Behavior of Metalloproteinases in Adipose Tissue, Liver and Arterial Wall: An Update of Extracellular Matrix Remodeling


{kind=link}
{kind=link}
Abstract
1. Introduction
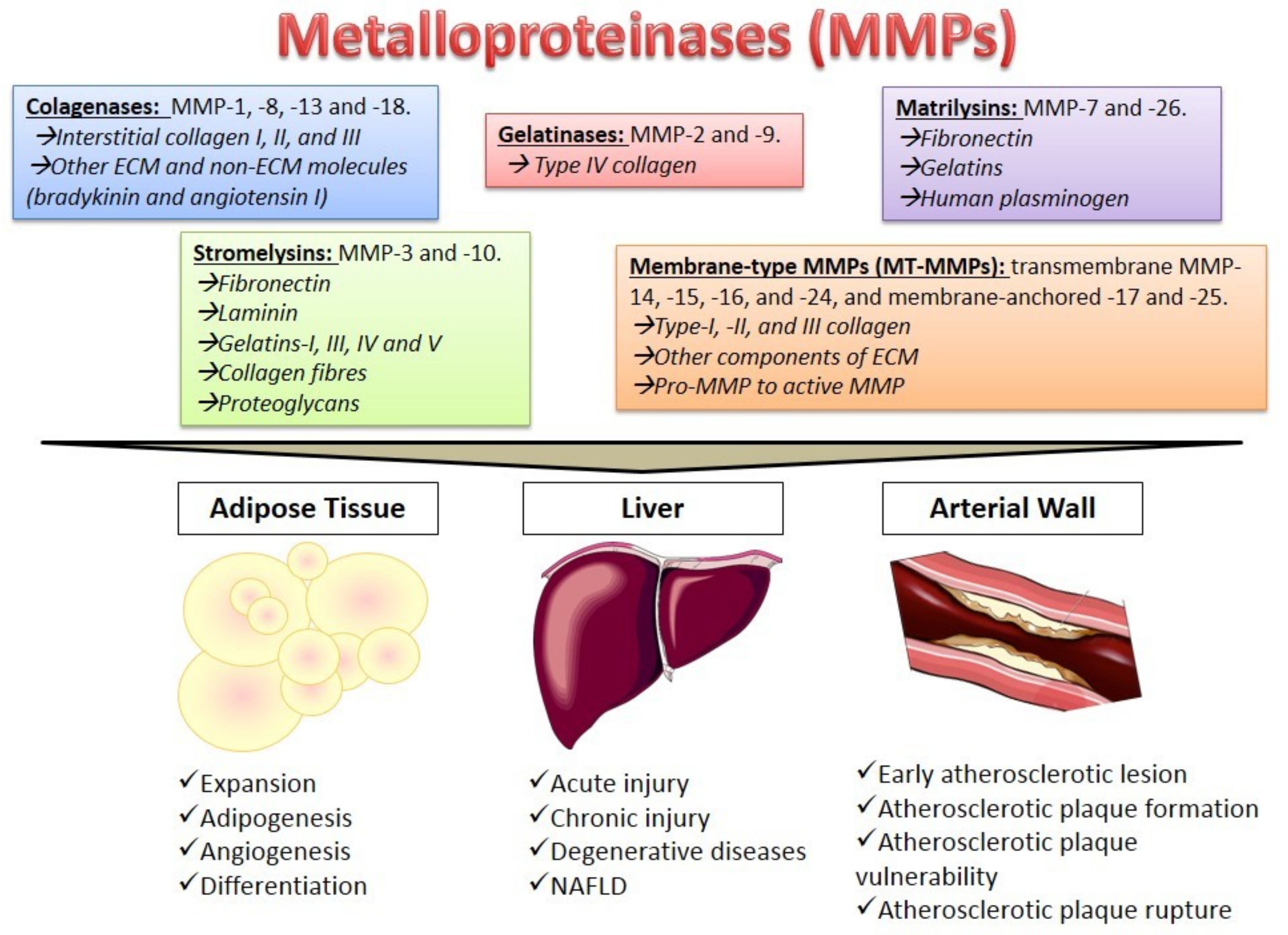
2. Metalloproteinases Characteristics
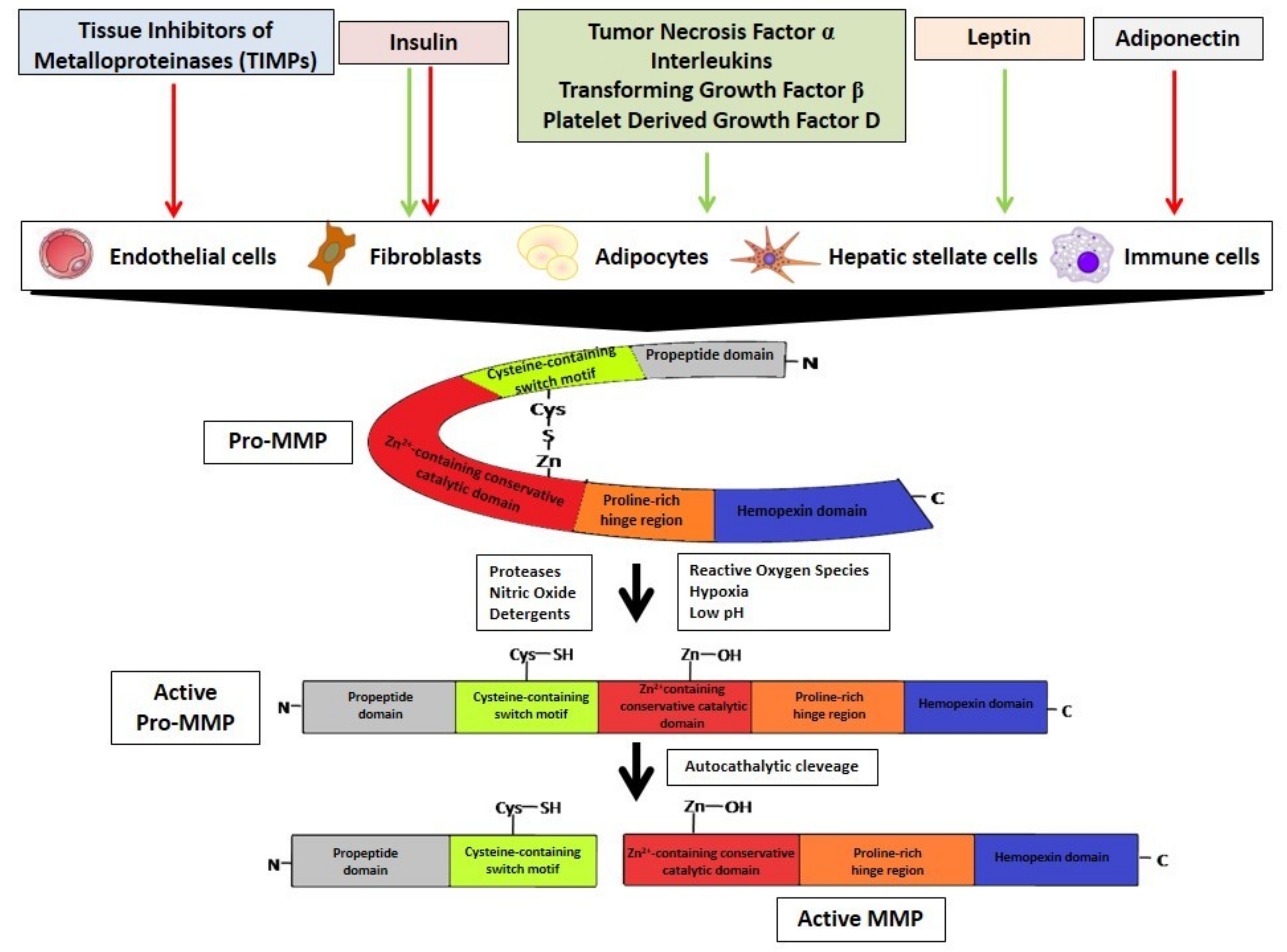
3. MMPs Regulation
4. The Role of MMPs in Different Tissues
4.1. Adipose Tissue
4.2. The Liver
4.3. The Arterial Wall
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Robert, S.; Gicquel, T.; Victoni, T.; Valença, S.; Barreto, E.; Bailly-Maître, B. Involvement of matrix metalloproteinases (MMPs) and inflammasome pathway in molecular mechanisms of fibrosis. Biosci. Rep. 2016, 36, e00360. [Google Scholar] [CrossRef]
- Yue, B. Biology of the extracellular matrix: An overview. J. Glaucoma 2014, 23, S20–S23. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, M.M.; Khalil, R.A. Matrix metalloproteinase inhibitors as investigative tools in the pathogenesis and management of vascular disease. Exp. Suppl. 2012, 103, 209–279. [Google Scholar] [PubMed]
- Chen, Q.; Jin, M.; Yang, F.; Zhu, J.; Xiao, Q.; Zhang, L. Matrix metalloproteinases: Inflammatory regulators of cell behaviors in vascular formation and remodeling. Mediat. Inflamm. 2013, 2013, 928315. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Miksztowicz, V.; Schreier, L. Metalloproteinases in metabolic syndrome. Clin. Chim. Acta 2011, 412, 1731–1739. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Mustafa, A.; Yerzhan, A.; Merzhakupova, D.; Yerlan, P.; Orakov, A.; Wang, X.; Huang, Y.; Miao, L. Nuclear matrix metalloproteinases: Functions resemble the evolution from the intracellular to the extracellular compartment. Cell Death Discov. 2017, 3, 17036. [Google Scholar] [CrossRef]
- Aldonyte, R.; Brantly, M.; Block, E.; Patel, J.; Zhang, J. Nuclear localization of active matrix metalloproteinase-2 in cigarette smoke-exposed apoptotic endothelial cells. Exp. Lung Res. 2009, 35, 59–75. [Google Scholar] [PubMed]
- Tsai, J.P.; Liou, J.H.; Kao, W.T.; Wang, S.C.; Lian, J.D.; Chang, H.R. Increased expression of intranuclear matrix metalloproteinase 9 in atrophic renal tubules is associated with renal fibrosis. PLoS ONE 2012, 7, e48164. [Google Scholar] [CrossRef] [PubMed]
- Roderfeld, M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol. 2018, 68–69, 452–462. [Google Scholar] [CrossRef]
