E6/E7 mouse model. Carcinogenesis 2016, 37, 1014–1025. [Google Scholar] [CrossRef]
- Czerninski, R.; Amornphimoltham, P.; Patel, V.; Molinolo, A.A.; Gutkind, J.S. Targeting mammalian target of rapamycin by rapamycin prevents tumor progression in an oral-specific chemical carcinogenesis model. Cancer Prev. Res. 2009, 2, 27–36. [Google Scholar] [CrossRef]
- Athar, M.; Kopelovich, L. Rapamycin and mTORC1 inhibition in the mouse: Skin cancer prevention. Cancer Prev. Res. 2011, 4, 957–961. [Google Scholar] [CrossRef]
- Sun, Z.J.; Zhang, L.; Zhang, W.; Hall, B.; Bian, Y.; Kulkarni, A.B. Inhibition of mTOR reduces anal carcinogenesis in transgenic mouse model. PLoS ONE 2013, 8, e74888. [Google Scholar] [CrossRef]
- Zhang, H.; Cohen, A.L.; Krishnakumar, S.; Wapnir, I.L.; Veeriah, S.; Deng, G.; Coram, M.A.; Piskun, C.M.; Longacre, T.A.; Herrler, M.; et al. Patient-derived xenografts of triple-negative breast cancer reproduce molecular features of patient tumors and respond to mTOR inhibition. Breast Cancer Res. 2014, 16, R36. [Google Scholar] [CrossRef]
- Sun, Z.J.; Zhang, L.; Hall, B.; Bian, Y.; Gutkind, J.S.; Kulkarni, A.B. Chemopreventive and chemotherapeutic actions of mTOR inhibitor in genetically defined head and neck squamous cell carcinoma mouse model. Clin. Cancer Res. 2012, 18, 5304–5313. [Google Scholar] [CrossRef]
- Bozec, A.; Ebran, N.; Radosevic-Robin, N.; Sudaka, A.; Monteverde, M.; Toussan, N.; Etienne-Grimaldi, M.C.; Nigro, C.L.; Merlano, M.; Penault-Llorca, F.; et al. Combination of mTOR and EGFR targeting in an orthotopic xenograft model of head and neck cancer. Laryngoscope 2016, 126, E156–E163. [Google Scholar] [CrossRef]
- Moore, E.C.; Cash, H.A.; Caruso, A.M.; Uppaluri, R.; Hodge, J.W.; Van Waes, C.; Allen, C.T. Enhanced Tumor Control with Combination mTOR and PD-L1 Inhibition in Syngeneic Oral Cavity Cancers. Cancer Immunol. Res. 2016, 4, 611–620. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef]
- Cosway, B.; Lovat, P. The role of autophagy in squamous cell carcinoma of the head and neck. Oral Oncol. 2016, 54, 1–6. [Google Scholar] [CrossRef]
- Liu, J.L.; Chen, F.F.; Lung, J.; Lo, C.H.; Lee, F.H.; Lu, Y.C.; Hung, C.H. Prognostic significance of p62/SQSTM1 subcellular localization and LC3B in oral squamous cell carcinoma. Br. J. Cancer 2014, 111, 944–954. [Google Scholar] [CrossRef]
- Tang, J.Y.; Hsi, E.; Huang, Y.C.; Hsu, N.C.; Chu, P.Y.; Chai, C.Y. High LC3 expression correlates with poor survival in patients with oral squamous cell carcinoma. Hum. Pathol. 2013, 44, 2558–2562. [Google Scholar] [CrossRef]
- Wright, T.J.; McKee, C.; Birch-Machin, M.A.; Ellis, R.; Armstrong, J.L.; Lovat, P.E. Increasing the therapeutic efficacy of docetaxel for cutaneous squamous cell carcinoma through the combined inhibition of phosphatidylinositol 3-kinase/AKT signalling and autophagy. Clin. Exp. Dermatol. 2013, 38, 421–423. [Google Scholar] [CrossRef]
- Zang, Y.; Thomas, S.M.; Chan, E.T.; Kirk, C.J.; Freilino, M.L.; DeLancey, H.M.; Grandis, J.R.; Li, C.; Johnson, D.E. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin. Cancer Res. 2012, 18, 5639–5649. [Google Scholar] [CrossRef]
- Jung, Y.S.; Najy, A.J.; Huang, W.; Sethi, S.; Snyder, M.; Sakr, W.; Dyson, G.; Huttemann, M.; Lee, I.; Ali-Fehmi, R.; et al. HPV-associated differential regulation of tumor metabolism in oropharyngeal head and neck cancer. Oncotarget 2017, 8, 51530–51541. [Google Scholar] [CrossRef]
- Swartz, J.E.; Pothen, A.J.; van Kempen, P.M.; Stegeman, I.; Formsma, F.K.; Cann, E.M.; Willems, S.M.; Grolman, W. Poor prognosis in human papillomavirus-positive oropharyngeal squamous cell carcinomas that overexpress hypoxia inducible factor-1alpha. Head Neck 2016, 38, 1338–1346. [Google Scholar] [CrossRef]
- Rodolico, V.; Arancio, W.; Amato, M.C.; Aragona, F.; Cappello, F.; Di Fede, O.; Pannone, G.; Campisi, G. Hypoxia inducible factor-1 alpha expression is increased in infected positive HPV16 DNA oral squamous cell carcinoma and positively associated with HPV16 E7 oncoprotein. Infect. Agents Cancer 2011, 6, 18. [Google Scholar] [CrossRef]
- Molinolo, A.A.; Marsh, C.; El Dinali, M.; Gangane, N.; Jennison, K.; Hewitt, S.; Patel, V.; Seiwert, T.Y.; Gutkind, J.S. mTOR as a molecular target in HPV-associated oral and cervical squamous carcinomas. Clin. Cancer Res. 2012, 18, 2558–2568. [Google Scholar] [CrossRef]
- Brand, T.M.; Hartmann, S.; Bhola, N.E.; Li, H.; Zeng, Y.; O’Keefe, R.A.; Ranall, M.V.; Bandyopadhyay, S.; Soucheray, M.; Krogan, N.J.; et al. Cross-talk Signaling between HER3 and HPV16 E6 and E7 Mediates Resistance to PI3K Inhibitors in Head and Neck Cancer. Cancer Res. 2018, 78, 2383–2395. [Google Scholar] [CrossRef]
- Madera, D.; Vitale-Cross, L.; Martin, D.; Schneider, A.; Molinolo, A.A.; Gangane, N.; Carey, T.E.; McHugh, J.B.; Komarck, C.M.; Walline, H.M.; et al. Prevention of tumor growth driven by PIK3CA and HPV oncogenes by targeting mTOR signaling with metformin in oral squamous carcinomas expressing OCT3. Cancer Prev. Res. 2015, 8, 197–207. [Google Scholar] [CrossRef]
- Goschzik, T.; Gessi, M.; Denkhaus, D.; Pietsch, T. PTEN mutations and activation of the PI3K/Akt/mTOR signaling pathway in papillary tumors of the pineal region. J. Neuropathol. Exp. Neurol. 2014, 73, 747–751. [Google Scholar] [CrossRef]
- Matsumoto, C.S.; Almeida, L.O.; Guimaraes, D.M.; Martins, M.D.; Papagerakis, P.; Papagerakis, S.; Leopoldino, A.M.; Castilho, R.M.; Squarize, C.H. PI3K-PTEN dysregulation leads to mTOR-driven upregulation of the core clock gene BMAL1 in normal and malignant epithelial cells. Oncotarget 2016, 7, 42393–42407. [Google Scholar] [CrossRef]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef]
- Wu, C.C.; Hou, S.; Orr, B.A.; Kuo, B.R.; Youn, Y.H.; Ong, T.; Roth, F.; Eberhart, C.G.; Robinson, G.W.; Solecki, D.J.; et al. mTORC1-Mediated Inhibition of 4EBP1 Is Essential for Hedgehog Signaling-Driven Translation and Medulloblastoma. Dev. Cell 2017, 43, 673–688 e5. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar]
