Autophagy as a Therapeutic Target to Enhance Aged Muscle Regeneration


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
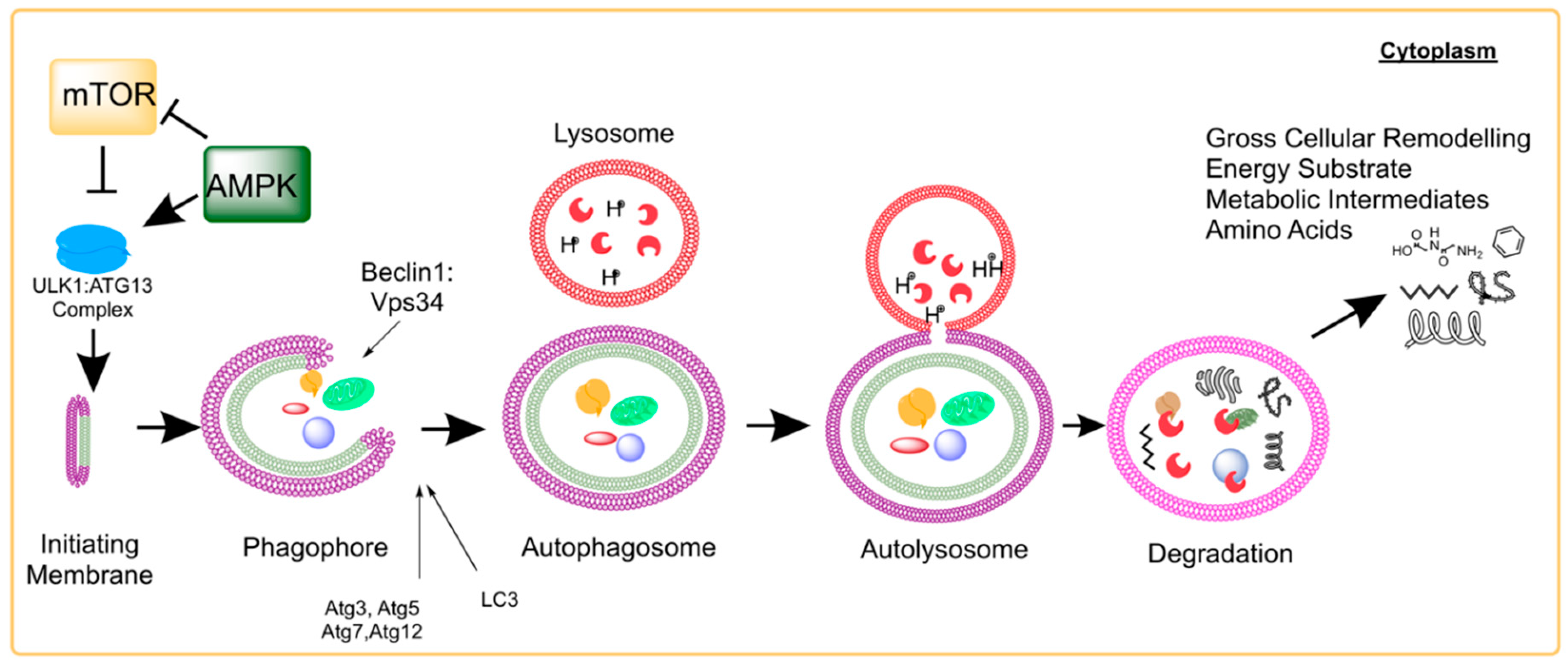
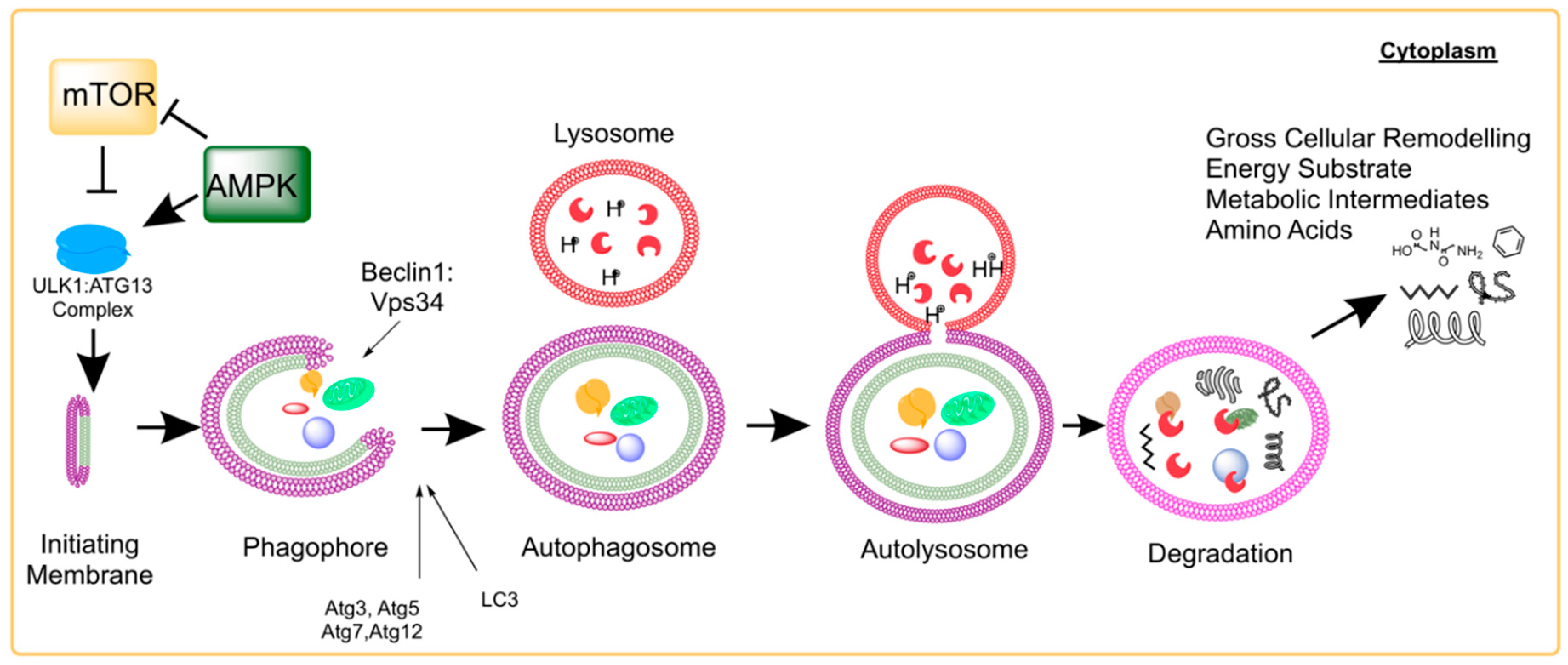
2. Molecular Process of Autophagy
3. Autophagy Effects on Skeletal Muscle Homeostasis, Regeneration, and Aging
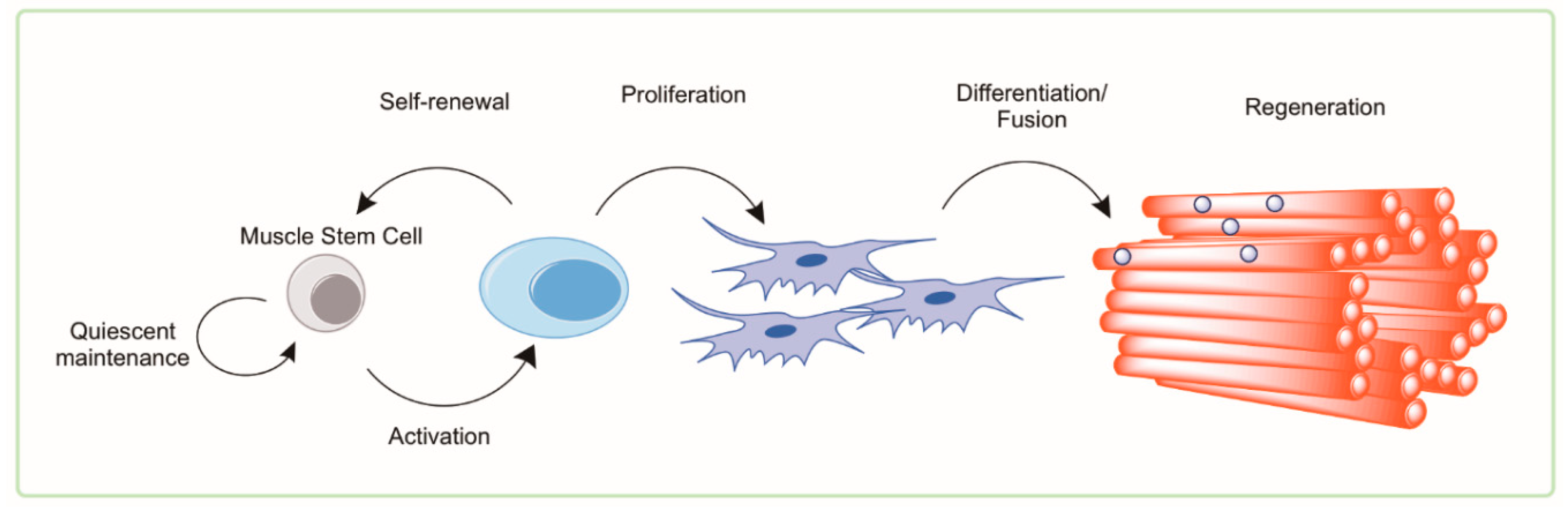
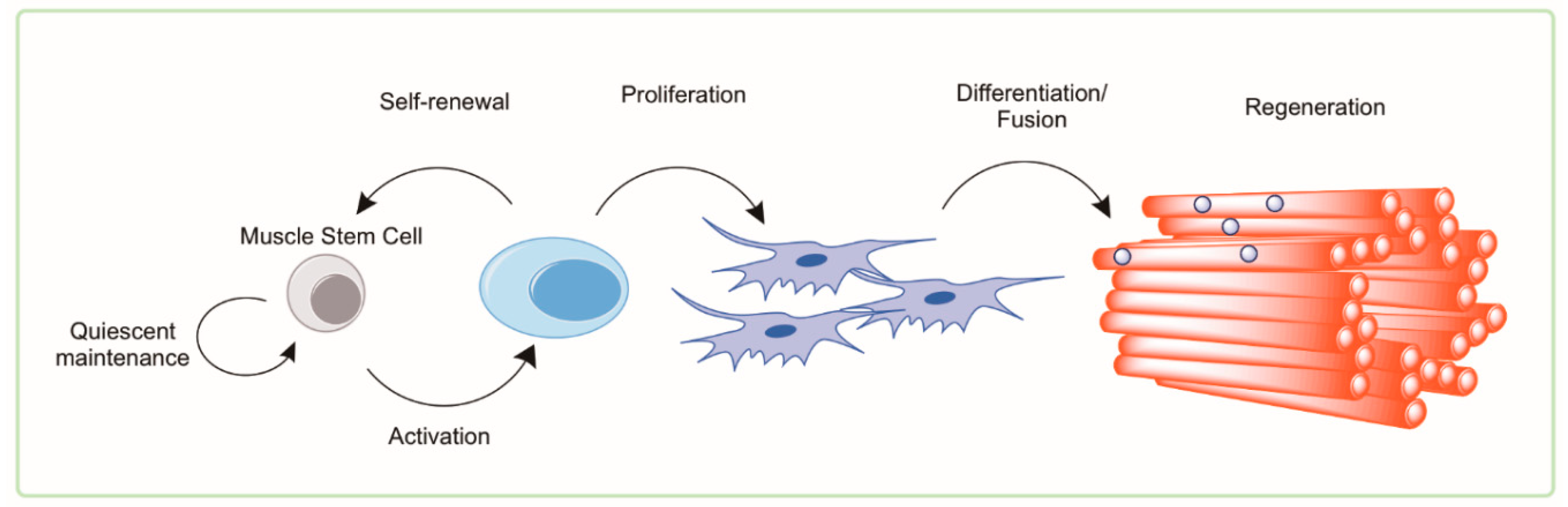
4. Muscle Satellite Cells
4.1. Satellite Cells in Health and Aging
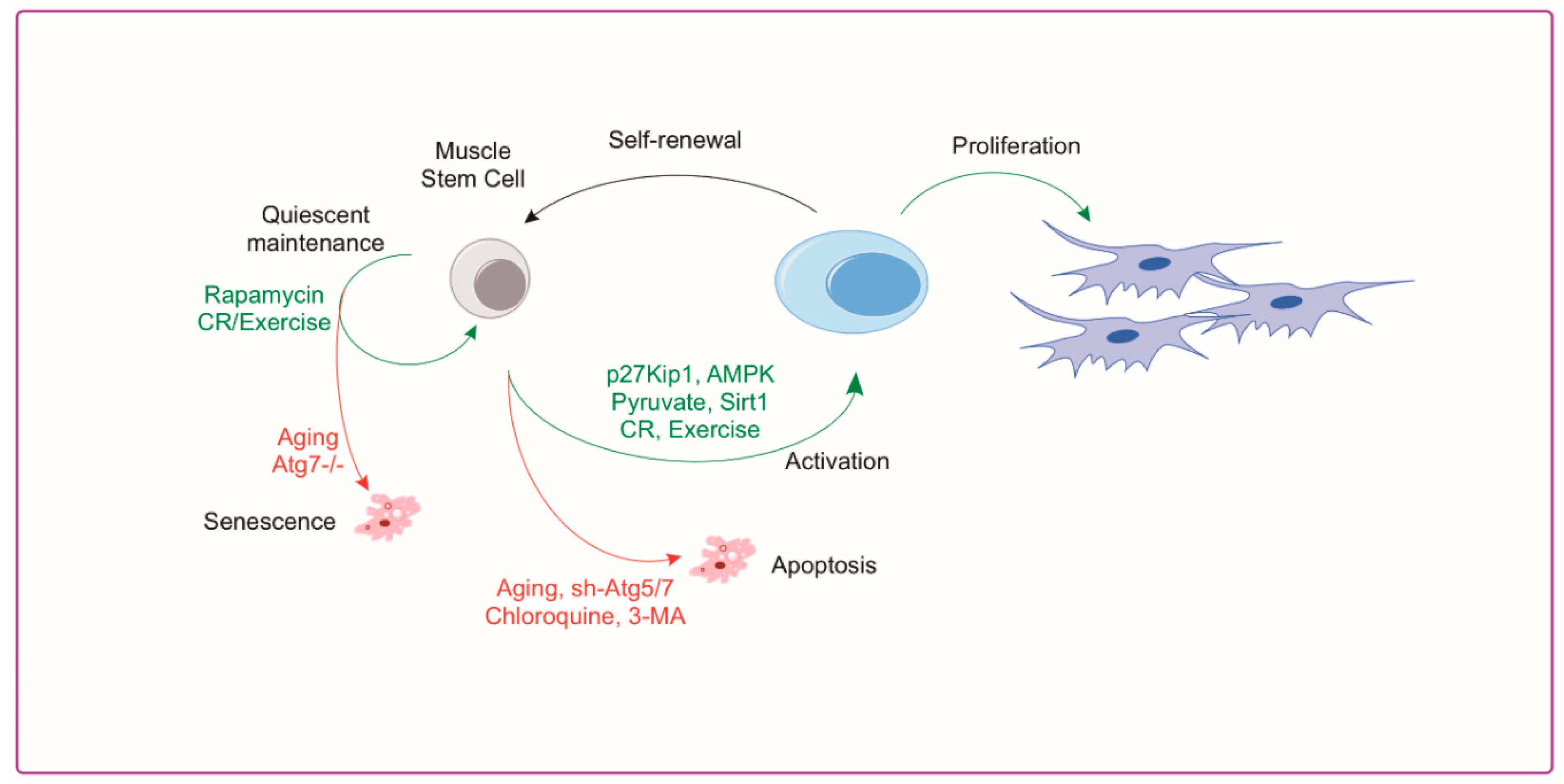
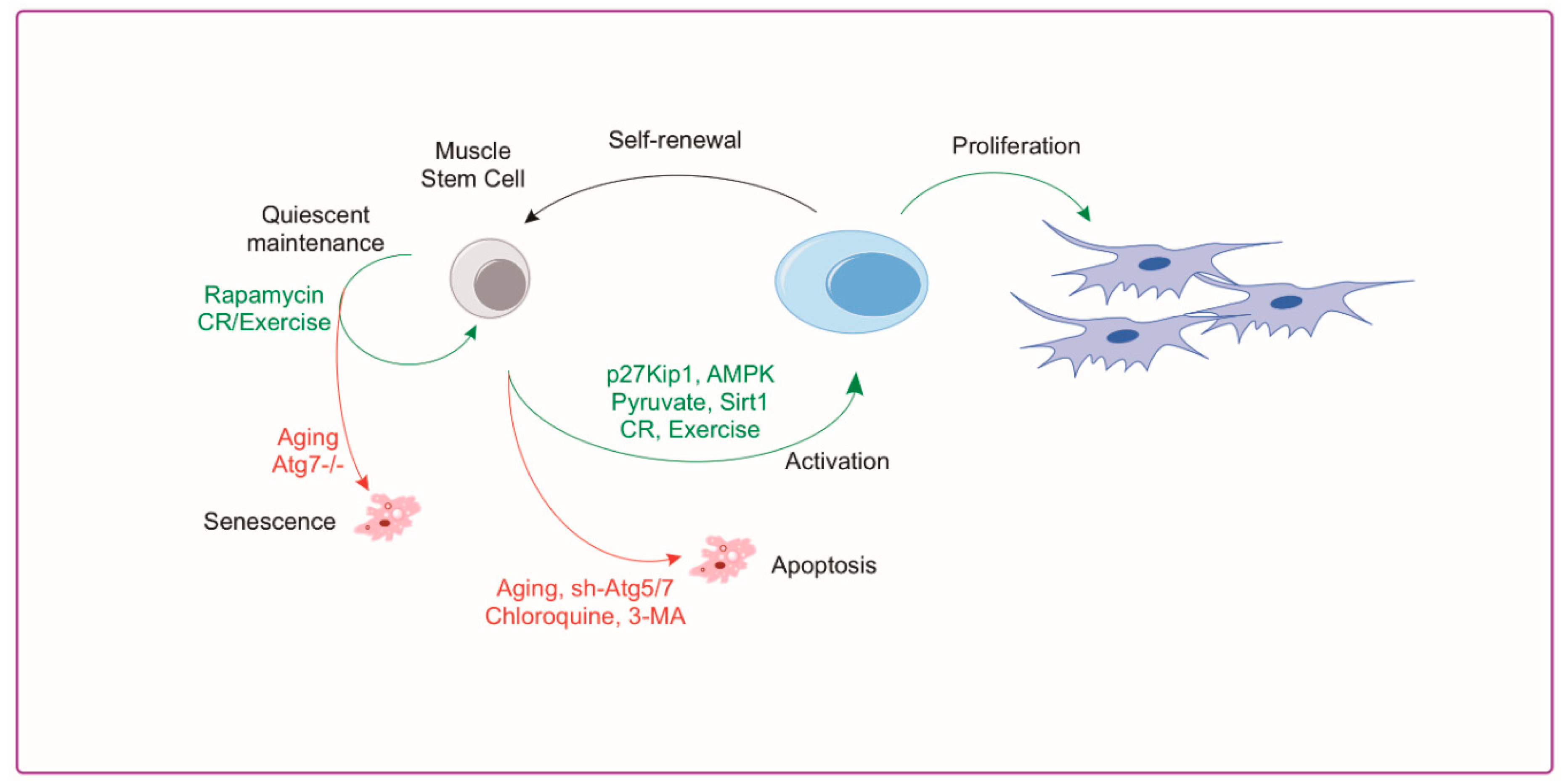
4.2. Autophagy and Muscle Satellite Cells
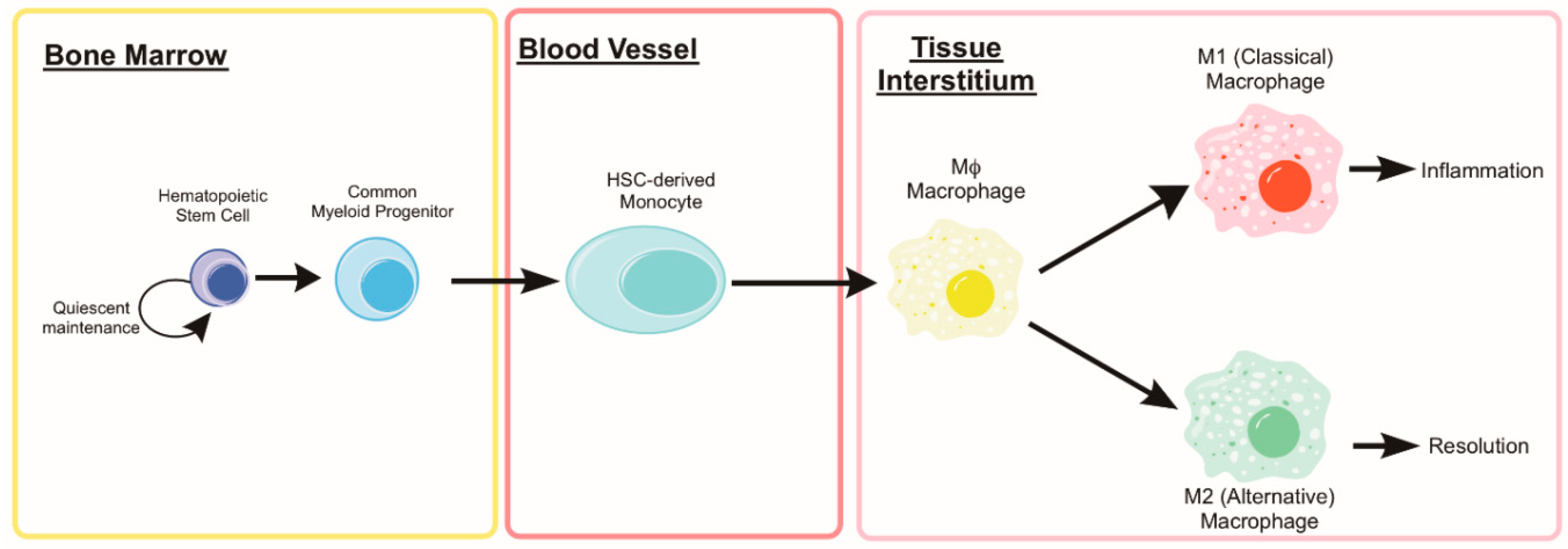
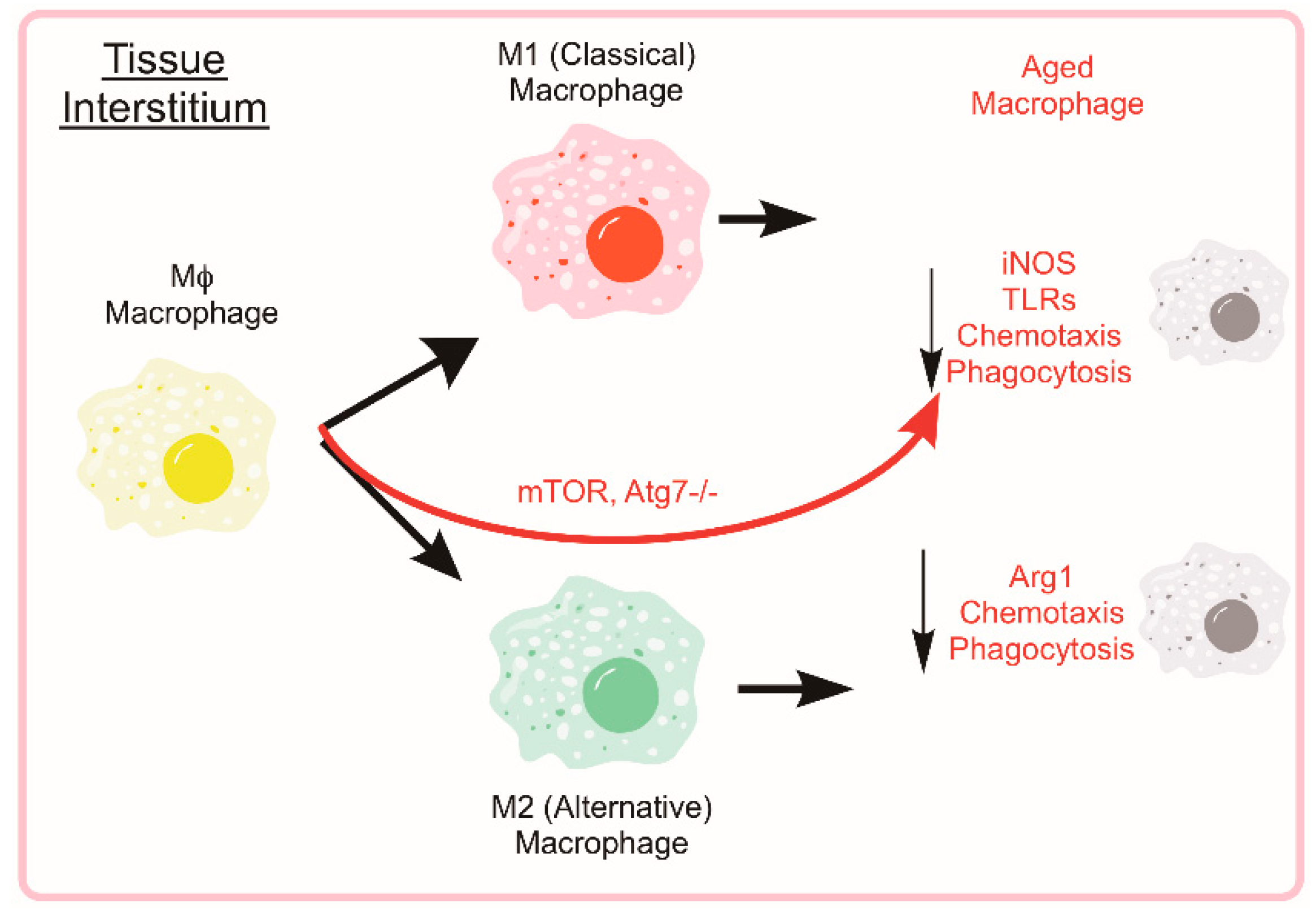
5. Monocytes and Macrophages
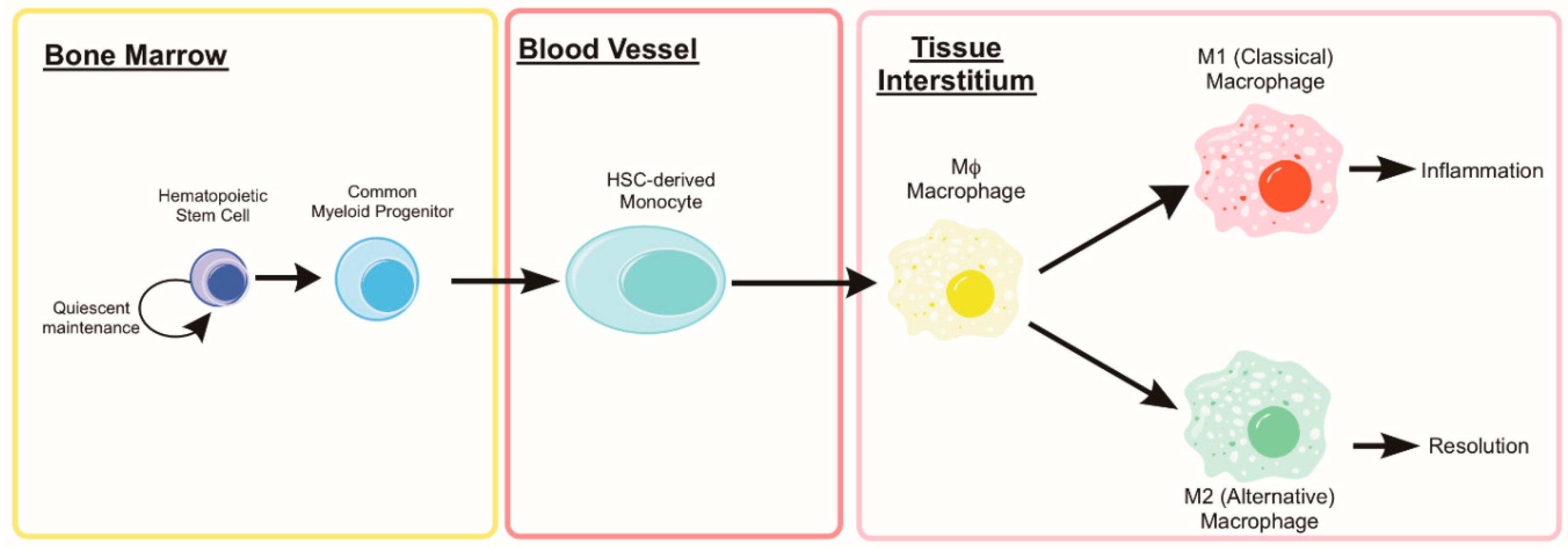
5.1. Autophagy in Monocyte Development and Macrophage Differentiation
5.2. Monocyte and Macrophage Heterogeneity
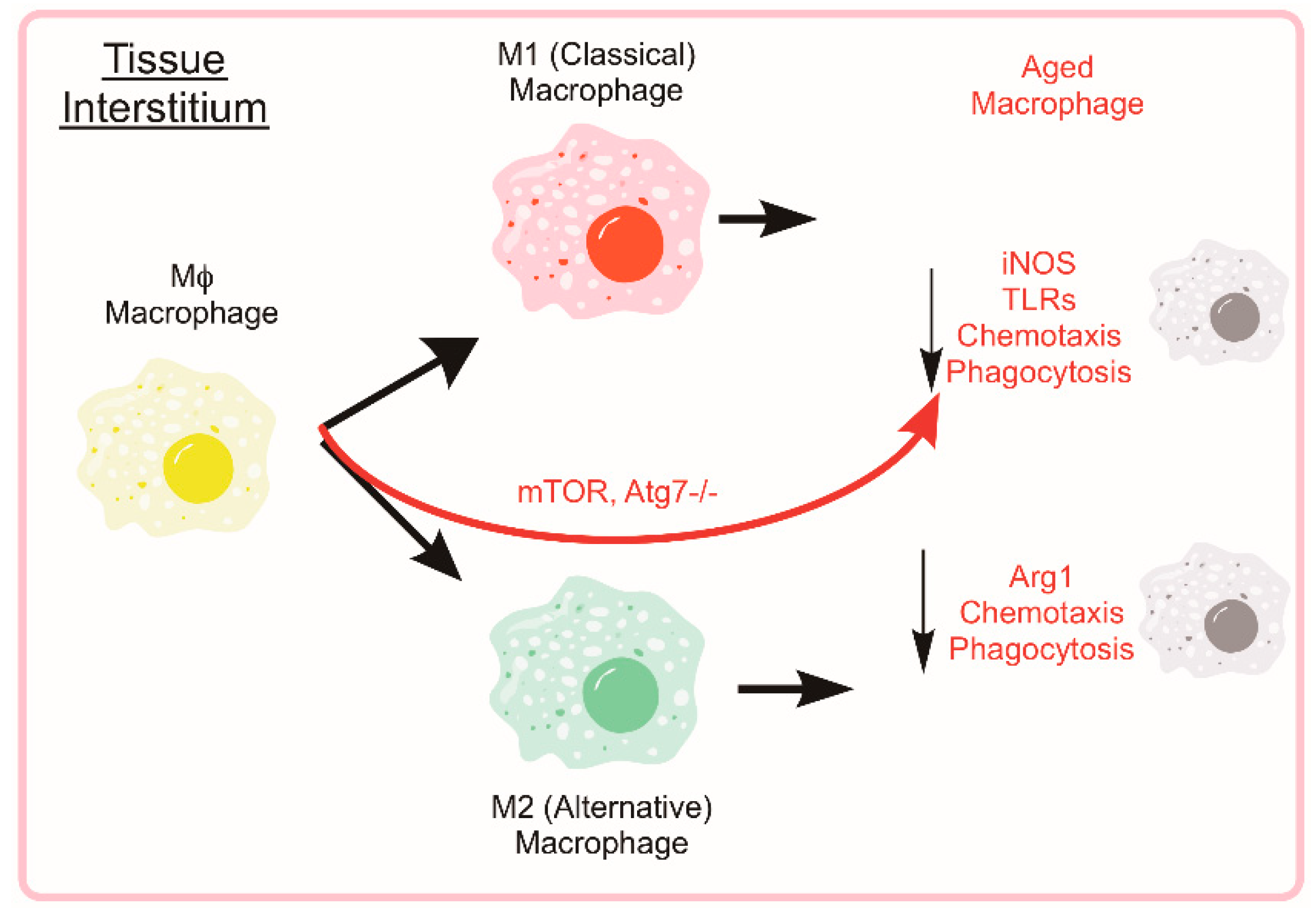
5.3. Autophagy and Macrophage Function and Phenotype
5.4. Age-Associated Suppression of Monocyte/Macrophage Function
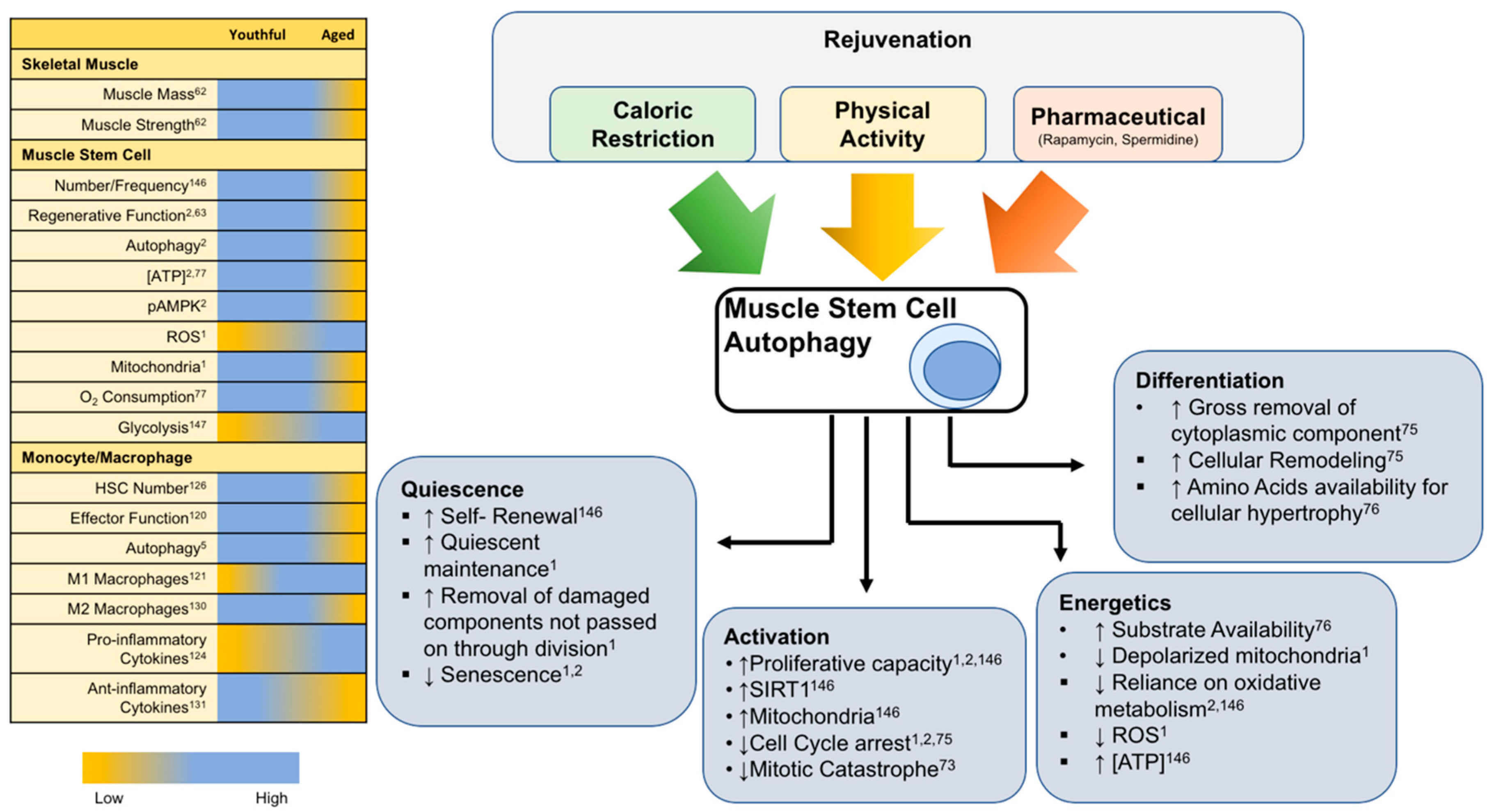
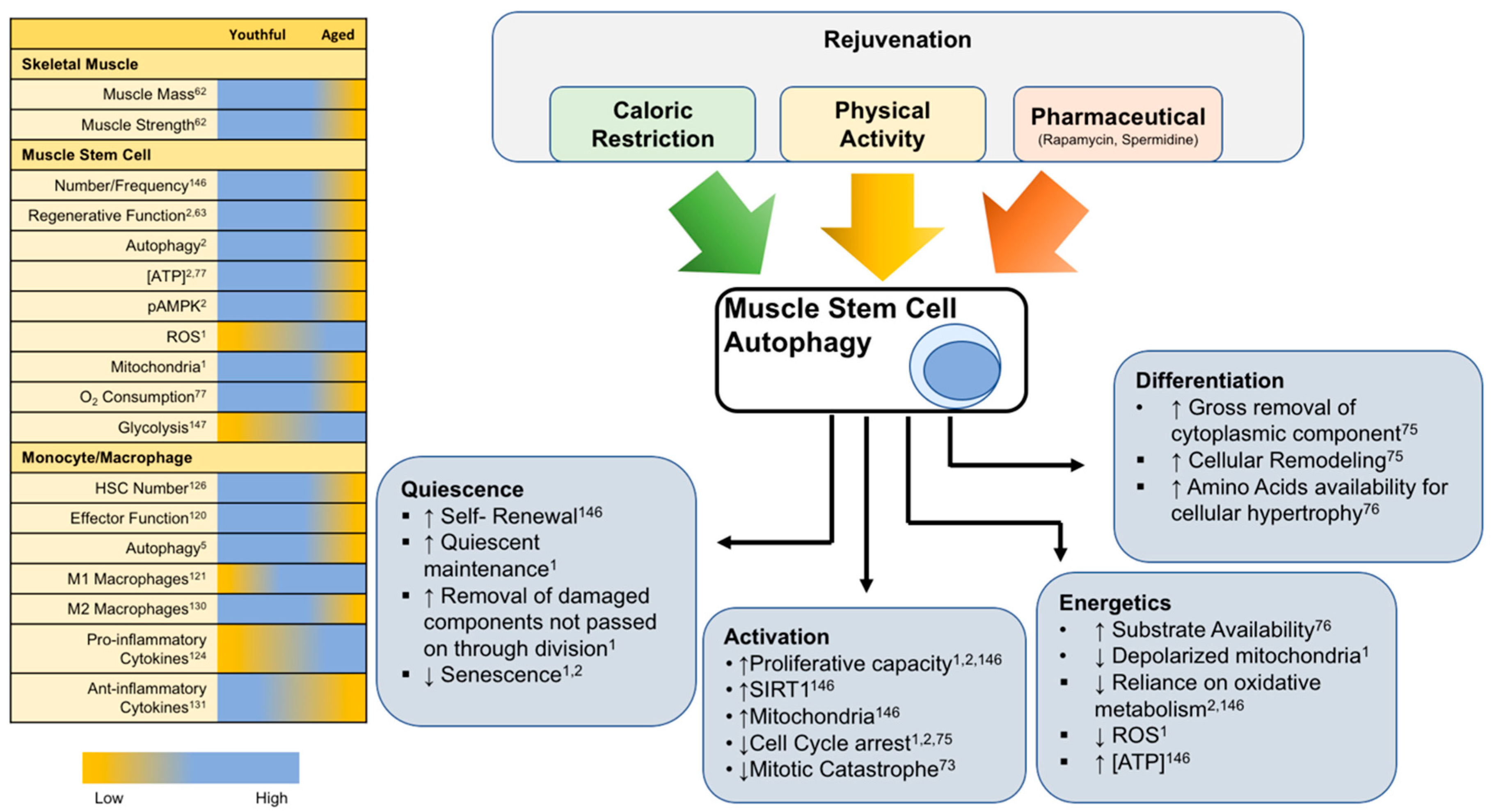
6. Rejuvenation of Autophagy for Muscle Regeneration
6.1. Rejuvenation of Satellite Cells through Exercise and CR
6.1.1. Exercise
6.1.2. Caloric Restriction
6.2. Rejuvenation of Macrophages through Exercise and CR
6.2.1. Exercise
6.2.2. Caloric Restriction
6.3. Pharmacological Inducers of Autophagy
7. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.P.; Billin, A.N.; Campbell, M.E.; Russell, A.J.; Huffman, K.M.; Kraus, W.E. The AMPK/p27(Kip1) Axis Regulates Autophagy/Apoptosis Decisions in Aged Skeletal Muscle Stem Cells. Stem Cell Rep. 2018, 11, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef]
- De Duve, C. The lysosome. Sci. Am. 1963, 208, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortimore, G.E.; Schworer, C.M. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 1977, 270, 174. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huang, J.; Geng, J.; Nair, U.; Klionsky, D.J.; Brodsky, J. Atg22 Recycles Amino Acids to Link the Degradative and Recycling Functions of Autophagy. Mol. Biol. Cell 2006, 17, 5094–5104. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Copetti, T.; Bertoli, C.; Dalla, E.; Demarchi, F.; Schneider, C. p65/RelA modulates BECN1 transcription and autophagy. Mol. Cell. Biol. 2009, 29, 2594–2608. [Google Scholar] [CrossRef] [PubMed]
- Bohensky, J.; Shapiro, I.M.; Leshinsky, S.; Terkhorn, S.P.; Adams, C.S.; Srinivas, V. HIF-1 regulation of chondrocyte apoptosis: Induction of the autophagic pathway. Autophagy 2007, 3, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Hypoxia, MTOR and autophagy: Converging on senescence or quiescence. Autophagy 2013, 9, 260–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Matecic, M.; Smith, D.L., Jr.; Pan, X.; Maqani, N.; Bekiranov, S.; Boeke, J.D.; Smith, J.S. A Microarray-Based Genetic Screen for Yeast Chronological Aging Factors. PLoS Genet. 2010, 6, e1000921. [Google Scholar] [CrossRef] [PubMed]
- Tóth, M.L.; Sigmond, T.; Borsos, É.; Barna, J.; Erdélyi, P.; Takács-Vellai, K.; Orosz, L.; Kovács, A.L.; Csikós, G.; Sass, M.; et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008, 4, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- De Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Man, C.D.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the Longevity-Associated Protein Sirtuin 1 in Insulin Resistance and Metabolic Syndrome: Potential Biochemical Mechanisms. Diabetes 2010, 59, 1006–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramés, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decuypere, J.P.; Monaco, G.; Missiaen, L.; De Smedt, H.; Parys, J.B.; Bultynck, G. IP(3) Receptors, Mitochondria, and Ca Signaling: Implications for Aging. J. Aging Res. 2011, 2011, 920178. [Google Scholar] [CrossRef]