- Liang, K.C.; Lee, C.W.; Lin, W.N.; Lin, C.C.; Wu, C.B.; Luo, S.F.; Yang, C.M. Interleukin-1 beta induces MMP-9 expression via p42/p44 MAPK, p38 MAPK, JNK, and nuclear factor-kappaB signaling pathways in human tracheal smooth muscle cells. J. Cell. Physiol. 2007, 211, 759–770. [Google Scholar] [CrossRef]
- Yamamoto, K.; Murphy, G.; Troeberg, L. Extracellular regulation of metalloproteinases. Matrix Biol. 2015, 44–46, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Miksztowicz, V. Metalloproteinases in the pathogenesis and progression of metabolic syndrome: Potential targets for improved outcomes. J. Metalloproteinases in Med. 2015, 2, 51–59. [Google Scholar] [CrossRef]
- Boden, G.; Song, W.; Pashko, L.; Kresge, K. In vivo effects of insulin and free fatty acids on matrix metalloproteinases in rat aorta. Diabetes 2008, 57, 476–483. [Google Scholar] [CrossRef]
- Boden, G.; Song, W.; Kresge, K.; Mozzoli, M.; Cheung, P. Effects of hyperinsulinemia on hepatic metalloproteinases and their tissue inhibitors. Am. J. Physiol. Endocrinol. Metab. 2008, 295, 692–697. [Google Scholar] [CrossRef]
- Moon, H.S.; Lee, H.G.; Seo, J.H.; Chung, C.S.; Guo, D.D.; Kim, T.G.; Choi, Y.J.; Cho, C.S. Leptin-induced matrix metalloproteinase-2 secretion is suppressed by trans-10, cis-12 conjugated linoleic acid. Biochem. Biophys. Res. Commun. 2007, 356, 955–956. [Google Scholar] [CrossRef]
- Schram, K.; Wong, M.M.; Palanivel, R.; No, E.K.; Dixon, I.M.; Sweeney, G. Increased expression and cell surface localization of MT1-MMP plays a role in stimulation of MMP-2 activity by leptin in neonatal rat cardiac myofibroblasts. J. Mol. Cell Cardiol. 2008, 44, 874–881. [Google Scholar] [CrossRef]
- Liu, R.; Chen, B.; Chen, J.; Lan, J. Leptin upregulates smooth muscle cell expression of MMP-9 to promote plaque destabilization by activating AP-1 via the leptin receptor/MAPK/ERK signaling pathways. Exp. Ther. Med. 2018, 16, 5327–5333. [Google Scholar] [CrossRef]
- Liberale, L.; Bonaventura, A.; Carbone, F.; Bertolotto, M.; Contini, P.; Scopinaro, N.; Camerini, G.B.; Papadia, F.S.; Cordera, R.; Camici, G.G.; et al. Early reduction of matrix metalloproteinase-8 serum levels is associated with leptin drop and predicts diabetes remission after bariatric surgery. Int. J. Cardiol. 2017, 245, 257–262. [Google Scholar] [CrossRef]
- Matsuzawa, Y. Adiponectin: Identification, physiology and clinical relevance in metabolic and vascular disease. Atheroscler. Suppl. 2005, 6, 7–14. [Google Scholar] [CrossRef]
- Hu, D.; Fukuhara, A.; Miyata, Y.; Yokoyama, C.; Otsuki, M.; Kihara, S.; Shimomura, I. Adiponectin regulates vascular endothelial growth factor-C expression in macrophages via Syk-ERK pathway. PLoS ONE 2013, 8, e56071. [Google Scholar] [CrossRef] [PubMed]