- Gkountakos, A.; Pilotto, S.; Mafficini, A.; Vicentini, C.; Simbolo, M.; Milella, M.; Tortora, G.; Scarpa, A.; Bria, E.; Corbo, V. Unmasking the impact of Rictor in cancer: Novel insights of mTORC2 complex. Carcinogenesis 2018. [Google Scholar] [CrossRef]
- Gupta, A.K.; McKenna, W.G.; Weber, C.N.; Feldman, M.D.; Goldsmith, J.D.; Mick, R.; Machtay, M.; Rosenthal, D.I.; Bakanauskas, V.J.; Cerniglia, G.J.; et al. Local recurrence in head and neck cancer: Relationship to radiation resistance and signal transduction. Clin. Cancer Res. 2002, 8, 885–892. [Google Scholar]
- Li, Z.; Yang, Z.; Passaniti, A.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H.C. A positive feedback loop involving EGFR/Akt/mTORC1 and IKK/NF-kB regulates head and neck squamous cell carcinoma proliferation. Oncotarget 2016, 7, 31892–31906. [Google Scholar]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef]
- Bigner, S.H.; Humphrey, P.A.; Wong, A.J.; Vogelstein, B.; Mark, J.; Friedman, H.S.; Bigner, D.D. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990, 50, 8017–8022. [Google Scholar]
- Sok, J.C.; Coppelli, F.M.; Thomas, S.M.; Lango, M.N.; Xi, S.; Hunt, J.L.; Freilino, M.L.; Graner, M.W.; Wikstrand, C.J.; Bigner, D.D.; et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin. Cancer Res. 2006, 12, 5064–5073. [Google Scholar] [CrossRef]
- Khattri, A.; Zuo, Z.; Bragelmann, J.; Keck, M.K.; El Dinali, M.; Brown, C.D.; Stricker, T.; Munagala, A.; Cohen, E.E.; Lingen, M.W.; et al. Rare occurrence of EGFRvIII deletion in head and neck squamous cell carcinoma. Oral Oncol. 2015, 51, 53–58. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. (Pozn) 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Hashmi, A.A.; Hussain, Z.F.; Aijaz, S.; Irfan, M.; Khan, E.Y.; Naz, S.; Faridi, N.; Khan, A.; Edhi, M.M. Immunohistochemical expression of epidermal growth factor receptor (EGFR) in South Asian head and neck squamous cell carcinoma: Association with various risk factors and clinico-pathologic and prognostic parameters. World J. Surg. Oncol. 2018, 16, 118. [Google Scholar] [CrossRef]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Ishii, G.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef]
- Chiang, C.T.; Demetriou, A.N.; Ung, N.; Choudhury, N.; Ghaffarian, K.; Ruderman, D.L.; Mumenthaler, S.M. mTORC2 contributes to the metabolic reprogramming in EGFR tyrosine-kinase inhibitor resistant cells in non-small cell lung cancer. Cancer Lett. 2018, 434, 152–159. [Google Scholar] [CrossRef]
- Cohen, E.E.; Davis, D.W.; Karrison, T.G.; Seiwert, T.Y.; Wong, S.J.; Nattam, S.; Kozloff, M.F.; Clark, J.I.; Yan, D.H.; Liu, W.; et al. Erlotinib and bevacizumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck: A phase I/II study. Lancet Oncol. 2009, 10, 247–257. [Google Scholar] [CrossRef]
- Sharp, H.; Morris, J.C.; Van Waes, C.; Gius, D.; Cooley-Zgela, T.; Singh, A.K. High incidence of oral dysesthesias on a trial of gefitinib, Paclitaxel, and concurrent external beam radiation for locally advanced head and neck cancers. Am. J. Clin. Oncol. 2008, 31, 557–560. [Google Scholar] [CrossRef]
- Xu, M.J.; Johnson, D.E.; Grandis, J.R. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 2017, 36, 463–473. [Google Scholar] [CrossRef]
- Porcelli, L.; Quatrale, A.E.; Mantuano, P.; Silvestris, N.; Rolland, J.F.; Biancolillo, L.; Paradiso, A.; Azzariti, A. Synergistic antiproliferative and antiangiogenic effects of EGFR and mTOR inhibitors. Curr. Pharm. Des. 2013, 19, 918–926. [Google Scholar] [CrossRef]
- Schmid, K.; Bago-Horvath, Z.; Berger, W.; Haitel, A.; Cejka, D.; Werzowa, J.; Filipits, M.; Herberger, B.; Hayden, H.; Sieghart, W. Dual inhibition of EGFR and mTOR pathways in small cell lung cancer. Br. J. Cancer 2010, 103, 622–628. [Google Scholar] [CrossRef]
- Boumahdi, S.; Driessens, G.; Lapouge, G.; Rorive, S.; Nassar, D.; Le Mercier, M.; Delatte, B.; Caauwe, A.; Lenglez, S.; Nkusi, E.; et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 2014, 511, 246–250. [Google Scholar] [CrossRef]
- Keysar, S.B.; Le, P.N.; Miller, B.; Jackson, B.C.; Eagles, J.R.; Nieto, C.; Kim, J.; Tang, B.; Glogowska, M.J.; Morton, J.J.; et al. Regulation of Head and Neck Squamous Cancer Stem Cells by PI3K and SOX2. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Huang, C.F.; Xu, X.R.; Wu, T.F.; Sun, Z.J.; Zhang, W.F. Correlation of ALDH1, CD44, OCT4 and SOX2 in tongue squamous cell carcinoma and their association with disease progression and prognosis. J. Oral Pathol. Med.: Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2014, 43, 492–498. [Google Scholar] [CrossRef]
- Suda, T.; Hama, T.; Kondo, S.; Yuza, Y.; Yoshikawa, M.; Urashima, M.; Kato, T.; Moriyama, H. Copy number amplification of the PIK3CA gene is associated with poor prognosis in non-lymph node metastatic head and neck squamous cell carcinoma. BMC Cancer 2012, 12, 416. [Google Scholar] [CrossRef]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef]
- Lau, C.E.; Tredwell, G.D.; Ellis, J.K.; Lam, E.W.; Keun, H.C. Metabolomic characterisation of the effects of oncogenic PIK3CA transformation in a breast epithelial cell line. Sci. Rep. 2017, 7, 46079. [Google Scholar] [CrossRef]
- Pedrero, J.M.; Carracedo, D.G.; Pinto, C.M.; Zapatero, A.H.; Rodrigo, J.P.; Nieto, C.S.; Gonzalez, M.V. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int. J. Cancer 2005, 114, 242–248. [Google Scholar] [CrossRef]
- Henderson, Y.C.; Wang, E.; Clayman, G.L. Genotypic analysis of tumor suppressor genes PTEN/MMAC1 and p53 in head and neck squamous cell carcinomas. Laryngoscope 1998, 108, 1553–1556. [Google Scholar] [CrossRef]
- Shao, X.; Tandon, R.; Samara, G.; Kanki, H.; Yano, H.; Close, L.G.; Parsons, R.; Sato, T. Mutational analysis of the PTEN gene in head and neck squamous cell carcinoma. Int. J. Cancer 1998, 77, 684–688. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Wilanowski, T.; Dworkin, S.; Auden, A.; Zhao, Q.; Rank, G.; Srivastava, S.; Finlay, M.J.; Papenfuss, A.T.; et al. Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto-oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell 2011, 20, 635–648. [Google Scholar] [CrossRef]