- Xiao, M.; Li, L.; Hu, Q.; Ma, L.; Liu, L.; Chu, W.; Zhang, H. Rapamycin reduces burn wound progression by enhancing autophagy in deep second-degree burn in rats. Wound Repair Regen. 2013, 21, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- De Palma, C.; Morisi, F.; Cheli, S.; Pambianco, S.; Cappello, V.; Vezzoli, M.; Rovere-Querini, P.; Moggio, M.; Ripolone, M.; Francolini, M.; et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 2012, 3, e418. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Kou, X.; Jia, S.; Yang, X.; Yang, Y.; Chen, N. Autophagy as a Potential Target for Sarcopenia. J. Cell. Physiol. 2016, 231, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Saera-Vila, A.; Kish, P.E.; Louie, K.W.; Grzegorski, S.J.; Klionsky, D.J.; Kahana, A. Autophagy regulates cytoplasmic remodeling during cell reprogramming in a zebrafish model of muscle regeneration. Autophagy 2016, 12, 1864–1875. [Google Scholar] [CrossRef]
- Brown, J.L.; Lee, D.E.; Rosa-Caldwell, M.E.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Huseman, K.; Sataranatarajan, K.; Van Remmen, H.; Washington, T.A.; et al. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J. CachexiaSarcopenia Muscle 2018, 9, 987–1002. [Google Scholar] [CrossRef]
- White, J.P.; Baynes, J.W.; Welle, S.L.; Kostek, M.C.; Matesic, L.E.; Sato, S.; Carson, J.A. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS ONE 2011, 6, e24650. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010, 24, 3052–3065. [Google Scholar] [CrossRef]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S.; Hoehn, K.L.; Yan, Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013, 27, 4184–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichenko, A.S.; Southern, W.M.; Atuan, M.; Luan, J.; Peissig, K.B.; Foltz, S.J.; Beedle, A.M.; Warren, G.L.; Call, J.A. Mitochondrial maintenance via autophagy contributes to functional skeletal muscle regeneration and remodeling. Am. J. Physiol. Cell Physiol. 2016, 311, C190–C200. [Google Scholar] [CrossRef] [PubMed]
- Fiacco, E.; Castagnetti, F.; Bianconi, V.; Madaro, L.; De Bardi, M.; Nazio, F.; D’Amico, A.; Bertini, E.; Cecconi, F.; Puri, P.L.; et al. Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death Differ. 2016, 23, 1839–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolini, A.; Omairi, S.; Mitchell, R.; Vaughan, D.; Matsakas, A.; Vaiyapuri, S.; Ricketts, T.; Rubinsztein, D.C.; Patel, K. Attenuation of autophagy impacts on muscle fibre development, starvation induced stress and fibre regeneration following acute injury. Sci. Rep. 2018, 8, 9062. [Google Scholar] [CrossRef] [PubMed]
- Call, J.A.; Wilson, R.J.; Laker, R.C.; Zhang, M.; Kundu, M.; Yan, Z. Ulk1-mediated autophagy plays an essential role in mitochondrial remodeling and functional regeneration of skeletal muscle. Am. J. Physiol. Cell Physiol. 2017, 312, C724–C732. [Google Scholar] [CrossRef]
- McMillan, E.M.; Quadrilatero, J. Autophagy is required and protects against apoptosis during myoblast differentiation. Biochem. J. 2014, 462, 267–277. [Google Scholar] [CrossRef]
- Chazaud, B. Inflammation during skeletal muscle regeneration and tissue remodeling: Application to exercise-induced muscle damage management. Immunol. Cell Biol. 2016, 94, 140–145. [Google Scholar] [CrossRef]
- Studitsky, A.N. FREE AUTO- AND HOMOGRAFTS OF MUSCLE TISSUE IN EXPERIMENTS ON ANIMALS. Ann. N. Y. Acad. Sci. 1964, 120, 789–801. [Google Scholar] [CrossRef]