- Miksztowicz, V.; Fernandez Machulsky, N.; Lucero, D.; Fassio, E.; Schreier, L.; Berg, G. Adiponectin predicts MMP-2 activity independently of obesity. Eur. J. Clin. Investig. 2014, 44, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Kou, H.; Deng, J.; Gao, D.; Song, A.; Han, Z.; Wei, J.; Jin, X.; Ma, R.; Zheng, Q. Relationship among adiponectin, insulin resistance and atherosclerosis in non-diabetic hypertensive patients and healthy adults. Clin. Exp. Hypertens. 2018, 40, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Wu, Y.; Fried, S.K. Adipose tissue remodeling in pathophysiology of obesity. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Maquoi, E.; Munaut, C.; Colige, A.; Collen, D.; Lijnen, H.R. Modulation of adipose tissue expression of murine matrix metalloproteinases and their tissue inhibitors with obesity. Diabetes 2002, 51, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Barchuk, M.; Morales, C.; Zago, V.; Friedman, S.; Schreier, L.; Miksztowicz, V.; Berg, G. Gelatinases behavior in adipose tissue, heart and liver in a diet induced obesity model. Medicina 2016, 76, 196. [Google Scholar]
- Miksztowicz, V.; Morales, C.; Zago, V.; Friedman, S.; Schreier, L.; Berg, G. Effect of insulin-resistance on circulating and adipose tissue MMP-2 and MMP-9 activity in rats fed a sucrose-rich diet. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 294–300. [Google Scholar] [CrossRef]
- Song, B.; Zhang, H.; Zhang, S. Toll-like receptor 2 mediates deposition of collagen I in adipose tissue of high fat diet-induced obese mice. Mol. Med. Rep. 2018, 17, 5958–5963. [Google Scholar] [CrossRef]
- Almalki, S.G.; Llamas Valle, Y.; Agrawal, D.K. MMP-2 and MMP-14 Silencing Inhibits VEGFR2 Cleavage and Induces the Differentiation of Porcine Adipose-Derived Mesenchymal Stem Cells to Endothelial Cells. Stem Cells Transl. Med. 2017, 6, 1385–1398. [Google Scholar] [CrossRef]
- Gummesson, A.; Hagg, D.; Olson, F.J.; Hulthe, J.; Carlsson, L.M.; Fagerberg, B. Adipose tissue is not an important source for matrix metalloproteinase-9 in the circulation. Scand. J. Clin. Lab. Investig. 2009, 69, 636–642. [Google Scholar] [CrossRef]