- Cangkrama, M.; Ting, S.B.; Darido, C. Stem cells behind the barrier. Int. J. Mol. Sci. 2013, 14, 13670–13686. [Google Scholar] [CrossRef]
- Cangkrama, M.; Darido, C.; Georgy, S.R.; Partridge, D.; Auden, A.; Srivastava, S.; Wilanowski, T.; Jane, S.M. Two Ancient Gene Families Are Critical for Maintenance of the Mammalian Skin Barrier in Postnatal Life. J. Investig. Dermatol. 2016, 136, 1438–1448. [Google Scholar] [CrossRef]
- Georgy, S.R.; Cangkrama, M.; Srivastava, S.; Partridge, D.; Auden, A.; Dworkin, S.; McLean, C.A.; Jane, S.M.; Darido, C. Identification of a Novel Proto-oncogenic Network in Head and Neck Squamous Cell Carcinoma. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Miles, L.B.; Dworkin, S.; Darido, C. Review article: Alternative splicing and start sites: Lessons from the Grainyhead-like family. Dev. Biol. 2017, 429, 12–19. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Jane, S.M. The role of barrier genes in epidermal malignancy. Oncogene 2016, 35, 5705–5712. [Google Scholar] [CrossRef]
- Youssef, M.; Cuddihy, A.; Darido, C. Long-Lived Epidermal Cancer-Initiating Cells. Int. J. Mol. Sci. 2017, 18, 1369. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Cullinane, C.; Partridge, D.D.; Walker, R.; Srivastava, S.; Roslan, S.; Carpinelli, M.R.; Dworkin, S.; Pearson, R.B.; et al. Stage-dependent therapeutic efficacy in PI3K/mTOR-driven squamous cell carcinoma of the skin. Cell Death Differ. 2018, 25, 1146–1159. [Google Scholar] [CrossRef]
- Lee, J.I.; Soria, J.C.; Hassan, K.A.; El-Naggar, A.K.; Tang, X.; Liu, D.D.; Hong, W.K.; Mao, L. Loss of PTEN expression as a prognostic marker for tongue cancer. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 1441–1445. [Google Scholar] [CrossRef]
- Squarize, C.H.; Castilho, R.M.; Santos Pinto, D., Jr. Immunohistochemical evidence of PTEN in oral squamous cell carcinoma and its correlation with the histological malignancy grading system. J. Oral Pathol. Med.: Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2002, 31, 379–384. [Google Scholar] [CrossRef]
- Bian, Y.; Hall, B.; Sun, Z.J.; Molinolo, A.; Chen, W.; Gutkind, J.S.; Waes, C.V.; Kulkarni, A.B. Loss of TGF-beta signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012, 31, 3322–3332. [Google Scholar] [CrossRef]
- Martin, P.L.; Yin, J.J.; Seng, V.; Casey, O.; Corey, E.; Morrissey, C.; Simpson, R.M.; Kelly, K. Androgen deprivation leads to increased carbohydrate metabolism and hexokinase 2-mediated survival in Pten/Tp53-deficient prostate cancer. Oncogene 2017, 36, 525–533. [Google Scholar] [CrossRef]
- Mathur, D.; Stratikopoulos, E.; Ozturk, S.; Steinbach, N.; Pegno, S.; Schoenfeld, S.; Yong, R.; Murty, V.V.; Asara, J.M.; Cantley, L.C.; et al. PTEN Regulates Glutamine Flux to Pyrimidine Synthesis and Sensitivity to Dihydroorotate Dehydrogenase Inhibition. Cancer Discov. 2017, 7, 380–390. [Google Scholar] [CrossRef]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.C.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef]
- Kiaris, H.; Spandidos, D.A.; Jones, A.S.; Vaughan, E.D.; Field, J.K. Mutations, expression and genomic instability of the H-ras proto-oncogene in squamous cell carcinomas of the head and neck. Br. J. Cance 1995, 72, 123–128. [Google Scholar] [CrossRef]
- Nakagaki, T.; Tamura, M.; Kobashi, K.; Omori, A.; Koyama, R.; Idogawa, M.; Ogi, K.; Hiratsuka, H.; Tokino, T.; Sasaki, Y. Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma. Tumour Biol. 2018, 40, 1010428318800180. [Google Scholar] [CrossRef]
- Su, S.C.; Lin, C.W.; Liu, Y.F.; Fan, W.L.; Chen, M.K.; Yu, C.P.; Yang, W.E.; Su, C.W.; Chuang, C.Y.; Li, W.H.; et al. Exome Sequencing of Oral Squamous Cell Carcinoma Reveals Molecular Subgroups and Novel Therapeutic Opportunities. Theranostics 2017, 7, 1088–1099. [Google Scholar] [CrossRef]
- Koumaki, D.; Kostakis, G.; Koumaki, V.; Papadogeorgakis, N.; Makris, M.; Katoulis, A.; Kamakari, S.; Koutsodontis, G.; Perisanidis, C.; Lambadiari, V.; et al. Novel mutations of the HRAS gene and absence of hotspot mutations of the BRAF genes in oral squamous cell carcinoma in a Greek population. Oncol. Rep. 2012, 27, 1555–1560. [Google Scholar] [CrossRef]
- Murugan, A.K.; Hong, N.T.; Cuc, T.T.; Hung, N.C.; Munirajan, A.K.; Ikeda, M.A.; Tsuchida, N. Detection of two novel mutations and relatively high incidence of H-RAS mutations in Vietnamese oral cancer. Oral Oncol. 2009, 45, e161–e166. [Google Scholar] [CrossRef]
- Zheng, W.; Tayyari, F.; Gowda, G.A.; Raftery, D.; McLamore, E.S.; Porterfield, D.M.; Donkin, S.S.; Bequette, B.; Teegarden, D. Altered glucose metabolism in Harvey-ras transformed MCF10A cells. Mol. Carcinog. 2015, 54, 111–120. [Google Scholar] [CrossRef]
- Ruicci, K.M.; Pinto, N.; Khan, M.I.; Yoo, J.; Fung, K.; MacNeil, D.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C. ERK-TSC2 signalling in constitutively-active HRAS mutant HNSCC cells promotes resistance to PI3K inhibition. Oral Oncol. 2018, 84, 95–103. [Google Scholar] [CrossRef]
- Hah, J.H.; Zhao, M.; Pickering, C.R.; Frederick, M.J.; Andrews, G.A.; Jasser, S.A.; Fooshee, D.R.; Milas, Z.L.; Galer, C.; Sano, D.; et al. HRAS mutations and resistance to the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in head and neck squamous cell carcinoma cells. Head & Neck 2014, 36, 1547–1554. [Google Scholar]
- Rampias, T.; Giagini, A.; Siolos, S.; Matsuzaki, H.; Sasaki, C.; Scorilas, A.; Psyrri, A. RAS/PI3K crosstalk and cetuximab resistance in head and neck squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 2933–2946. [Google Scholar] [CrossRef] [PubMed]
- Pyronnet, S.; Imataka, H.; Gingras, A.C.; Fukunaga, R.; Hunter, T.; Sonenberg, N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 1999, 18, 270–279. [Google Scholar] [CrossRef]
- Raught, B.; Gingras, A.C.; Gygi, S.P.; Imataka, H.; Morino, S.; Gradi, A.; Aebersold, R.; Sonenberg, N. Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J. 2000, 19, 434–444. [Google Scholar] [CrossRef]