- Clevers, H. STEM CELLS. What is an adult stem cell? Science 2015, 350, 1319–1320. [Google Scholar] [CrossRef]
- Moss, F.P.; Leblond, C.P. Satellite cells as the source of nuclei in muscles of growing rats. Anat. Rec. 1971, 170, 421–435. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J.; Mula, J.; Miyazaki, M.; Erfani, R.; Garrison, K.; Farooqui, A.B.; Srikuea, R.; Lawson, B.A.; Grimes, B.; Keller, C.; et al. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development 2011, 138, 3657–3666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.J.; Hutcheson, D.A.; Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138, 3625–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepper, C.; Partridge, T.A.; Fan, C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 2011, 138, 3639–3646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Schultz, E.; Lipton, B.H. Skeletal muscle satellite cells: Changes in proliferation potential as a function of age. Mech. Ageing Dev. 1982, 20, 377–383. [Google Scholar] [CrossRef]
- Conboy, I.M.; Conboy, M.J.; Smythe, G.M.; Rando, T.A. Notch-mediated restoration of regenerative potential to aged muscle. Science 2003, 302, 1575–1577. [Google Scholar] [CrossRef]
- Blau, H.M.; Cosgrove, B.D.; Ho, A.T. The central role of muscle stem cells in regenerative failure with aging. Nat. Med. 2015, 21, 854–862. [Google Scholar] [CrossRef] [Green Version]
- Relaix, F.; Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns centre stage. Development 2012, 139, 2845–2856. [Google Scholar] [CrossRef]
- Rathbone, C.R.; Wenke, J.C.; Warren, G.L.; Armstrong, R.B. Importance of satellite cells in the strength recovery after eccentric contraction-induced muscle injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R1490–R1495. [Google Scholar] [CrossRef]
- Lowe, D.A.; Alway, S.E. Stretch-induced myogenin, MyoD, and MRF4 expression and acute hypertrophy in quail slow-tonic muscle are not dependent upon satellite cell proliferation. Cell Tissue Res. 1999, 296, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Fielding, R.A.; Vellas, B.; Evans, W.J.; Bhasin, S.; Morley, J.E.; Newman, A.B.; Abellan van Kan, G.; Andrieu, S.; Bauer, J.; Breuille, D.; et al. Sarcopenia: An undiagnosed condition in older adults. Current consensus definition: Prevalence, etiology, and consequences. International working group on sarcopenia. J. Am. Med Dir. Assoc. 2011, 12, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Lee, J.D.; Mula, J.; Kirby, T.J.; Jackson, J.R.; Liu, F.; Yang, L.; Mendias, C.L.; Dupont-Versteegden, E.E.; McCarthy, J.J.; et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 2015, 21, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Shefer, G.; Rauner, G.; Yablonka-Reuveni, Z.; Benayahu, D. Reduced satellite cell numbers and myogenic capacity in aging can be alleviated by endurance exercise. PLoS ONE 2010, 5, e13307. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.A.; Zammit, P.S.; Ruiz, A.P.; Morgan, J.E.; Partridge, T.A. A population of myogenic stem cells that survives skeletal muscle aging. Stem Cells (Dayt. Ohio) 2007, 25, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Brooks, N.E.; Schuenke, M.D.; Hikida, R.S. No change in skeletal muscle satellite cells in young and aging rat soleus muscle. J. Physiol. Sci. 2009, 59, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Hikida, R.S. Aging changes in satellite cells and their functions. Curr. Aging Sci. 2011, 4, 279–297. [Google Scholar] [CrossRef]
