Extracellular Matrix and Fibrocyte Accumulation in BALB/c Mouse Lung upon Transient Overexpression of Oncostatin M



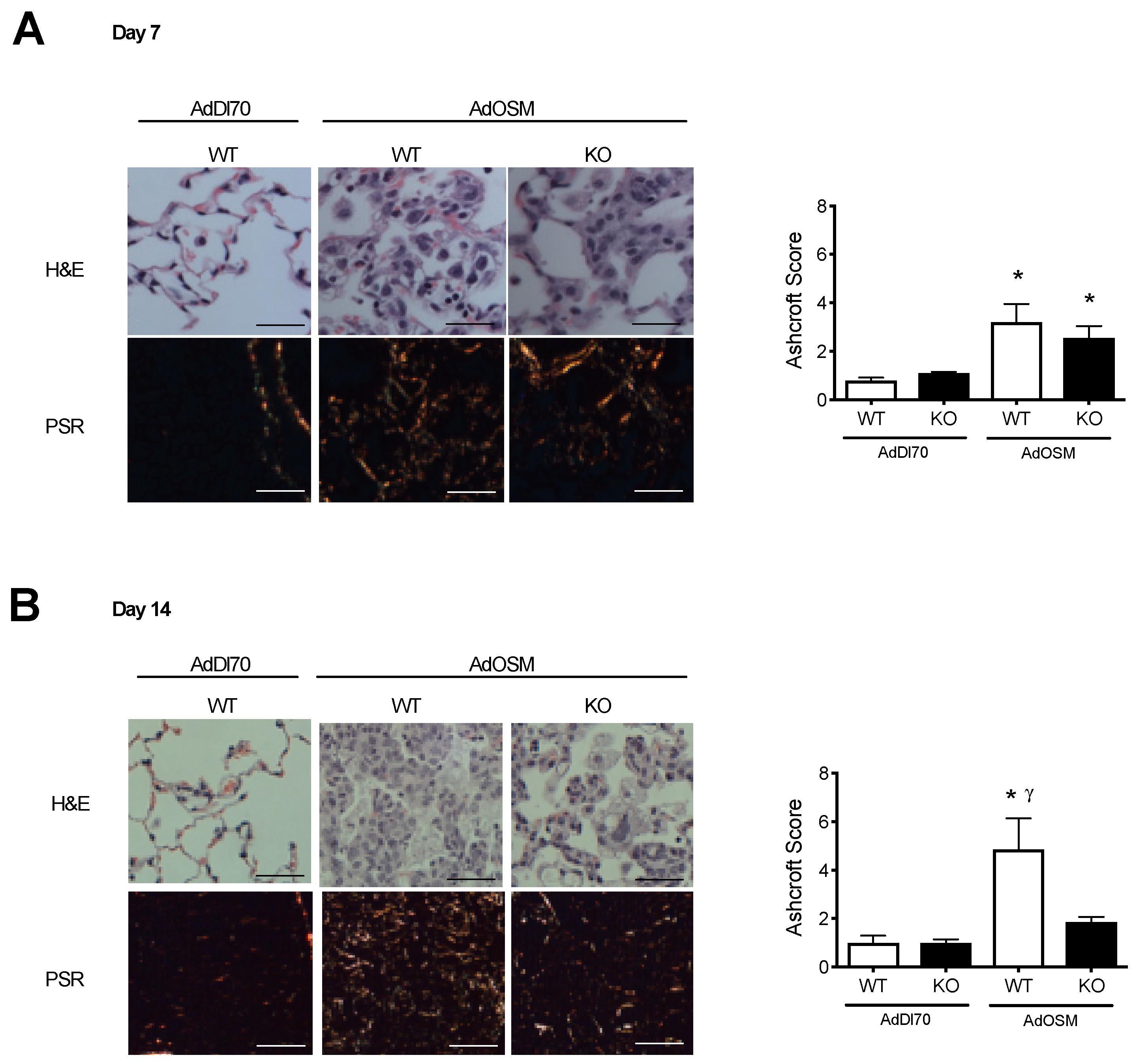{kind=link}
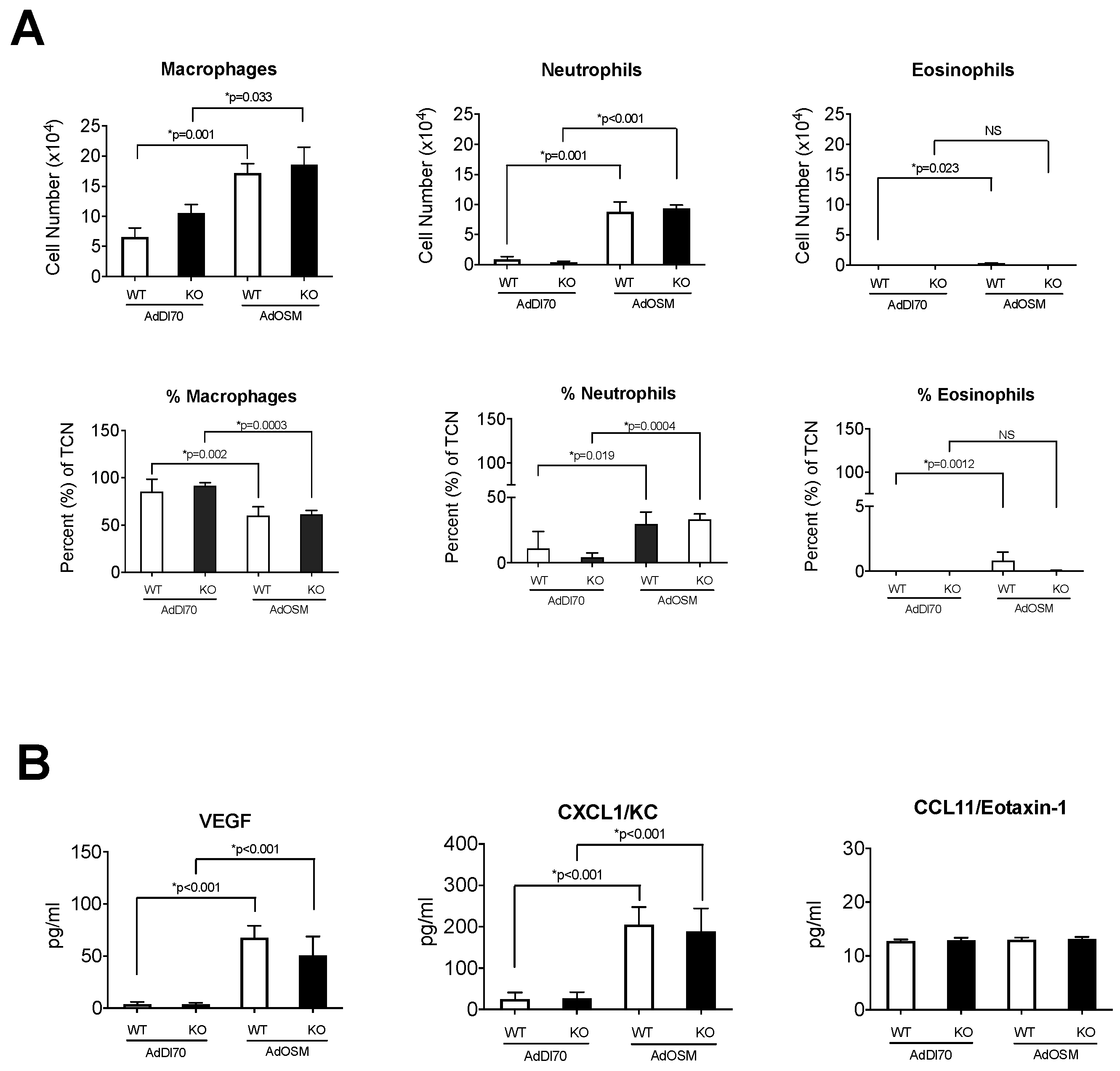{kind=link}
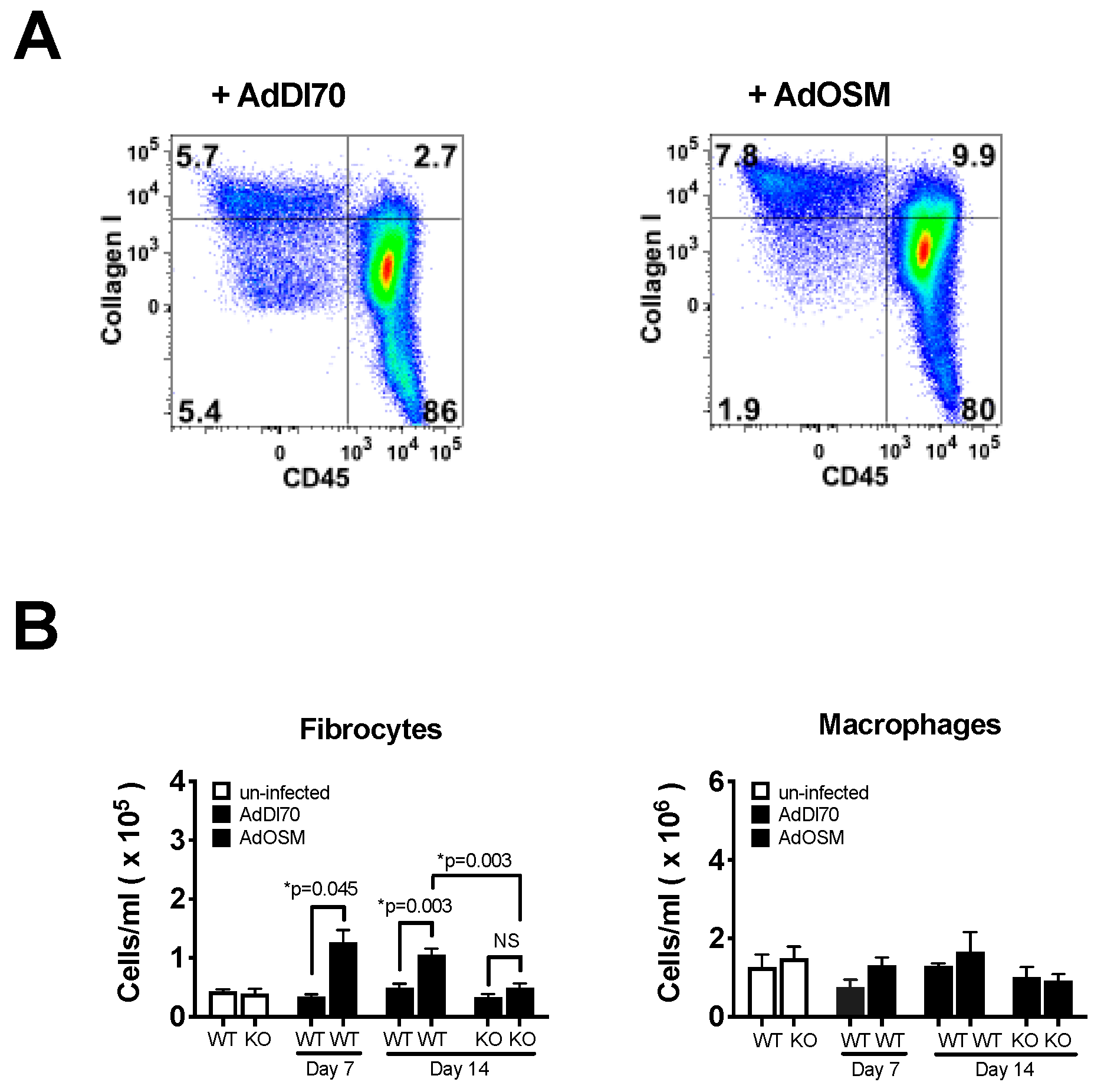{kind=link}
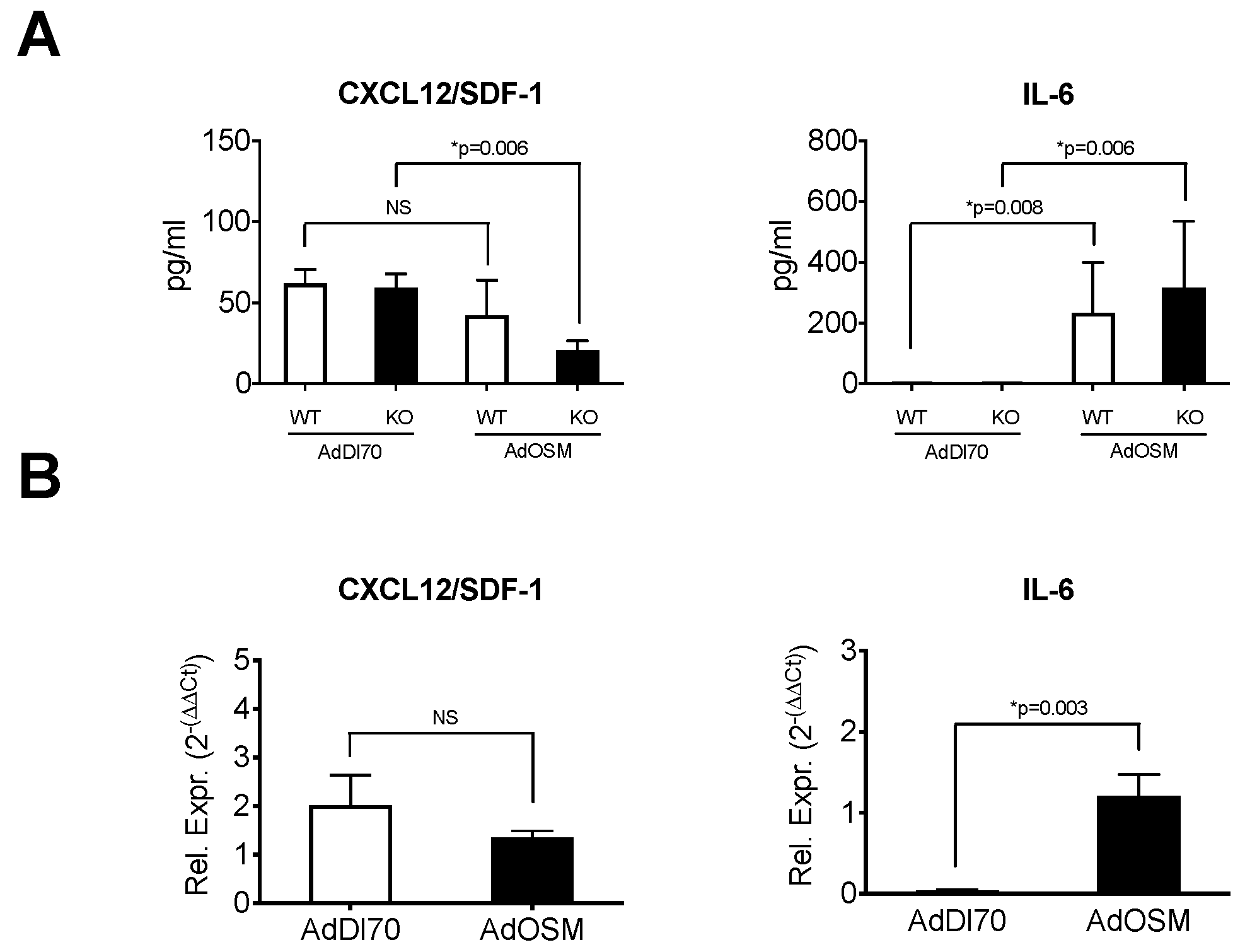{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Administration of Adenovirus Constructs and Sample Collection
2.3. Reagents and Cell Culture
2.4. ELISA and Extracellular Matrix Analysis
2.5. RNA Extraction
2.6. Isolation of Lung Mononuclear Cells and Flow Cytometric Analysis
2.7. Statistics
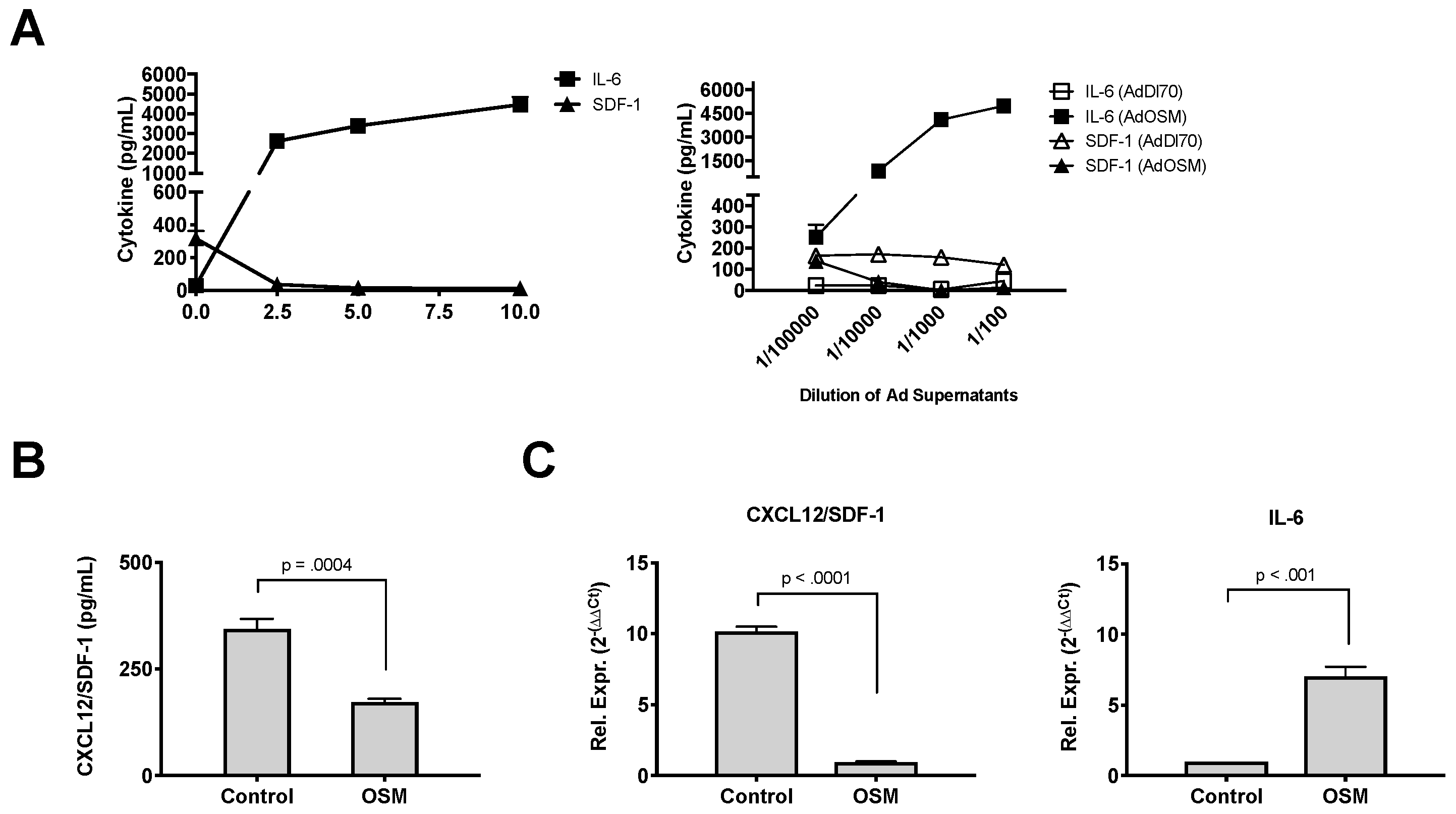
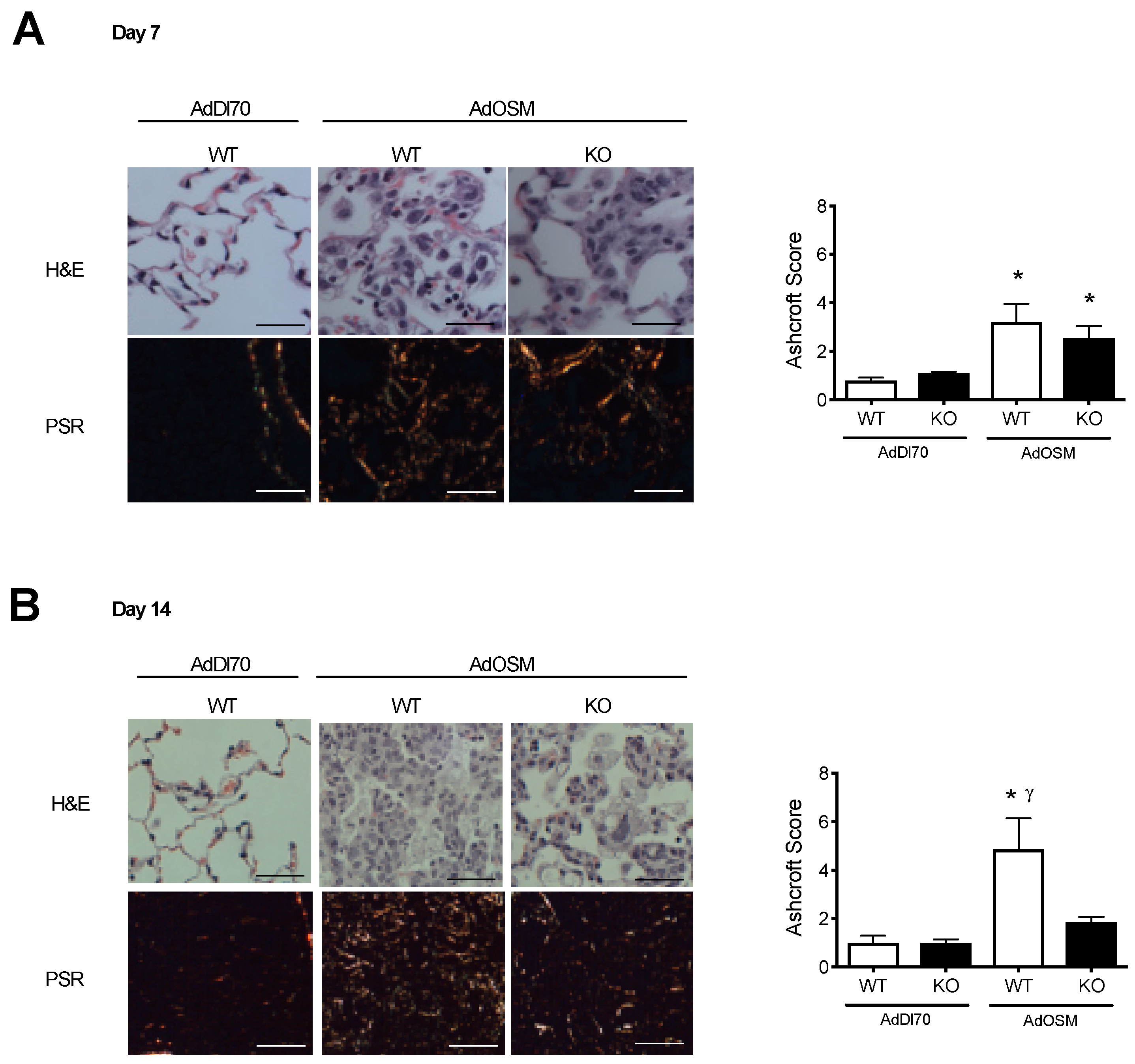
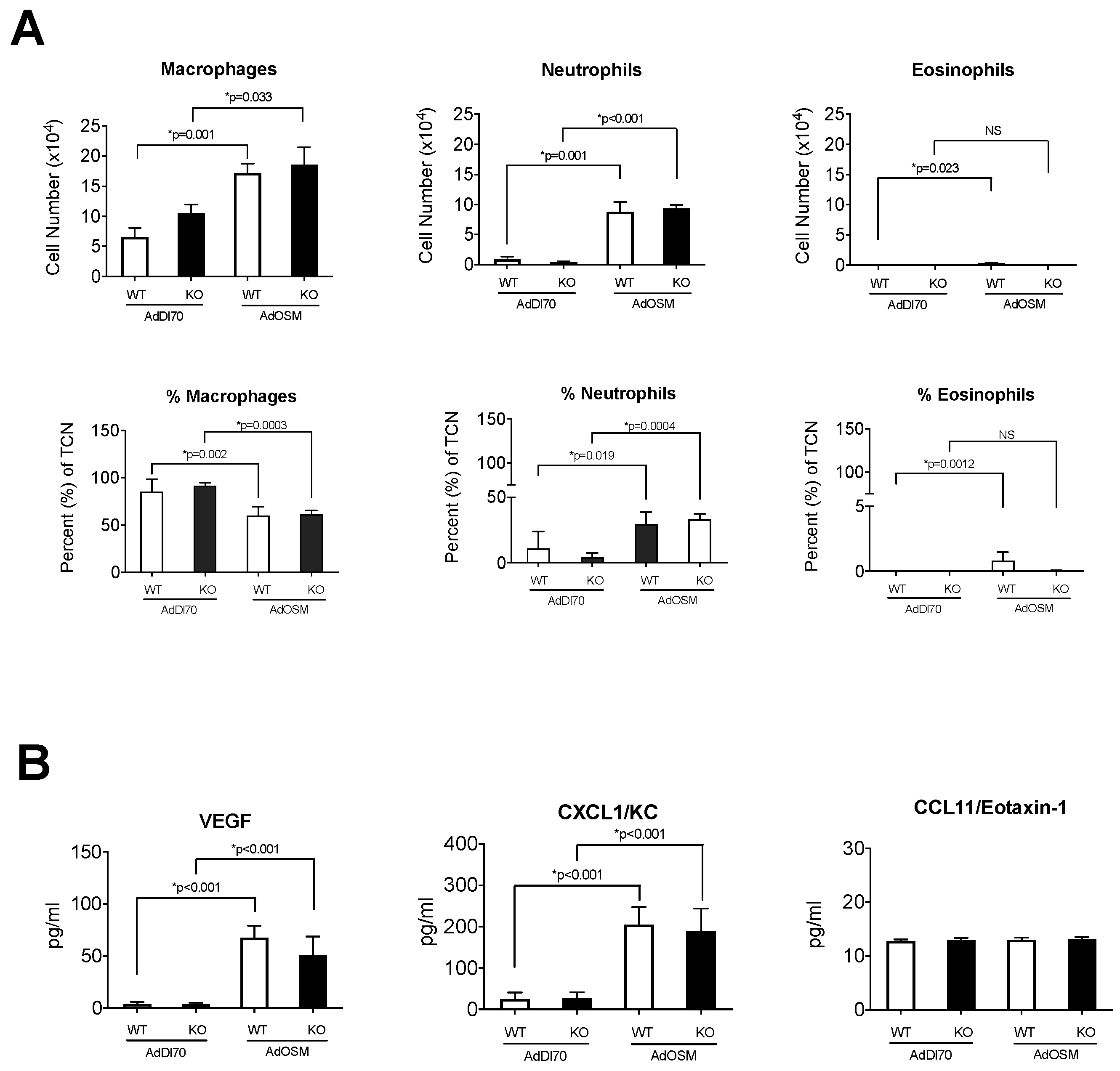
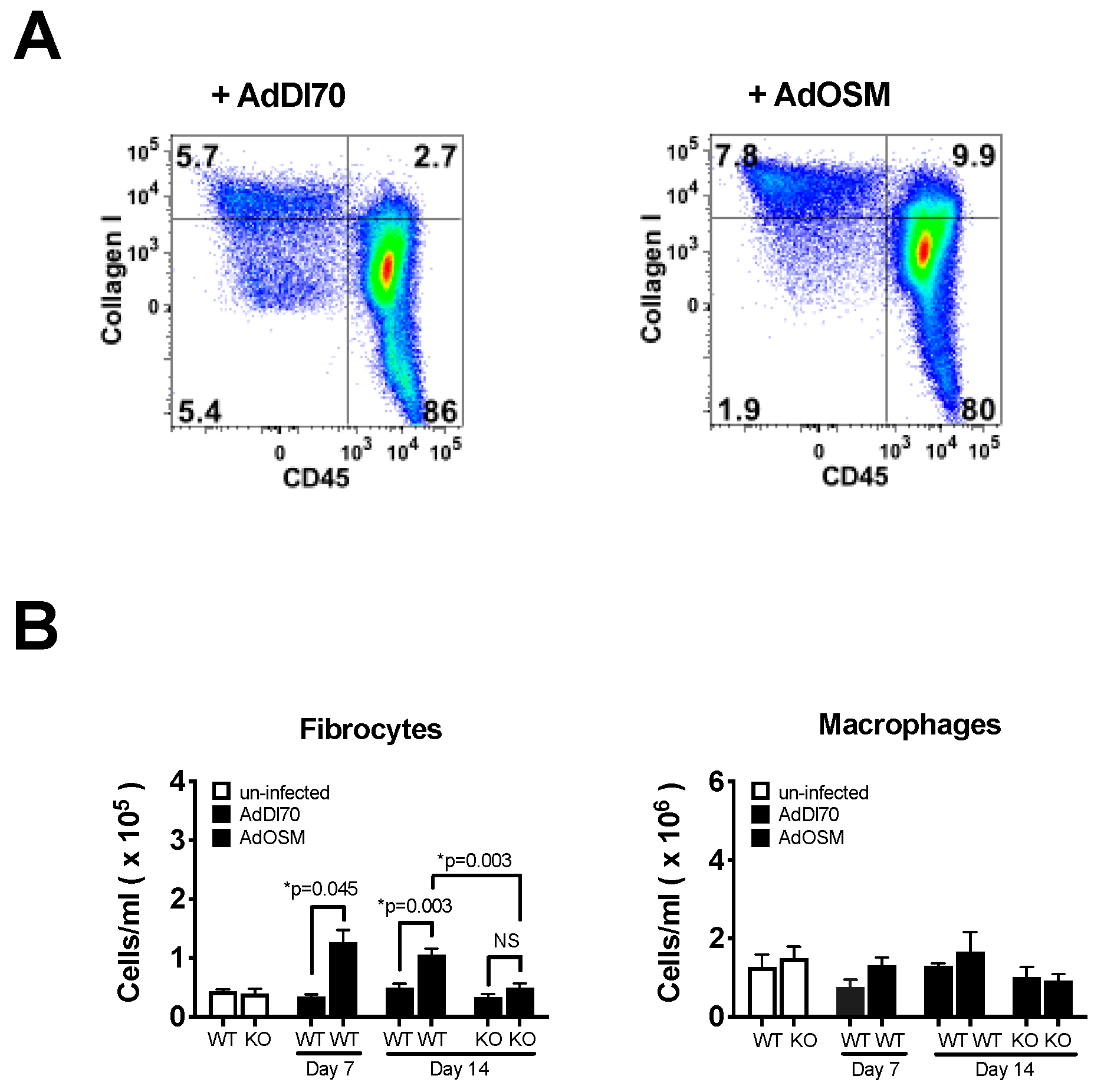
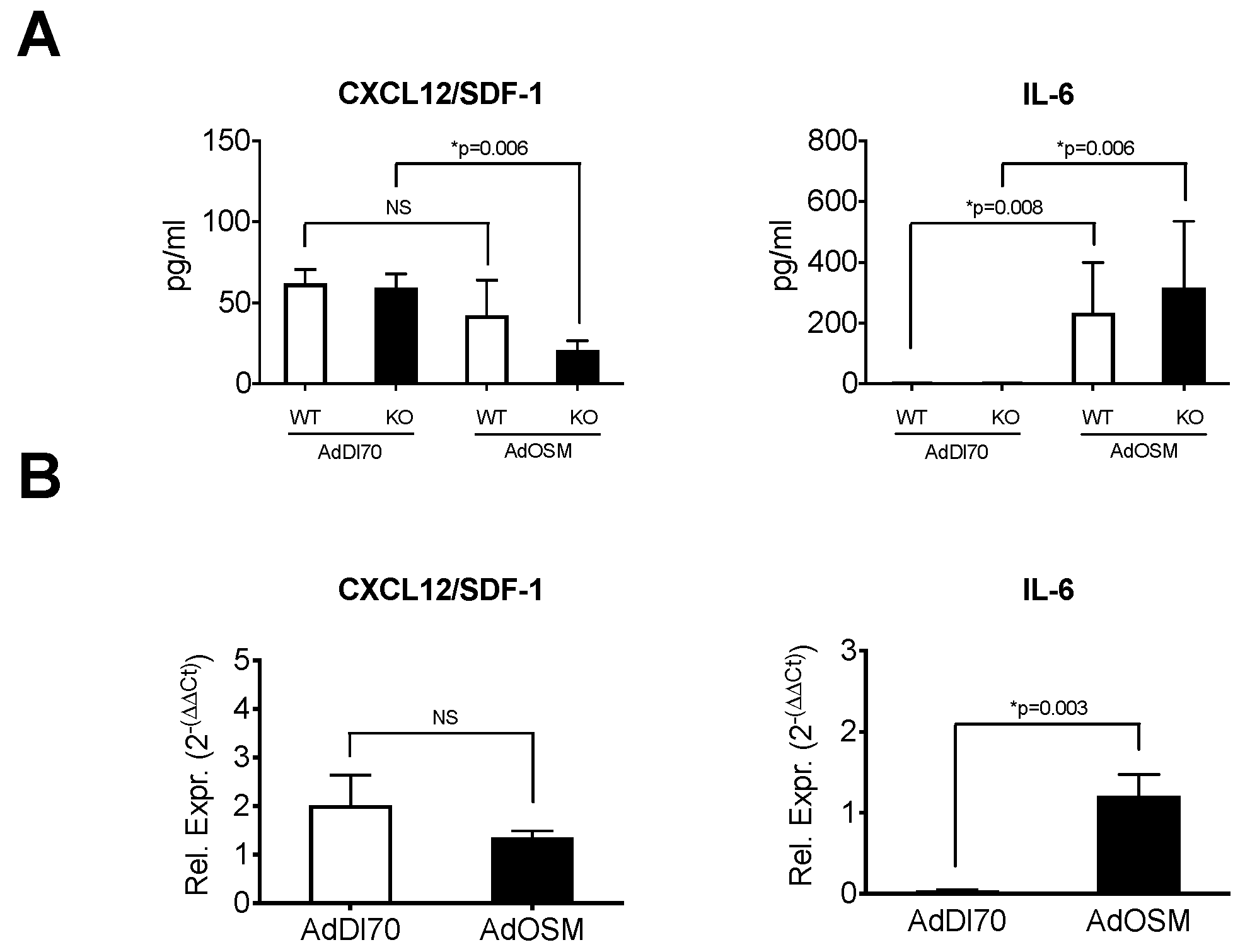
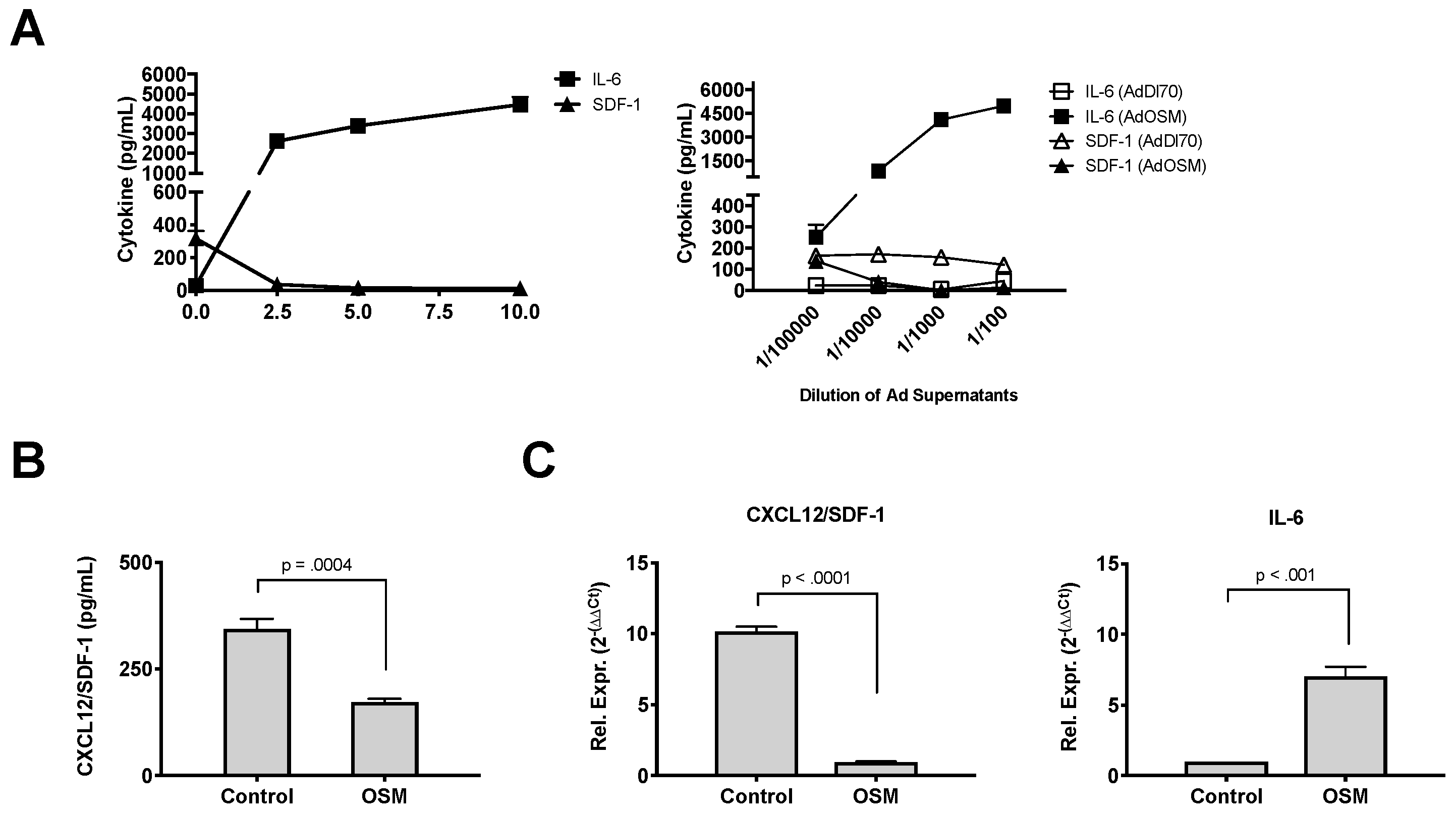
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Gauldie, J.; Bonniaud, P.; Sime, P.; Ask, K.; Kolb, M. TGF-beta, Smad3 and the process of progressive fibrosis. Biochem. Soc. Trans. 2007, 35, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.S.; Hunter, C.A. gp130 at the nexus of inflammation, autoimmunity, and cancer. J. Leukoc. Biol. 2010, 88, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, C., III; Homer, R.J.; Zhu, Z.; Ward, N.; Flavell, R.A.; Geba, G.P.; Elias, J.A. Airway hyperresponsiveness and airway obstruction in transgenic mice. Morphologic correlates in mice overexpressing interleukin (IL)-11 and IL-6 in the lung. Am. J. Respir. Cell Mol. Biol. 2000, 22, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Geba, G.P.; Zheng, T.; Ray, P.; Homer, R.J.; Kuhn, C.; Flavell, R.A.; Elias, J.A. Targeted expression of IL-11 in the murine airway causes lymphocytic inflammation, bronchial remodeling, and airways obstruction. J. Clin. Investig. 