- Marcotrigiano, J.; Gingras, A.C.; Sonenberg, N.; Burley, S.K. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol. Cell 1999, 3, 707–716. [Google Scholar] [CrossRef]
- Yang, H.S.; Jansen, A.P.; Komar, A.A.; Zheng, X.; Merrick, W.C.; Costes, S.; Lockett, S.J.; Sonenberg, N.; Colburn, N.H. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol. Cell Biol. 2003, 23, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Comtesse, N.; Keller, A.; Diesinger, I.; Bauer, C.; Kayser, K.; Huwer, H.; Lenhof, H.P.; Meese, E. Frequent overexpression of the genes FXR1, CLAPM1 and EIF4G located on amplicon 3q26-27 in squamous cell carcinoma of the lung. Int. J. Cancer 2007, 120, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Silvera, D.; Arju, R.; Darvishian, F.; Levine, P.H.; Zolfaghari, L.; Goldberg, J.; Hochman, T.; Formenti, S.C.; Schneider, R.J. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat. Cell Biol. 2009, 11, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zhou, Y.; Chen, Y.; Ke, G.; Wen, H.; Wu, X. Decreased expression of EIF4A1 after preoperative brachytherapy predicts better tumor-specific survival in cervical cancer. Int. J. Gynecol. Cancer 2014, 24, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Liu, Z.; He, X.; He, Y.; Yang, H.; Jiang, Q.; Xie, S.; Xiao, G.; Li, X.; Yao, K.; et al. Over-expression of eukaryotic translation initiation factor 4 gamma 1 correlates with tumor progression and poor prognosis in nasopharyngeal carcinoma. Mol. Cancer 2010, 9, 78. [Google Scholar] [CrossRef]
- Ramirez-Valle, F.; Braunstein, S.; Zavadil, J.; Formenti, S.C.; Schneider, R.J. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J. Cell Biol. 2008, 181, 293–307. [Google Scholar] [CrossRef]
- Moerke, N.J.; Aktas, H.; Chen, H.; Cantel, S.; Reibarkh, M.Y.; Fahmy, A.; Gross, J.D.; Degterev, A.; Yuan, J.; Chorev, M.; et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [Google Scholar] [CrossRef]
- Chen, L.; Aktas, B.H.; Wang, Y.; He, X.; Sahoo, R.; Zhang, N.; Denoyelle, S.; Kabha, E.; Yang, H.; Freedman, R.Y.; et al. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 2012, 3, 869–881. [Google Scholar] [CrossRef]
- Papadopoulos, E.; Jenni, S.; Kabha, E.; Takrouri, K.J.; Yi, T.; Salvi, N.; Luna, R.E.; Gavathiotis, E.; Mahalingam, P.; Arthanari, H.; et al. Structure of the eukaryotic translation initiation factor eIF4E in complex with 4EGI-1 reveals an allosteric mechanism for dissociating eIF4G. Proc. Natl. Acad. Sci. USA 2014, 111, E3187–E3195. [Google Scholar] [CrossRef]
- Cencic, R.; Hall, D.R.; Robert, F.; Du, Y.; Min, J.; Li, L.; Qui, M.; Lewis, I.; Kurtkaya, S.; Dingledine, R.; et al. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc. Natl. Acad. Sci. USA 2011, 108, 1046–1051. [Google Scholar] [CrossRef]
- Feng, Y.; Pinkerton, A.B.; Hulea, L.; Zhang, T.; Davies, M.A.; Grotegut, S.; Cheli, Y.; Yin, H.; Lau, E.; Kim, H.; et al. SBI-0640756 Attenuates the Growth of Clinically Unresponsive Melanomas by Disrupting the eIF4F Translation Initiation Complex. Cancer Res. 2015, 75, 5211–5218. [Google Scholar] [CrossRef]
- Miyakawa, S.; Oguro, A.; Ohtsu, T.; Imataka, H.; Sonenberg, N.; Nakamura, Y. RNA aptamers to mammalian initiation factor 4G inhibit cap-dependent translation by blocking the formation of initiation factor complexes. RNA 2006, 12, 1825–1834. [Google Scholar] [CrossRef]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Agarwal, N.K.; Chen, C.H.; Cho, H.; Boulbes, D.R.; Spooner, E.; Sarbassov, D.D. Rictor regulates cell migration by suppressing RhoGDI2. Oncogene 2013, 32, 2521–2526. [Google Scholar] [CrossRef]
- Morrison Joly, M.; Williams, M.M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Young, C.D.; Sarbassov, D.D.; Muller, W.J.; Brantley-Sieders, D.; Cook, R.S. Two distinct mTORC2-dependent pathways converge on Rac1 to drive breast cancer metastasis. Breast Cancer Res. 2017, 19, 74. [Google Scholar] [CrossRef]
- Saci, A.; Cantley, L.C.; Carpenter, C.L. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol. Cell 2011, 42, 50–61. [Google Scholar] [CrossRef]
- Hampsch, R.A.; Shee, K.; Bates, D.; Lewis, L.D.; Desire, L.; Leblond, B.; Demidenko, E.; Stefan, K.; Huang, Y.H.; Miller, T.W. Therapeutic sensitivity to Rac GTPase inhibition requires consequential suppression of mTORC1, AKT, and MEK signaling in breast cancer. Oncotarget 2017, 8, 21806–21817. [Google Scholar] [CrossRef]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef]
- Liu, S.Y.; Yen, C.Y.; Yang, S.C.; Chiang, W.F.; Chang, K.W. Overexpression of Rac-1 small GTPase binding protein in oral squamous cell carcinoma. J. Oral Maxillofac. Surg. 2004, 62, 702–707. [Google Scholar] [CrossRef]
- Kamai, T.; Yamanishi, T.; Shirataki, H.; Takagi, K.; Asami, H.; Ito, Y.; Yoshida, K. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin. Cancer Res. 2004, 10, 4799–4805. [Google Scholar] [CrossRef]
- Engers, R.; Ziegler, S.; Mueller, M.; Walter, A.; Willers, R.; Gabbert, H.E. Prognostic relevance of increased Rac GTPase expression in prostate carcinomas. Endocr. Relat. Cancer 2007, 14, 245–256. [Google Scholar] [CrossRef]
- Gulhati, P.; Bowen, K.A.; Liu, J.; Stevens, P.D.; Rychahou, P.G.; Chen, M.; Lee, E.Y.; Weiss, H.L.; O’Connor, K.L.; Gao, T.; et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011, 71, 3246–3256. [Google Scholar] [CrossRef]
- Choi, U.J.; Jee, B.K.; Lim, Y.; Lee, K.H. KAI1/CD82 decreases Rac1 expression and cell proliferation through PI3K/Akt/mTOR pathway in H1299 lung carcinoma cells. Cell Biochem. Funct. 2009, 27, 40–47. [Google Scholar] [CrossRef]
- Skvortsov, S.; Jimenez, C.R.; Knol, J.C.; Eichberger, P.; Schiestl, B.; Debbage, P.; Skvortsova, I.; Lukas, P. Radioresistant head and neck squamous cell carcinoma cells: Intracellular signaling, putative biomarkers for tumor recurrences and possible therapeutic targets. Radiother. Oncol. 2011, 101, 177–182. [Google Scholar] [CrossRef]