- Domienik-Karłowicz, J.; Rymarczyk, Z.; Dzikowska-Diduch, O.; Lisik, W.; Chmura, A.; Demkow, U.; Pruszczyk, P. Emerging markers of atherosclerosis before and after bariatric surgery. Obes. Surg. 2015, 25, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.J.; Ser, K.H.; Lin, M.T.; Nien, H.C.; Chen, C.N.; Yang, W.S.; Lee, W.J. Diabetes Associated Markers After Bariatric Surgery: Fetuin-A, but Not Matrix Metalloproteinase-7, Is Reduced. Obes. Surg. 2015, 25, 2328–2334. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Aron-Wisnewsky, J.; Marcelin, G.; Genser, L.; Le Naour, G.; Torcivia, A.; Bauvois, B.; Bouchet, S.; Pelloux, V.; Sasso, M.; et al. Accumulation and Changes in Composition of Collagens in Subcutaneous Adipose Tissue After Bariatric Surgery. J. Clin. Endocrinol. Metab. 2016, 101, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Djaberi, R.; Schuijf, J.D; van Werkhoven, J.M.; Nucifora, G.; Jukema, J.W.; Bax, J.J. Relation of epicardial adipose tissue to coronary atherosclerosis. Am. J. Cardiol. 2008, 102, 1602–1607. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Lonn, E.; Lamy, A.; Singh, N.; Sharma, A.M. Epicardial fat thickness and CAD correlate independently of obesity. Int. J. Cardiol. 2011, 146, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Miksztowicz, V.; Morales, C.; Barchuk, M.; López, G.; Póveda, R.; Gelpi, R.; Schreier, L.; Rubio, M.; Berg, G. Metalloproteinase 2 and 9 Activity Increase in Epicardial Adipose Tissue of Patients with Coronary Artery Disease. Curr. Vasc. Pharmacol. 2017, 15, 135–143. [Google Scholar] [CrossRef]
- McKenney, M.L.; Schultz, K.A.; Boyd, J.H.; Byrd, J.P.; Alloosh, M.; Teague, S.D.; Arce-Esquivel, A.A.; Fain, J.N.; Laughlin, M.H.; Sacks, H.S.; et al. Epicardial adipose excision slows the progression of porcine coronary atherosclerosis. J. Cardiothorac. Surg. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Qorri, B.; Kalaydina, R.V.; Velickovic, A.; Kaplya, Y.; Decarlo, A.; Szewczuk, M.R. Agonist-Biased Signaling via Matrix Metalloproteinase-9 Promotes Extracellular Matrix Remodeling. Cells 2018, 7, 117. [Google Scholar] [CrossRef]
- Mazurek, T.; Zhang, L.; Zalewski, A.; Mannion, J.D.; Diehl, J.T.; Arafat, H.; Sarov-Blat, L.; O’Brien, S.; Keiper, E.A.; Johnson, A.G.; et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 2003, 108, 2460–2466. [Google Scholar] [CrossRef]
- Friedman, S.L.; Maher, J.J.; Bissell, D.M. Mechanisms and therapy of hepatic fibrosis: Report of the AASLD Single Topic Basic Research Conference. Hepatology 2000, 32, 1403–1408. [Google Scholar] [CrossRef]