1996, 98, 2845–2853. [Google Scholar] [CrossRef]
- Mozaffarian, A.; Brewer, A.W.; Trueblood, E.S.; Luzina, I.R.; Todd, N.W.; Atamas, S.P.; Arnett, H.A. Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis. J. Immunol. 2008, 181, 7243–7253. [Google Scholar] [CrossRef]
- O’Donoghue, R.J.; Knight, D.A.; Richards, C.D.; Prêle, C.M.; Lau, H.L.; Jarnicki, A.G.; Jones, J.; Bozinovski, S.; Vlahos, R.; Thiem, S.; et al. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 2012, 4, 939–951. [Google Scholar] [CrossRef]
- Tanaka, M.; Miyajima, A. Oncostatin M, A multifunctional cytokine. Rev. Physiol. Biochem. Pharmacol. 2003, 149, 39–52. [Google Scholar]
- Chen, S.H.; Benveniste, E.N. Oncostatin M: A pleiotropic cytokine in the central nervous system. Cytokine Growth Factor Rev. 2004, 15, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Komori, T.; Tanaka, M.; Senba, E.; Miyajima, A.; Morikawa, Y. Lack of Oncostatin M Receptor beta Leads to Adipose Tissue Inflammation and Insulin Resistance by Switching Macrophage Phenotype. J. Biol. Chem. 2013, 288, 21861–21875. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, M.; Hara, T.; Kim, H.; Murate, T.; Miyajima, A. Oncostatin M and leukemia inhibitory factor do not use the same functional receptor in mice. Blood 1997, 90, 165–173. [Google Scholar] [PubMed]