- Patel, V.; Rosenfeldt, H.M.; Lyons, R.; Servitja, J.M.; Bustelo, X.R.; Siroff, M.; Gutkind, J.S. Persistent activation of Rac1 in squamous carcinomas of the head and neck: Evidence for an EGFR/Vav2 signaling axis involved in cell invasion. Carcinogenesis 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernandez, E.; Humphries-Bickley, T.; De la Mota-Peynado, A.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef]
- Hagiwara, N.; Watanabe, M.; Iizuka-Ohashi, M.; Yokota, I.; Toriyama, S.; Sukeno, M.; Tomosugi, M.; Sowa, Y.; Hongo, F.; Mikami, K.; et al. Mevalonate pathway blockage enhances the efficacy of mTOR inhibitors with the activation of retinoblastoma protein in renal cell carcinoma. Cancer Lett. 2018, 431, 182–189. [Google Scholar] [CrossRef]
- Frankel, W.N.; Yang, Y.; Mahaffey, C.L.; Beyer, B.J.; O’Brien, T.P. Szt2, a novel gene for seizure threshold in mice. Genes Brain Behav. 2009, 8, 568–576. [Google Scholar] [CrossRef]
- Venkatesan, C.; Angle, B.; Millichap, J.J. Early-life epileptic encephalopathy secondary to SZT2 pathogenic recessive variants. Epileptic Disord. 2016, 18, 195–200. [Google Scholar]
- Pizzino, A.; Whitehead, M.; Sabet Rasekh, P.; Murphy, J.; Helman, G.; Bloom, M.; Evans, S.H.; Murnick, J.G.; Conry, J.; Taft, R.J.; et al. Mutations in SZT2 result in early-onset epileptic encephalopathy and leukoencephalopathy. Am. J. Med. Genet. A 2018, 176, 1443–1448. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Wyant, G.A.; Gu, X.; Orozco, J.M.; Shen, K.; Condon, K.J.; Petri, S.; Kedir, J.; Scaria, S.M.; et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017, 543, 438–442. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Li, M.O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017, 543, 433–437. [Google Scholar] [CrossRef]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef]
- Yoon, M.S.; Sun, Y.; Arauz, E.; Jiang, Y.; Chen, J. Phosphatidic acid activates mammalian target of rapamycin complex 1 (mTORC1) kinase by displacing FK506 binding protein 38 (FKBP38) and exerting an allosteric effect. J. Biol. Chem. 2011, 286, 29568–29574. [Google Scholar] [CrossRef]
- Winter, J.N.; Fox, T.E.; Kester, M.; Jefferson, L.S.; Kimball, S.R. Phosphatidic acid mediates activation of mTORC1 through the ERK signaling pathway. Am. J. Physiol. Cell Physiol. 2010, 299, C335–C344. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Pentecost, B.T.; Marconi, S.A.; Ricketts-Loriaux, R.S.; Otis, C.N.; Arcaro, K.F. PLD1 is overexpressed in an ER-negative MCF-7 cell line variant and a subset of phospho-Akt-negative breast carcinomas. Br. J. Cancer 2007, 97, 809–817. [Google Scholar] [CrossRef]
- Hu, J.; Hu, H.; Hang, J.J.; Yang, H.Y.; Wang, Z.Y.; Wang, L.; Chen, D.H.; Wang, L.W. Simultaneous high expression of PLD1 and Sp1 predicts a poor prognosis for pancreatic ductal adenocarcinoma patients. Oncotarget 2016, 7, 78557–78565. [Google Scholar] [CrossRef]
- Kang, D.W.; Lee, B.H.; Suh, Y.A.; Choi, Y.S.; Jang, S.J.; Kim, Y.M.; Choi, K.Y.; Min, D.S. Phospholipase D1 Inhibition Linked to Upregulation of ICAT Blocks Colorectal Cancer Growth Hyperactivated by Wnt/beta-Catenin and PI3K/Akt Signaling. Clin. Cancer Res. 2017, 23, 7340–7350. [Google Scholar] [CrossRef]
- Ohguchi, K.; Banno, Y.; Nakagawa, Y.; Akao, Y.; Nozawa, Y. Negative regulation of melanogenesis by phospholipase D1 through mTOR/p70 S6 kinase 1 signaling in mouse B16 melanoma cells. J. Cell Physiol. 2005, 205, 444–451. [Google Scholar] [CrossRef]
- Tang, W.; Liang, R.; Duan, Y.; Shi, Q.; Liu, X.; Liao, Y. PLD1 overexpression promotes invasion and migration and function as a risk factor for Chinese glioma patients. Oncotarget 2017, 8, 57039–57046. [Google Scholar] [CrossRef]
- Xiao, J.; Sun, Q.; Bei, Y.; Zhang, L.; Dimitrova-Shumkovska, J.; Lv, D.; Yang, Y.; Cao, Y.; Zhao, Y.; Song, M.; et al. Therapeutic inhibition of phospholipase D1 suppresses hepatocellular carcinoma. Clin. Sci. (Lond.) 2016, 130, 1125–1136. [Google Scholar] [CrossRef]
- Lewis, J.A.; Scott, S.A.; Lavieri, R.; Buck, J.R.; Selvy, P.E.; Stoops, S.L.; Armstrong, M.D.; Brown, H.A.; Lindsley, C.W. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg. Med. Chem. Lett. 2009, 19, 1916–1920. [Google Scholar]
- Monovich, L.; Mugrage, B.; Quadros, E.; Toscano, K.; Tommasi, R.; LaVoie, S.; Liu, E.; Du, Z.; LaSala, D.; Boyar, W.; et al. Optimization of halopemide for phospholipase D2 inhibition. Bioorg. Med. Chem. Lett. 2007, 17, 2310–2311. [Google Scholar] [CrossRef]
- Grunwald, V.; Keilholz, U.; Boehm, A.; Guntinas-Lichius, O.; Hennemann, B.; Schmoll, H.J.; Ivanyi, P.; Abbas, M.; Lehmann, U.; Koch, A.; et al. TEMHEAD: A single-arm multicentre phase II study of temsirolimus in platin- and cetuximab refractory recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN) of the German SCCHN Group (AIO). Ann. Oncol. 2015, 26, 561–567. [Google Scholar] [CrossRef]
- Dunn, L.A.; Fury, M.G.; Xiao, H.; Baxi, S.S.; Sherman, E.J.; Korte, S.; Pfister, C.; Haque, S.; Katabi, N.; Ho, A.L.; et al. A phase II study of temsirolimus added to low-dose weekly carboplatin and paclitaxel for patients with recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC). Ann. Oncol. 2017, 28, 2533–2538. [Google Scholar] [CrossRef]
- Keam, B.; Kim, S.; Ahn, Y.O.; Kim, T.M.; Lee, S.H.; Kim, D.W.; Heo, D.S. In vitro anticancer activity of PI3K alpha selective inhibitor BYL719 in head and neck cancer. AntiCancer Res. 2015, 35, 175–182. [Google Scholar]
- Mehibel, M.; Ortiz-Martinez, F.; Voelxen, N.; Boyers, A.; Chadwick, A.; Telfer, B.A.; Mueller-Klieser, W.; West, C.M.; Critchlow, S.E.; Williams, K.J.; et al. Statin-induced metabolic reprogramming in head and neck cancer: A biomarker for targeting monocarboxylate transporters. Sci. Rep. 2018, 8, 16804. [Google Scholar] [CrossRef]
- Polanski, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef]
- Bola, B.M.; Chadwick, A.L.; Michopoulos, F.; Blount, K.G.; Telfer, B.A.; Williams, K.J.; Smith, P.D.; Critchlow, S.E.; Stratford, I.J. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol. Cancer Ther. 2014, 13, 2805–2816. [Google Scholar] [CrossRef]
Figure 1.