- Schuppan, D.; Ruehl, M.; Somasundaram, R.; Hahn, E.G. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Roderfeld, M.; Geier, A.; Dietrich, C.G.; Siewert, E.; Jansen, B.; Gartung, C.; Roeb, E. Cytokine blockade inhibits hepatic tissue inhibitor of metalloproteinase-1 expression and upregulates matrix metalloproteinase-9 in toxic liver injury. Liver Int. 2006, 26, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Kurzepa, J.; Mądro, A.; Czechowska, G.; Kurzepa, J.; Celiński, K.; Kazmierak, W.; Slomka, M. Role of MMP-2 and MMP-9 and their natural inhibitors in liver fibrosis, chronic pancreatitis and non-specific inflammatory bowel diseases. Hepatobiliary Pancreat Dis. Int. 2014, 13, 570–579. [Google Scholar] [CrossRef]
- Duarte, S.; Shen, X.D.; Fondevila, C.; Busuttil, R.W.; Coito, A.J. Fibronectin-α4β1 interactions in hepatic cold ischemia and reperfusion injury: Regulation of MMP-9 and MT1-MMP via the p38 MAPK pathway. Am. J. Transplant. 2012, 12, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.P.; Zhou, L.; Wang, J.; Xiong, S.; Garner, W.L.; French, S.W.; Tsukamoto, H. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J. Biol. Chem. 2004, 279, 4820–4828. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.J.; Huang, T.J.; Zhang, Q.Q.; Zhang, H.Y.; Guo, X.H.; Fan, H.Q.; Li, R.K.; Liu, L.X. Insulin-like growth factor binding protein related protein 1 knockdown attenuates hepatic fibrosis via the regulation of MMPs/TIMPs in mice. Hepatobiliary Pancreat. Dis. Int. 2018. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Kumada, T.; Kiriyama, S.; Tanikawa, M.; Hisanaga, Y.; Kanamori, A.; Tada, T.; Murakami, Y. Higher hepatic gene expression and serum levels of matrix metalloproteinase-2 are associated with steatohepatitis in non-alcoholic fatty liver diseases. Biomarkers 2013, 18, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Forgione, A.; La Torre, G.; Vero, V.; Cefalo, C.; Racco, S.; Vellone, V.G.; Vecchio, F.M.; Gasbarrini, G.; Rapaccini, G.L.; et al. Serum levels of hyaluronic acid and tissue metalloproteinase inhibitor-1 combined with age predict the presence of nonalcoholic steatohepatitis in a pilot cohort of subjects with nonalcoholic fatty liver disease. Transl. Res. 2009, 154, 194–201. [Google Scholar] [CrossRef]
- Barchuk, M.; Schreier, L.; Berg, G.; Miksztowicz, V. Metalloproteinases in non-alcoholic fatty liver disease and their behavior in liver fibrosis. Horm. Mol. Biol. Clin. Investig. 2018. [Google Scholar] [CrossRef]
- Hemmann, S.; Graf, J.; Roderfeld, M.; Roeb, E. Expression of MMPs and TIMPs in liver fibrosis a systematic review with special emphasis on anti-fibrotic strategies. J. Hepatol. 2007, 46, 955–975. [Google Scholar] [CrossRef]