- Lindberg, R.A.; Juan, T.S.; Welcher, A.A.; Sun, Y.; Cupples, R.; Guthrie, B.; Fletcher, F.A. Cloning and characterization of a specific receptor for mouse oncostatin M. Mol. Cell. Biol. 1998, 18, 3357–3367. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Hara, T.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Miyajima, A. Reconstitution of the functional mouse oncostatin M (OSM) receptor: Molecular cloning of the mouse OSM receptor beta subunit. Blood 1999, 93, 804–815. [Google Scholar] [PubMed]
- Cawston, T.; Billington, C.; Cleaver, C.; Elliott, S.; Hul, W.; Koshy, P.; Shingleton, B.; Rowan, A. The regulation of MMPs and TIMPs in cartilage turnover. Ann. N. Y. Acad. Sci. 1999, 878, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Rowan, A.D.; Koshy, P.J.; Shingleton, W.D.; Degnan, B.A.; Heath, J.K.; Vernallis, A.B.; Spaull, J.R.; Life, P.F.; Hudson, K.; Cawston, T.E. Synergistic effects of glycoprotein 130 binding cytokines in combination with interleukin-1 on cartilage collagen breakdown. Arthritis Rheum. 2001, 44, 1620–1632. [Google Scholar] [CrossRef]
- Langdon, C.; Kerr, C.; Hassen, M.; Hara, T.; Arsenault, A.L.; Richards, C.D. Murine oncostatin M stimulates mouse synovial fibroblasts in vitro and induces inflammation and destruction in mouse joints in vivo. Am. J. Pathol. 2000, 157, 1187–1196. [Google Scholar] [CrossRef]
- Finelt, N.; Gazel, A.; Gorelick, S.; Blumenberg, M. Transcriptional responses of human epidermal keratinocytes to Oncostatin-M. Cytokine 2005, 31, 305–313. [Google Scholar] [CrossRef]
- Boniface, K.; Diveu, C.; Morel, F.; Pedretti, N.; Froger, J.; Ravon, E.; Garcia, M.; Venereau, E.; Preisser, L.; Guignouard, E.; et al. Oncostatin M secreted by skin infiltrating T lymphocytes is a potent keratinocyte activator involved in skin inflammation. J. Immunol. 2007, 178, 4615–4622. [Google Scholar] [CrossRef]