- Bernet, J.D.; Doles, J.D.; Hall, J.K.; Kelly Tanaka, K.; Carter, T.A.; Olwin, B.B. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Collins-Hooper, H.; Woolley, T.E.; Dyson, L.; Patel, A.; Potter, P.; Baker, R.E.; Gaffney, E.A.; Maini, P.K.; Dash, P.R.; Patel, K. Age-related changes in speed and mechanism of adult skeletal muscle stem cell migration. Stem Cells (Dayt. Ohio) 2012, 30, 1182–1195. [Google Scholar] [CrossRef]
- Chakkalakal, J.V.; Jones, K.M.; Basson, M.A.; Brack, A.S. The aged niche disrupts muscle stem cell quiescence. Nature 2012, 490, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, B.D.; Gilbert, P.M.; Porpiglia, E.; Mourkioti, F.; Lee, S.P.; Corbel, S.Y.; Llewellyn, M.E.; Delp, S.L.; Blau, H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014, 20, 255–264. [Google Scholar] [CrossRef]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Charville, G.W.; Cheung, T.H.; Yoo, B.; Santos, P.J.; Schroeder, M.; Rando, T.A. Impaired Notch Signaling Leads to a Decrease in p53 Activity and Mitotic Catastrophe in Aged Muscle Stem Cells. Cell Stem Cell 2018. [Google Scholar] [CrossRef]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef]
- Wen, X.; Klionsky, D.J. Autophagy is a key factor in maintaining the regenerative capacity of muscle stem cells by promoting quiescence and preventing senescence. Autophagy 2016, 12, 617–618. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.H.; Rando, T.A. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 2014, 33, 2782–2797. [Google Scholar] [CrossRef] [Green Version]
- Ryall, J.G. Simultaneous Measurement of Mitochondrial and Glycolytic Activity in Quiescent Muscle Stem Cells. Methods Mol. Biol. (Clifton N.J.) 2017, 1556, 245–253. [Google Scholar] [CrossRef]
- Jejurikar, S.S.; Henkelman, E.A.; Cederna, P.S.; Marcelo, C.L.; Urbanchek, M.G.; Kuzon, W.M., Jr. Aging increases the susceptibility of skeletal muscle derived satellite cells to apoptosis. Exp. Gerontol. 2006, 41, 828–836. [Google Scholar] [CrossRef]
- Godwin, J.W.; Pinto, A.R.; Rosenthal, N.A. Macrophages are required for adult salamander limb regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 9415–9420. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yan, B.; Shi, Y.Q.; Zhang, W.Q.; Wen, Z.L. Live imaging reveals differing roles of macrophages and neutrophils during zebrafish tail fin regeneration. J. Biol. Chem. 2012, 287, 25353–25360. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [Green Version]
- van Amerongen, M.J.; Harmsen, M.C.; van Rooijen, N.; Petersen, A.H.; van Luyn, M.J. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am. J. Pathol. 2007, 170, 818–829. [Google Scholar] [CrossRef]
- Sieweke, M.H.; Allen, J.E. Beyond stem cells: Self-renewal of differentiated macrophages. Science 2013, 342, 1242974. [Google Scholar] [CrossRef]
- Saclier, M.; Cuvellier, S.; Magnan, M.; Mounier, R.; Chazaud, B. Monocyte/macrophage interactions with myogenic precursor cells during skeletal muscle regeneration. FEBS J. 2013, 280, 4118–4130. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Macian, F. Autophagy and the immune function in aging. Curr. Opin. Immunol. 2014, 29, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Mohyeldin, A.; Garzon-Muvdi, T.; Quinones-Hinojosa, A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.L.; Simon, A.K.; Prescott, M.; Menendez, J.A.; Liu, F.; Wang, F.; Wang, C.; Wolvetang, E.; Vazquez-Martin, A.; Zhang, J. Autophagy in stem cells. Autophagy 2013, 9, 830–849. [Google Scholar] [CrossRef] [Green Version]
- Salemi, S.; Yousefi, S.; Constantinescu, M.A.; Fey, M.F.; Simon, H.U. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 2012, 22, 432–435. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.B.; Aifantis, I. Regulation of hematopoietic stem cell fate by the ubiquitin proteasome system. Trends Immunol. 2012, 33, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegue, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [Green Version]