Biological functions of the mammalian target of rapamycin (mTOR): (A) mTOR complex 1 (mTORC1) and (B) mTOR complex 2 (mTORC2).
Figure 1.
Biological functions of the mammalian target of rapamycin (mTOR): (A) mTOR complex 1 (mTORC1) and (B) mTOR complex 2 (mTORC2).

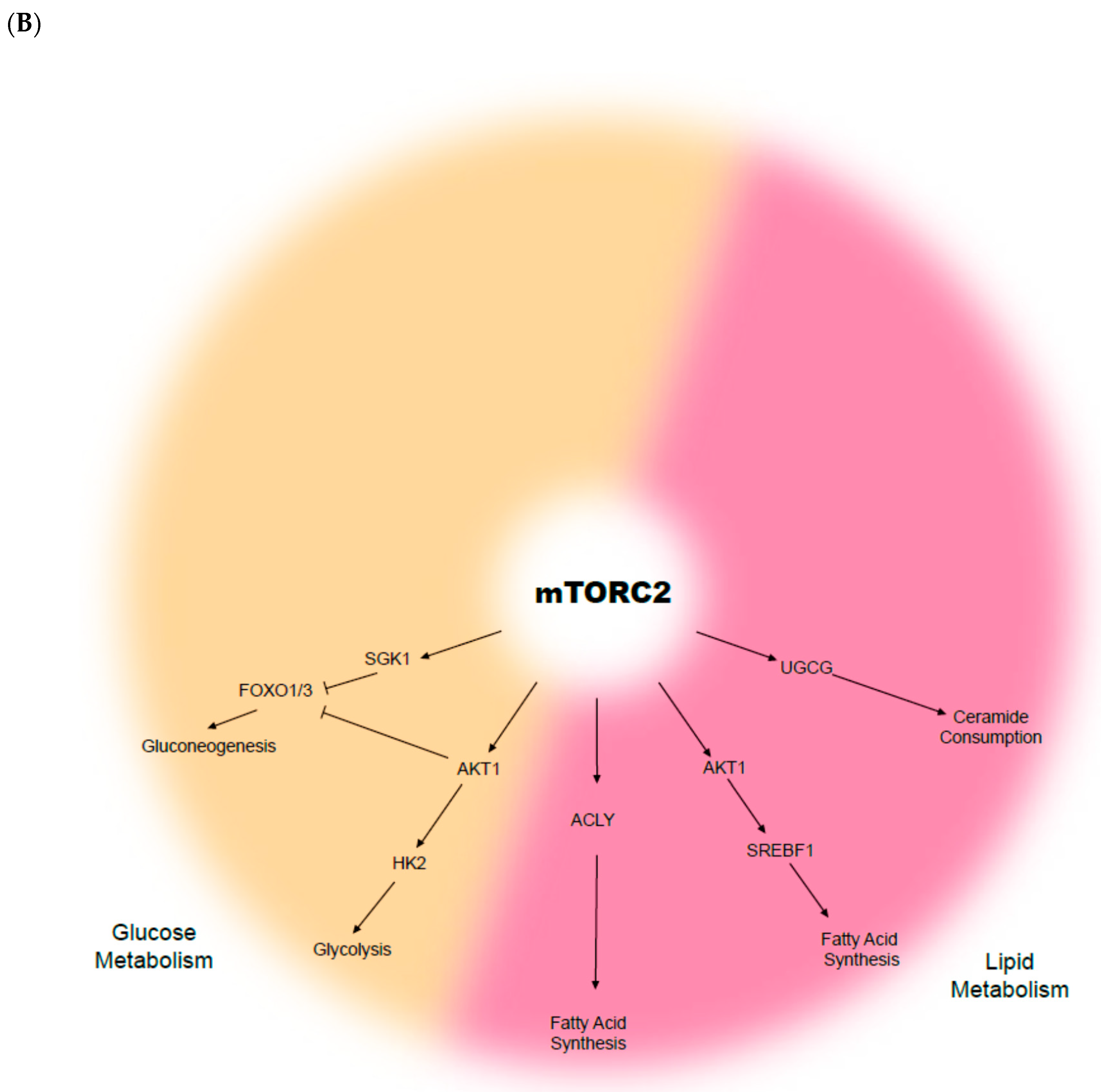
Figure 2.
Validated and proposed mutant genes found to deregulate mTOR signaling in head and neck squamous cell carcinoma (HNSCC). Percentages represent frequency of mutations per gene based on The Cancer Genome Atlas (TCGA) dataset (n = 297).
Figure 2.
Validated and proposed mutant genes found to deregulate mTOR signaling in head and neck squamous cell carcinoma (HNSCC). Percentages represent frequency of mutations per gene based on The Cancer Genome Atlas (TCGA) dataset (n = 297).
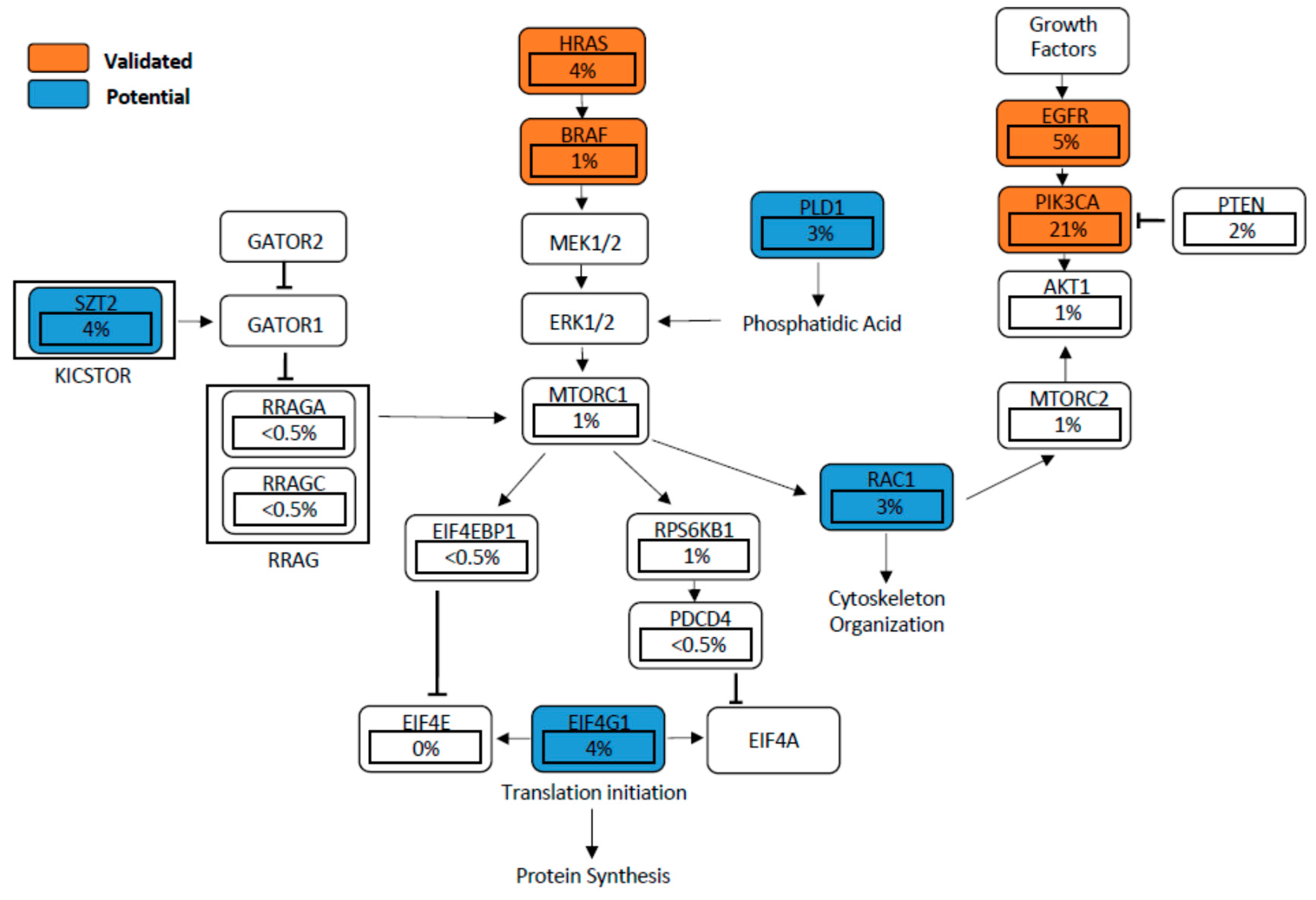