- Sims, N.A.; Walsh, N.C. GP130 cytokines and bone remodelling in health and disease. BMB Rep. 2010, 43, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Guihard, P.; Danger, Y.; Brounais, B. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells 2012, 30, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Fritz, D.K.; Kerr, C.; Fattouh, R.; Llop-Guevara, A.; Khan, W.I.; Jordana, M.; Richards, C.D. A mouse model of airway disease: Oncostatin M-induced pulmonary eosinophilia, goblet cell hyperplasia, and airway hyperresponsiveness are STAT6 dependent, and interstitial pulmonary fibrosis is STAT6 independent. J. Immunol. 2011, 186, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Botelho, F.M.; Rodrigues, R.M.; Richards, C.D. Oncostatin M overexpression induces matrix deposition, STAT3 activation, and SMAD1 Dysregulation in lungs of fibrosis-resistant BALB/c mice. Lab. Investig. 2014, 94, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Baines, K.J.; Boyle, M.J.; Scott, R.J.; Gibson, P.G. Oncostatin M (OSM) is increased in asthma with incompletely reversible airflow obstruction. Exp. Lung Res. 2009, 35, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Fritz, D.K.; Kerr, C.; Tong, L.; Smyth, D.; Richards, C.D. Oncostatin-M up-regulates VCAM-1 and synergizes with IL-4 in eotaxin expression: Involvement of STAT6. J. Immunol. 2006, 176, 4352–4360. [Google Scholar] [CrossRef] [PubMed]