- Phadwal, K.; Alegre-Abarrategui, J.; Watson, A.S.; Pike, L.; Anbalagan, S.; Hammond, E.M.; Wade-Martins, R.; McMichael, A.; Klenerman, P.; Simon, A.K. A novel method for autophagy detection in primary cells: Impaired levels of macroautophagy in immunosenescent T cells. Autophagy 2012, 8, 677–689. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, Y.; Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2009, 2, ra75. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.L.; Liu, Y.; Zheng, P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef]
- Kharas, M.G.; Okabe, R.; Ganis, J.J.; Gozo, M.; Khandan, T.; Paktinat, M.; Gilliland, D.G.; Gritsman, K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010, 115, 1406–1415. [Google Scholar] [CrossRef]
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z.G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905. [Google Scholar] [CrossRef] [Green Version]
- Jacquel, A.; Obba, S.; Boyer, L.; Dufies, M.; Robert, G.; Gounon, P.; Lemichez, E.; Luciano, F.; Solary, E.; Auberger, P. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 2012, 119, 4527–4531. [Google Scholar] [CrossRef] [Green Version]
- Jacquel, A.; Obba, S.; Solary, E.; Auberger, P. Proper macrophagic differentiation requires both autophagy and caspase activation. Autophagy 2012, 8, 1141–1143. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Lemos, D.R.; Babaeijandaghi, F.; Low, M.; Chang, C.K.; Lee, S.T.; Fiore, D.; Zhang, R.H.; Natarajan, A.; Nedospasov, S.A.; Rossi, F.M. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 2015, 21, 786–794. [Google Scholar] [CrossRef]
- Varga, T.; Mounier, R.; Horvath, A.; Cuvellier, S.; Dumont, F.; Poliska, S.; Ardjoune, H.; Juban, G.; Nagy, L.; Chazaud, B. Highly Dynamic Transcriptional Signature of Distinct Macrophage Subsets during Sterile Inflammation, Resolution, and Tissue Repair. J. Immunol. 2016, 196, 4771–4782. [Google Scholar] [CrossRef] [Green Version]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef]
- Chang, C.P.; Su, Y.C.; Lee, P.H.; Lei, H.Y. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy 2013, 9, 619–621. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.P.; Su, Y.C.; Hu, C.W.; Lei, H.Y. TLR2-dependent selective autophagy regulates NF-kappaB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. 2013, 20, 515–523. [Google Scholar] [CrossRef]
- Chen, W.; Ma, T.; Shen, X.N.; Xia, X.F.; Xu, G.D.; Bai, X.L.; Liang, T.B. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012, 72, 1363–1372. [Google Scholar] [CrossRef]
- Chen, P.; Cescon, M.; Bonaldo, P. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy 2014, 10, 192–200. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Stranks, A.J.; Hansen, A.L.; Panse, I.; Mortensen, M.; Ferguson, D.J.; Puleston, D.J.; Shenderov, K.; Watson, A.S.; Veldhoen, M.; Phadwal, K.; et al. Autophagy Controls Acquisition of Aging Features in Macrophages. J. Innate Immun. 2015, 7, 375–391. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.; Cunha, L.D.; Park, S.; Yang, M.; Lu, Q.; Orchard, R.; Li, Q.Z.; Yan, M.; Janke, L.; Guy, C.; et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 2016, 533, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Yokoyama, C.C.; Williams, J.W.; Baldridge, M.T.; Jin, X.; DesRochers, B.; Bricker, T.; Wilen, C.B.; Bagaitkar, J.; Loginicheva, E.; et al. Homeostatic Control of Innate Lung Inflammation by Vici Syndrome Gene Epg5 and Additional Autophagy Genes Promotes Influenza Pathogenesis. Cell Host Microbe 2016, 19, 102–113. [Google Scholar] [CrossRef]