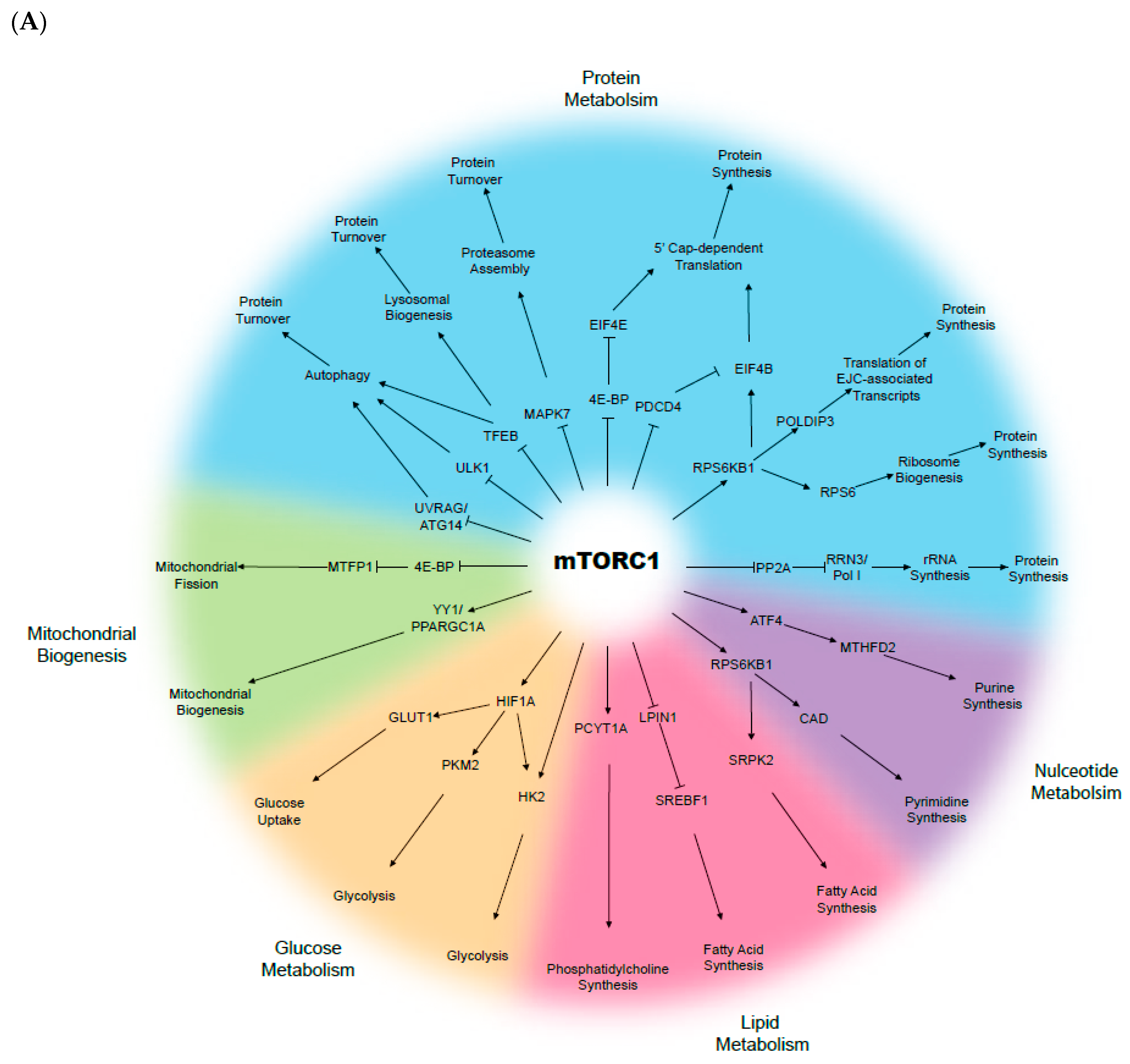{kind=link}
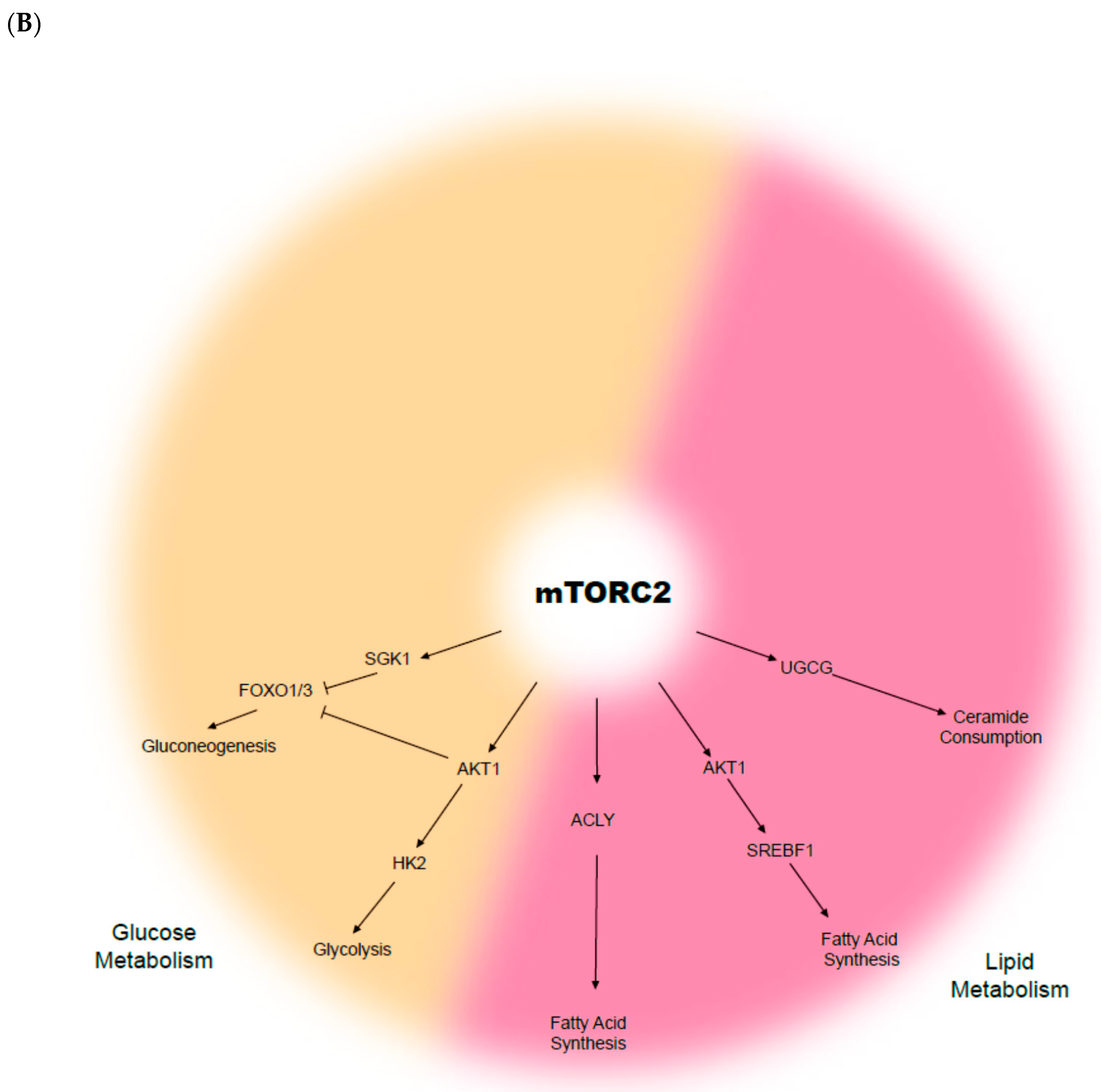{kind=link}
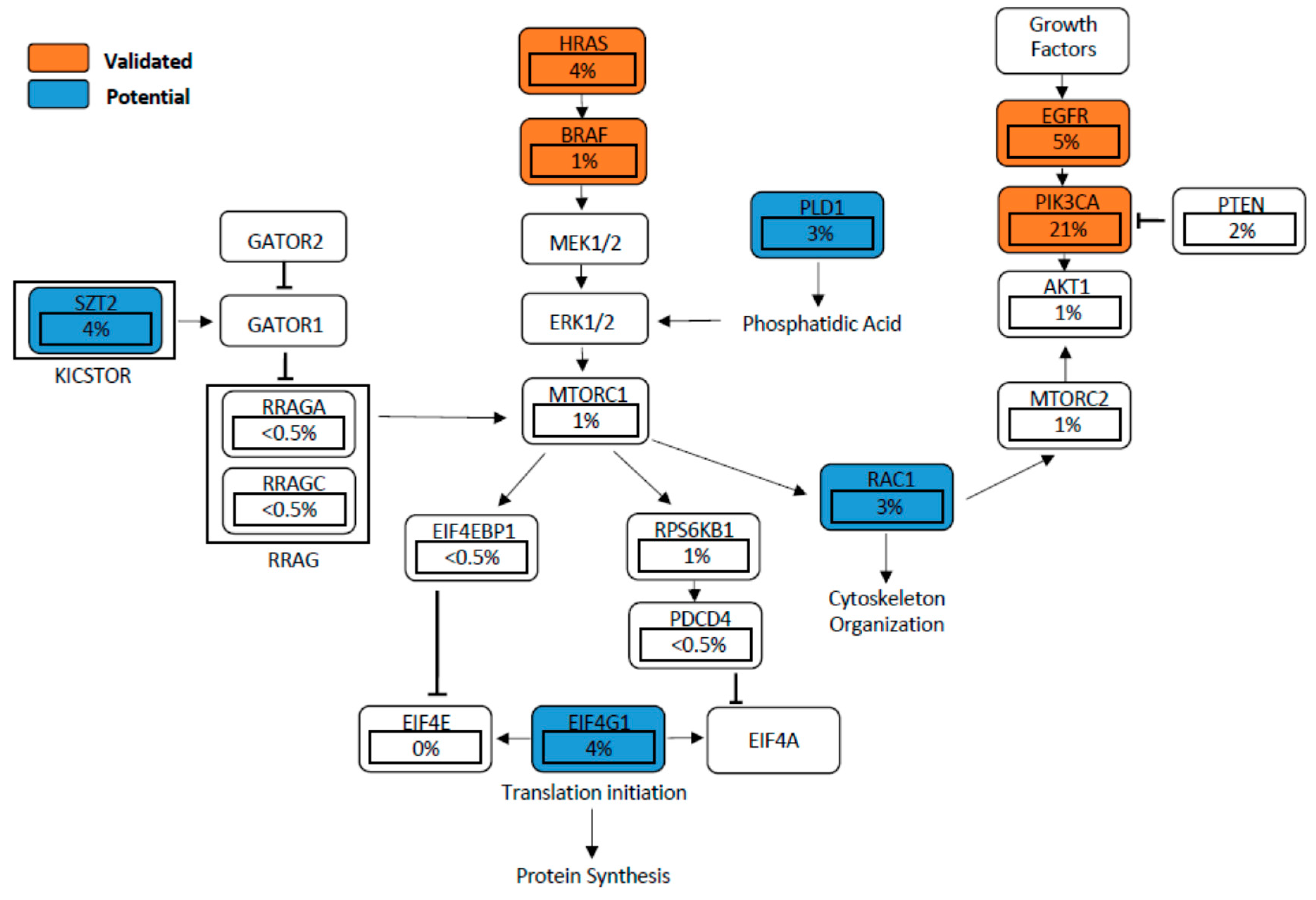{kind=link}
Table 1.
Summary of clinical trials targeting mTOR in HNSCC tumours.
| Inhibitor | Phase | Status | Targeted Pathway | Targeted Tumour | Reference |
|---|---|---|---|---|---|
| Palbociclib + Gedatolisib | I | Recruiting | CDK4/6 + mTOR | Advanced HNSCC | NCT03065062 |
| BYL719 | II | Recruiting | PI3K | Recurrent or Metastatic HNSCC | NCT02145312 |
| Everolimus + Palbociclib + Trametinib | I | Recruiting | mTOR + CDK4/6 + MEK | Malignant neoplasms of Oral cavity | NCT03065387 |
| CC-115 | I | Active, not recruiting | Dual DNA-PK and TOR kinase | Advanced HNSCC | NCT01353625 |
| PQR 309 | I | Active, not recruiting | PI3K/mTOR/AKT | Advanced HNSCC | NCT02483858 |
| Temsirolimus | II | Completed | mTOR | HNSCSC | NCT01172769 |
| Sirolimus | I/II | Completed | mTOR | Advanced HNSCC | NCT01195922 |
| Temsirolimus + Paclitaxel + Carboplatin | I/II | Completed | mTOR | Recurrent or Metastatic HNSCC | NCT01016769 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tan, F.H.; Bai, Y.; Saintigny, P.; Darido, C. mTOR Signalling in Head and Neck Cancer: Heads Up. Cells 2019, 8, 333. https://doi.org/10.3390/cells8040333
AMA Style
Tan FH, Bai Y, Saintigny P, Darido C. mTOR Signalling in Head and Neck Cancer: Heads Up. Cells. 2019; 8(4):333. https://doi.org/10.3390/cells8040333
Chicago/Turabian StyleTan, Fiona H., Yuchen Bai, Pierre Saintigny, and Charbel Darido. 2019. "mTOR Signalling in Head and Neck Cancer: Heads Up" Cells 8, no. 4: 333. https://doi.org/10.3390/cells8040333
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.