- Faffe, D.S.; Flynt, L.; Mellema, M.; Moore, P.E.; Silverman, E.S.; Subramaniam, V.; Jones, M.R.; Mizgerd, J.P.; Whitehead, T.; Imrich, A.; et al. Oncostatin M causes eotaxin-1 release from airway smooth muscle: Synergy with IL-4 and IL-13. J. Allergy Clin. Immunol. 2005, 115, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Loewen, G.M.; Tracy, E.; Blanchard, F.; Tan, D.; Yu, J.; Raza, S.; Matsui, S.; Baumann, H. Transformation of human bronchial epithelial cells alters responsiveness to inflammatory cytokines. BMC Cancer 2005, 5, 145. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Tracy, E.; Liang, P.; Robledo, O.; Rose-John, S.; Baumann, H. Interleukin-31 and oncostatin-M mediate distinct signaling reactions and response patterns in lung epithelial cells. J. Biol. Chem. 2007, 282, 3014–3026. [Google Scholar] [CrossRef]
- Sallenave, J.M.; Tremblay, G.M.; Gauldie, J.; Richards, C.D. Oncostatin M, but not interleukin-6 or leukemia inhibitory factor, stimulates expression of alpha1-proteinase inhibitor in A549 human alveolar epithelial cells. J. Interferon Cytokine Res. 1997, 17, 337–346. [Google Scholar] [CrossRef]
- Quan, T.E.; Cowper, S.; Wu, S.P.; Bockenstedt, L.K.; Bucala, R. Circulating fibrocytes: Collagen-secreting cells of the peripheral blood. Int. J. Biochem. Cell Biol. 2004, 36, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, S.S.; Baroke, E.; Gauldie, J.; Kolb, M.R. Fibrocytes in chronic lung disease--facts and controversies. Pulm. Pharmacol. Ther. 2012, 25, 263–267. [Google Scholar] [CrossRef]