- Kang, Y.H.; Cho, M.H.; Kim, J.Y.; Kwon, M.S.; Peak, J.J.; Kang, S.W.; Yoon, S.Y.; Song, Y. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget 2016, 7, 35577–35591. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Hallowell, R.W.; Collins, S.L.; Craig, J.M.; Zhang, Y.; Oh, M.; Illei, P.B.; Chan-Li, Y.; Vigeland, C.L.; Mitzner, W.; Scott, A.L.; et al. mTORC2 signalling regulates M2 macrophage differentiation in response to helminth infection and adaptive thermogenesis. Nat. Commun. 2017, 8, 14208. [Google Scholar] [CrossRef] [Green Version]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 2834. [Google Scholar] [CrossRef] [Green Version]
- Pawelec, G.; Larbi, A.; Derhovanessian, E. Senescence of the human immune system. J. Comp. Pathol. 2010, 142 (Suppl. 1), S39–S44. [Google Scholar] [CrossRef]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Koike, E.; Kobayashi, T.; Mochitate, K.; Murakami, M. Effect of aging on nitric oxide production by rat alveolar macrophages. Exp. Gerontol. 1999, 34, 889–894. [Google Scholar] [CrossRef]
- Renshaw, M.; Rockwell, J.; Engleman, C.; Gewirtz, A.; Katz, J.; Sambhara, S. Cutting edge: impaired Toll-like receptor expression and function in aging. J. Immunol. 2002, 169, 4697–4701. [Google Scholar] [CrossRef]
- Herrero, C.; Marques, L.; Lloberas, J.; Celada, A. IFN-gamma-dependent transcription of MHC class II IA is impaired in macrophages from aged mice. J. Clin. Investig. 2001, 107, 485–493. [Google Scholar] [CrossRef]
- Henry, C.J.; Huang, Y.; Wynne, A.M.; Godbout, J.P. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. BrainBehav. Immun. 2009, 23, 309–317. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- De Martinis, M.; Franceschi, C.; Monti, D.; Ginaldi, L. Inflammation markers predicting frailty and mortality in the elderly. Exp. Mol. Pathol. 2006, 80, 219–227. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Witkowski, J.M.; McElhaney, J.; Loeb, M.; Mitnitski, A.; Pawelec, G. Aging, frailty and age-related diseases. Biogerontology 2010, 11, 547–563. [Google Scholar] [CrossRef]
- Effros, R.B.; Svoboda, K.; Walford, R.L. Influence of age and caloric restriction on macrophage IL-6 and TNF production. Lymphokine Cytokine Res. 1991, 10, 347–351. [Google Scholar]
- Mahbub, S.; Deburghgraeve, C.R.; Kovacs, E.J. Advanced Age Impairs Macrophage Polarization. J. Interferon Cytokine Res. 2012, 32, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Sandmand, M.; Bruunsgaard, H.; Kemp, K.; Andersen-Ranberg, K.; Pedersen, A.N.; Skinhøj, P. Is ageing associated with a shift in the balance between Type 1 and Type 2 cytokines in humans? Clin. Exp. Immunol. 2002, 127, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Dethlefsen, M.M.; Kristensen, C.M.; Tondering, A.S.; Lassen, S.B.; Ringholm, S.; Pilegaard, H. Impact of liver PGC-1alpha on exercise and exercise training-induced regulation of hepatic autophagy and mitophagy in mice on HFF. Physiol. Rep. 2018, 6, e13731. [Google Scholar] [CrossRef]
- He, C.; Sumpter, R., Jr.; Levine, B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012, 8, 1548–1551. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Licastro, D.; Cava, E.; Veronese, N.; Spelta, F.; Rizza, W.; Bertozzi, B.; Villareal, D.T.; Hotamisligil, G.S.; Holloszy, J.O.; et al. Long-Term Calorie Restriction Enhances Cellular Quality-Control Processes in Human Skeletal Muscle. Cell Rep. 2016, 14, 422–428. [Google Scholar] [CrossRef] [Green Version]