- Munsterman, I.D.; Kendall, T.J.; Khelil, N.; Popa, M.; Lomme, R.; Drenth, J.P.H.; Tjwa, E.T.T. Extracellular matrix components indicate remodelling activity in different fibrosis stages of human non-alcoholic fatty liver disease. Histopathology 2018, 73, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Eren, F. Serum biomarkers of fibrosis and extracellular matrix remodeling in patients with nonalcoholic fatty liver disease: Association with liver histology. Eur. J. Gastroenterol. Hepatol. 2019, 31, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Al-Hashem, F.; Al-Humayed, S.; Amin, S.N.; Kamar, S.S.; Mansy, S.S.; Hassan, S.; Abdel-Salam, L.O.; Ellatif, M.A.; Alfaifi, M.; Haidara, M.A.; et al. Metformin inhibits mTOR-HIF-1α axis and profibrogenic and inflammatory biomarkers in thioacetamide-induced hepatic tissue alterations. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Kaji, K.; Yoshiji, H.; Ikenaka, Y.; Noguchi, R.; Aihara, Y.; Douhara, A.; Moriya, K.; Kawaratani, H.; Shirai, Y.; Yoshii, J.; et al. Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats. J. Gastroenterol. 2014, 49, 481–491. [Google Scholar] [CrossRef]
- Newby, A.C. Metalloproteinases promote plaque rupture and myocardial infarction: A persuasive concept waiting for clinical translation. Matrix Biol. 2015, 44–46, 157–166. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.; Bond, M.; Crook, M.F.; Newby, A.C. Role of nuclear factor-κB activation in metalloproteinase-1, -3 and -9 secretion by human macrophages in vitro and rabbit foam cells produced in vivo. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 765–771. [Google Scholar] [CrossRef]
- Aikawa, M.; Rabkin, E.; Sugiyama, S.; Voglic, S.J.; Fukumoto, Y.; Furukawa, Y.; Shiomi, M.; Schoen, F.J.; Libby, P. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001, 103, 276–283. [Google Scholar] [CrossRef]
- Chen, M.; Masaki, T.; Sawamura, T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ishibashi, T.; Sawamura, T.; Inoue, N.; Kamioka, M.; Uekita, H.; Ohkawara, H.; Sakamoto, T.; Sakamoto, N.; Okamoto, Y.; et al. LOX-1-MT1-MMP axis is crucial for RhoA and Rac1 activation induced by oxidized low-density lipoprotein in endothelial cells. Cardiovasc. Res. 2009, 84, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc. Med. 2007, 17, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Jenkins, N.P.; Huang, W.C.; Di Gregoli, K.; Sala-Newby, G.B.; Scholtes, V.P.; Moll, F.L.; Pasterkamp, G.; Newby, A.C. Relationship of MMP-14 and TIMP-3 expression with macrophage activation and human atherosclerotic plaque vulnerability. Mediators Inflamm. 