- Gomperts, B.N.; Strieter, R.M. Fibrocytes in lung disease. J. Leukoc. Biol. 2007, 82, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Lama, V.N.; Phan, S.H. The extrapulmonary origin of fibroblasts: Stem/progenitor cells and beyond. Proc. Am. Thorac. Soc. 2006, 3, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.J.; Burdick, M.D.; Hong, K. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Investig. 2004, 114, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Rabach, L.; Noble, P.; Zheng, T.; Lee, C.G.; Homer, R.J.; Elias, J.A. IL-11 receptor alpha in the pathogenesis of IL-13-induced inflammation and remodeling. J. Immunol. 2005, 174, 2305–2313. [Google Scholar] [CrossRef]
- Therien, A.G.; Bernier, V.; Weicker, S.; Tawa, P.; Falgueyret, J.; Mathieu, M.; Honsberger, J.; Pomerleau, V.; Robichaud, A.; Stocco, R.; et al. Adenovirus IL-13-induced airway disease in mice: A corticosteroid-resistant model of severe asthma. Am. J. Respir. Cell Mol. Biol. 2008, 39, 26–35. [Google Scholar] [CrossRef]
- Botelho, F.M.; Rangel-Moreno, J.; Fritz, D.; Randall, T.D.; Xing, Z.; Richards, C.D. Pulmonary Expression of Oncostatin M (OSM) Promotes Inducible BALT Formation Independently of IL-6, Despite a Role for IL-6 in OSM-Driven Pulmonary Inflammation. J. Immunol. 2013, 191, 1453–1464. [Google Scholar] [CrossRef]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1998, 41, 467–470. [Google Scholar] [CrossRef]
- Dubey, A.; Izakelian, L.; Ayaub, E.A.; Ho, L.; Stephenson, K.; Wong, S.; Kwofie, K.; Austin, R.C.; Botelho, F.; Ask, K.; et al. Separate roles of IL-6 and oncostatin M in mouse macrophage polarization in vitro and in vivo. Immunol. Cell Biol. 2018, 96, 257–272. [Google Scholar] [CrossRef]