- Milton-Laskibar, I.; Aguirre, L.; Etxeberria, U.; Milagro, F.I.; Martinez, J.A.; Portillo, M.P. Involvement of autophagy in the beneficial effects of resveratrol in hepatic steatosis treatment. A comparison with energy restriction. Food Funct. 2018, 9, 4207–4215. [Google Scholar] [CrossRef]
- Holloszy, J.O. Mortality rate and longevity of food-restricted exercising male rats: A reevaluation. J. Appl. Physiol. 1997, 82, 399–403. [Google Scholar] [CrossRef]
- McKiernan, S.H.; Colman, R.J.; Lopez, M.; Beasley, T.M.; Aiken, J.M.; Anderson, R.M.; Weindruch, R. Caloric restriction delays aging-induced cellular phenotypes in rhesus monkey skeletal muscle. Exp. Gerontol. 2011, 46, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, S.; Pietrangelo, L.; Loefler, S.; Fruhmann, H.; Vogelauer, M.; Burggraf, S.; Pond, A.; Grim-Stieger, M.; Cvecka, J.; Sedliak, M.; et al. Lifelong physical exercise delays age-associated skeletal muscle decline. J. Gerontol. Ser. ABiol. Sci. Med Sci. 2015, 70, 163–173. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Schechtman, K.B. Interaction between exercise and food restriction: effects on longevity of male rats. J. Appl. Physiol. 1991, 70, 1529–1535. [Google Scholar] [CrossRef]
- Goh, J.; Ladiges, W.C. Exercise enhances wound healing and prevents cancer progression during aging by targeting macrophage polarity. Mech. Ageing Dev. 2014, 139, 41–48. [Google Scholar] [CrossRef]
- Shefer, G.; Rauner, G.; Stuelsatz, P.; Benayahu, D.; Yablonka-Reuveni, Z. Moderate-intensity treadmill running promotes expansion of the satellite cell pool in young and old mice. FEBS J. 2013, 280, 4063–4073. [Google Scholar] [CrossRef]
- Fujimaki, S.; Hidaka, R.; Asashima, M.; Takemasa, T.; Kuwabara, T. Wnt protein-mediated satellite cell conversion in adult and aged mice following voluntary wheel running. J. Biol. Chem. 2014, 289, 7399–7412. [Google Scholar] [CrossRef]
- Vanfleteren, J.R.; Braeckman, B.P. Mechanisms of life span determination in Caenorhabditis elegans. Neurobiol. Aging 1999, 20, 487–502. [Google Scholar] [CrossRef]
- Il’yasova, D.; Fontana, L.; Bhapkar, M.; Pieper, C.F.; Spasojevic, I.; Redman, L.M.; Das, S.K.; Huffman, K.M.; Kraus, W.E. Effects of 2 years of caloric restriction on oxidative status assessed by urinary F2-isoprostanes: The CALERIE 2 randomized clinical trial. Aging Cell 2018, 17. [Google Scholar] [CrossRef]
- Mazzoccoli, G.; Tevy, M.F.; Borghesan, M.; Delle Vergini, M.R.; Vinciguerra, M. Caloric restriction and aging stem cells: The stick and the carrot? Exp. Gerontol. 2014, 50, 137–148. [Google Scholar] [CrossRef]
- Cerletti, M.; Jang, Y.C.; Finley, L.W.; Haigis, M.C.; Wagers, A.J. Short-term calorie restriction enhances skeletal muscle stem cell function. Cell Stem Cell 2012, 10, 515–519. [Google Scholar] [CrossRef]
- Pala, F.; Di Girolamo, D.; Mella, S.; Yennek, S.; Chatre, L.; Ricchetti, M.; Tajbakhsh, S. Distinct metabolic states govern skeletal muscle stem cell fates during prenatal and postnatal myogenesis. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Fehr, H.-G.; Lötzerich, H.; Michna, H. Human macrophage function and physical exercise: Phagocytic and histochemical studies. Eur. J. Appl. Physiol. Occup. Physiol. 1989, 58, 613–617. [Google Scholar] [CrossRef]
- Bartlett, D.B.; Shepherd, S.O.; Wilson, O.J.; Adlan, A.M.; Wagenmakers, A.J.M.; Shaw, C.S.; Lord, J.M. Neutrophil and Monocyte Bactericidal Responses to 10 Weeks of Low-Volume High-Intensity Interval or Moderate-Intensity Continuous Training in Sedentary Adults. Oxid. Med. Cell. Longev. 2017, 2017, 8148742. [Google Scholar] [CrossRef]