2014, 2014, 276457. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Metalloproteinase production from macrophages—A perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp. Physiol. 2016, 101, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Gregan, S.M.; Navin, T.J.; Foxwell, B.M.; Davies, A.H.; Feldmann, M. Toll-like receptor-2 mediates inflammation and matrix degradation in human atherosclerosis. Circulation 2009, 120, 2462–2469. [Google Scholar] [CrossRef] [PubMed]
- Schaper, F.; Rose-John, S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor 2015, 26, 475–487. [Google Scholar] [CrossRef]
- Tsaousi, A.; Hayes, E.M.; Di Gregoli, K.; Bond, A.R.; Bevan, L.; Thomas, A.C.; Newby, A.C. Plaque Size Is Decreased but M1 Macrophage Polarization and Rupture Related Metalloproteinase Expression Are Maintained after Deleting T-Bet in ApoE Null Mice. PLoS ONE 2016, 11, e0148873. [Google Scholar] [CrossRef]
- Sluimer, J.C.; Gasc, J.M; van Wanroij, J.L.; Kisters, N.; Groeneweg, M.; Sollewijn Gelpke, M.D.; Cleutjens, J.P.; van den Akker, L.H.; Corvol, P.; Wouters, B.G.; et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J. Am. Coll. Cardiol. 2008, 51, 1258–1265. [Google Scholar] [CrossRef]
- Fang, H.Y.; Hughes, R.; Murdoch, C.; Coffelt, S.B.; Biswas, S.K.; Harris, A.L.; Johnson, R.S.; Imityaz, H.Z.; Simon, M.C.; Fredlund, E.; et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 2009, 114, 844–859. [Google Scholar] [CrossRef]
- Lee, Y.A.; Choi, H.M.; Lee, S.H.; Hong, S.J.; Yang, H.I.; Yoo, M.C.; Kim, K.S. Hypoxia differentially affects IL-1β-stimulated MMP-1 and MMP-13 expression of fibroblast-like synoviocytes in an HIF-1α-dependent manner. Rheumatology 2012, 51, 443–450. [Google Scholar] [CrossRef]
- Gao, W.; McCormick, J.; Connolly, M.; Balogh, E.; Veale, D.J.; Fearon, U. Hypoxia and STAT3 signalling interactions regulate pro-inflammatory pathways in rheumatoid arthritis. Ann. Rheum Dis. 2015, 74, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Souissi, I.J.; Billiet, L.; Cuaz-Pérolin, C.; Slimane, M.N.; Rouis, M. Matrix metalloproteinase-12 gene regulation by a PPAR alpha agonist in human monocyte-derived macrophages. Exp. Cell Res. 2008, 314, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