- Langdon, C.; Kerr, C.; Tong, L.; Richards, C.D. Oncostatin M regulates eotaxin expression in fibroblasts and eosinophilic inflammation in C57BL/6 mice. J. Immunol. 2003, 170, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Grenier, A.; Combaux, D.; Chastre, J.; Gougerot-Pocidalo, M.A.; Gibert, C.; Dehoux, M.; Chollet-Martin, S. Oncostatin M production by blood and alveolar neutrophils during acute lung injury. Lab. Investig. 2001, 81, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.; Gilpin, S.E.; Ask, K.; Cox, G.; Cook, D.; Gauldie, J.; Margetts, P.J.; Farkas, L.; Dobranowski, J.; Boylan, C.; et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Bellini, A.; Marini, M.A.; Bianchetti, L.; Barczyk, M.; Schmidt, M.; Mattoli, S. Interleukin (IL)-4, IL-13 and IL-17A differentially affect the profibrotic an dproinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol. 2012, 5, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Hohensinner, P.J.; Kaun, C.; Rychli, K.; Nlessner, A.; Pfaffenberger, S.; Rega, G.; Furnkranz, A.; Uhrin, P.; Zaujec, J.; Afonyushkin, T.; et al. The inflammatory mediator oncostatin M induces stromal derived factor-1 in human adult cardiac cells. FASEB J. 2009, 23, 774–782. [Google Scholar] [CrossRef]
- Moore, B.B.; Kolodsick, J.E.; Thannickal, V.J.; Cooke, K.; Moore, T.A.; Hogaboam, C.; Wilke, C.A.; Toews, G.B. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am. J. Pathol. 2005, 166, 675–684. [Google Scholar] [CrossRef]
- Moore, B.B.; Murray, L.; Das, A.; Wilke, C.A.; Herrygers, A.B.; Toews, G.B. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2006, 35, 175–181. [Google Scholar] [CrossRef]