- Bartlett, D.B.; Fox, O.; McNulty, C.L.; Greenwood, H.L.; Murphy, L.; Sapey, E.; Goodman, M.; Crabtree, N.; Thogersen-Ntoumani, C.; Fisher, J.P.; et al. Habitual physical activity is associated with the maintenance of neutrophil migratory dynamics in healthy older adults. Brain Behav. Immun. 2016, 56, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Terra, R.; Alves, P.J.; Goncalves da Silva, S.A.; Salerno, V.P.; Dutra, P.M. Exercise improves the Th1 response by modulating cytokine and NO production in BALB/c mice. Int. J. Sports Med. 2013, 34, 661–666. [Google Scholar] [CrossRef]
- Kawanishi, N.; Yano, H.; Yokogawa, Y.; Suzuki, K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc. Immunol. Rev. 2010, 16, 105–118. [Google Scholar]
- Fabbiano, S.; Suarez-Zamorano, N.; Rigo, D.; Veyrat-Durebex, C.; Stevanovic Dokic, A.; Colin, D.J.; Trajkovski, M. Caloric Restriction Leads to Browning of White Adipose Tissue through Type 2 Immune Signaling. Cell Metab. 2016, 24, 434–446. [Google Scholar] [CrossRef] [Green Version]
- Neff, F.; Flores-Dominguez, D.; Ryan, D.P.; Horsch, M.; Schroder, S.; Adler, T.; Afonso, L.C.; Aguilar-Pimentel, J.A.; Becker, L.; Garrett, L.; et al. Rapamycin extends murine lifespan but has limited effects on aging. J. Clin. Investig. 2013, 123, 3272–3291. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Buttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef]
- Fan, J.; Yang, X.; Li, J.; Shu, Z.; Dai, J.; Liu, X.; Li, B.; Jia, S.; Kou, X.; Yang, Y.; et al. Spermidine coupled with exercise rescues skeletal muscle atrophy from D-gal-induced aging rats through enhanced autophagy and reduced apoptosis via AMPK-FOXO3a signal pathway. Oncotarget 2017, 8, 17475–17490. [Google Scholar] [CrossRef] [Green Version]
- Chrisam, M.; Pirozzi, M.; Castagnaro, S.; Blaauw, B.; Polishchuck, R.; Cecconi, F.; Grumati, P.; Bonaldo, P. Reactivation of autophagy by spermidine ameliorates the myopathic defects of collagen VI-null mice. Autophagy 2015, 11, 2142–2152. [Google Scholar] [CrossRef] [Green Version]
- Borzi, R.M.; Guidotti, S.; Minguzzi, M.; Facchini, A.; Platano, D.; Trisolino, G.; Filardo, G.; Cetrullo, S.; D’Adamo, S.; Stefanelli, C.; et al. Polyamine delivery as a tool to modulate stem cell differentiation in skeletal tissue engineering. Amino Acids 2014, 46, 717–728. [Google Scholar] [CrossRef]
- Zhang, L.; Gong, H.; Sun, Q.; Zhao, R.; Jia, Y. Spermidine-Activated Satellite Cells Are Associated with Hypoacetylation in ACVR2B and Smad3 Binding to Myogenic Genes in Mice. J. Agric. Food Chem. 2018, 66, 540–550. [Google Scholar] [CrossRef]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Zheng, C.; Cao, J.; Cao, G.; Shou, P.; Lin, L.; Velletri, T.; Jiang, M.; Chen, Q.; Han, Y.; et al. Spermidine alleviates experimental autoimmune encephalomyelitis through inducing inhibitory macrophages. Cell Death Differ. 2016, 23, 1850–1861. [Google Scholar] [CrossRef] [Green Version]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.E.; Bareja, A.; Bartlett, D.B.; White, J.P. Autophagy as a Therapeutic Target to Enhance Aged Muscle Regeneration. Cells 2019, 8, 183. https://doi.org/10.3390/cells8020183
Lee DE, Bareja A, Bartlett DB, White JP. Autophagy as a Therapeutic Target to Enhance Aged Muscle Regeneration. Cells. 2019; 8(2):183. https://doi.org/10.3390/cells8020183
Chicago/Turabian StyleLee, David E., Akshay Bareja, David B. Bartlett, and James P. White. 2019. "Autophagy as a Therapeutic Target to Enhance Aged Muscle Regeneration" Cells 8, no. 2: 183. https://doi.org/10.3390/cells8020183
APA StyleLee, D. E., Bareja, A., Bartlett, D. B., & White, J. P. (2019). Autophagy as a Therapeutic Target to Enhance Aged Muscle Regeneration. Cells, 8(2), 183. https://doi.org/10.3390/cells8020183