- Traylor, M.; Mäkelä, K.M.; Kilarski, L.L.; Holliday, E.G.; Devan, W.J.; Nalls, M.A.; Wiggins, K.L.; Zhao, W.; Cheng, Y.C.; Achterberg, S.; et al. A novel MMP12 locus is associated with large artery atherosclerotic stroke using a genome-wide age-at-onset informed approach. PLoS Genet. 2014, 10, e1004469. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Vlachopoulou, E.; Havulinna, A.S.; Tervahartiala, T.; Sattler, W.; Lokki, M.L.; Nieminen, M.S.; Perola, M.; Salomaa, V.; Sinisalo, J.; et al. Genetic Variants Contributing to Circulating Matrix Metalloproteinase 8 Levels and Their Association with Cardiovascular Diseases: A Genome-Wide Analysis. Circ. Cardiovasc. Genet. 2017, 10, e001731. [Google Scholar] [CrossRef] [PubMed]
- Hohensinner, P.J.; Baumgartner, J.; Ebenbauer, B.; Thaler, B.; Fischer, M.B.; Huber, K.; Speidl, W.S.; Wojta, J. Statin treatment reduces matrix degradation capacity of proinflammatory polarized macrophages. Vascul. Pharmacol. 2018, 110, 49–54. [Google Scholar] [CrossRef]
- Eilenberg, W.; Stojkovic, S.; Kaider, A.; Kozakowski, N.; Domenig, C.M.; Burghuber, C.; Nanobachvili, J.; Huber, K.; Klinger, M.; Neumayer, C.; et al. NGAL and MMP-9/NGAL as biomarkers of plaque vulnerability and targets of statins in patients with carotid atherosclerosis. Clin. Chem Lab. Med. 2017, 56, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Colon, E.; Klych-Ratuszny, A.; Kosowska, A.; Garczorz, W.; Aghdam, M.; Wozniak, M.; Francuz, T. Exenatide modulates metalloproteinase expression in human cardiac smooth muscle cells via the inhibition of Akt signaling pathway. Pharmacol. Rep. 2018, 70, 178–183. [Google Scholar]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berg, G.; Barchuk, M.; Miksztowicz, V. Behavior of Metalloproteinases in Adipose Tissue, Liver and Arterial Wall: An Update of Extracellular Matrix Remodeling. Cells 2019, 8, 158. https://doi.org/10.3390/cells8020158
Berg G, Barchuk M, Miksztowicz V. Behavior of Metalloproteinases in Adipose Tissue, Liver and Arterial Wall: An Update of Extracellular Matrix Remodeling. Cells. 2019; 8(2):158. https://doi.org/10.3390/cells8020158
Chicago/Turabian StyleBerg, Gabriela, Magalí Barchuk, and Verónica Miksztowicz. 2019. "Behavior of Metalloproteinases in Adipose Tissue, Liver and Arterial Wall: An Update of Extracellular Matrix Remodeling" Cells 8, no. 2: 158. https://doi.org/10.3390/cells8020158
APA StyleBerg, G., Barchuk, M., & Miksztowicz, V. (2019). Behavior of Metalloproteinases in Adipose Tissue, Liver and Arterial Wall: An Update of Extracellular Matrix Remodeling. Cells, 8(2), 158. https://doi.org/10.3390/cells8020158