- Sun, L.; Louie, M.C.; Vannella, K.M.; Wilke, C.A.; LeVine, A.M.; Moore, B.B.; Shanley, T.P. New concepts of IL-10-induced lung fibrosis: Fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L341–L353. [Google Scholar] [CrossRef]
- Ayaub, E.A.; Dubey, A.; Imani, J.; Botelho, F.; Kolb, M.R.J.; Richards, C.D.; Ask, K. Overexpression of OSM and IL-6 impacts the polarization of pro-fibrotic macrophages and the development of bleomycin-induced lung. Sci. Rep. 2017, 7, 13281. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botelho, F.M.; Rodrigues, R.; Guerette, J.; Wong, S.; Fritz, D.K.; Richards, C.D. Extracellular Matrix and Fibrocyte Accumulation in BALB/c Mouse Lung upon Transient Overexpression of Oncostatin M. Cells 2019, 8, 126. https://doi.org/10.3390/cells8020126
Botelho FM, Rodrigues R, Guerette J, Wong S, Fritz DK, Richards CD. Extracellular Matrix and Fibrocyte Accumulation in BALB/c Mouse Lung upon Transient Overexpression of Oncostatin M. Cells. 2019; 8(2):126. https://doi.org/10.3390/cells8020126
Chicago/Turabian StyleBotelho, Fernando M., Rebecca Rodrigues, Jessica Guerette, Steven Wong, Dominik K. Fritz, and Carl D. Richards. 2019. "Extracellular Matrix and Fibrocyte Accumulation in BALB/c Mouse Lung upon Transient Overexpression of Oncostatin M" Cells 8, no. 2: 126. https://doi.org/10.3390/cells8020126
APA StyleBotelho, F. M., Rodrigues, R., Guerette, J., Wong, S., Fritz, D. K., & Richards, C. D. (2019). Extracellular Matrix and Fibrocyte Accumulation in BALB/c Mouse Lung upon Transient Overexpression of Oncostatin M. Cells, 8(2), 126. https://doi.org/10.3390/cells8020126