Mast Cells in Liver Fibrogenesis



Abstract
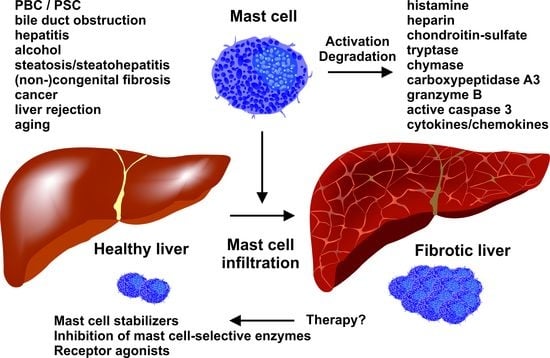
1. Introduction
1.1. Mast Cell Development
1.2. Different Roles of Mast Cells in Physiology and Pathophysiology
1.3. Mast Cell Mediators
1.4. Murine Models to Study Mast Cell Involvement
2. Fibrosis: Some General Aspects
2.1. Common Mediators in Inflammation and Fibrogenesis
2.2. Liver Fibrogenesis: A Number of Different Cell Types Contributing to the Initiation and Progression of Fibrosis
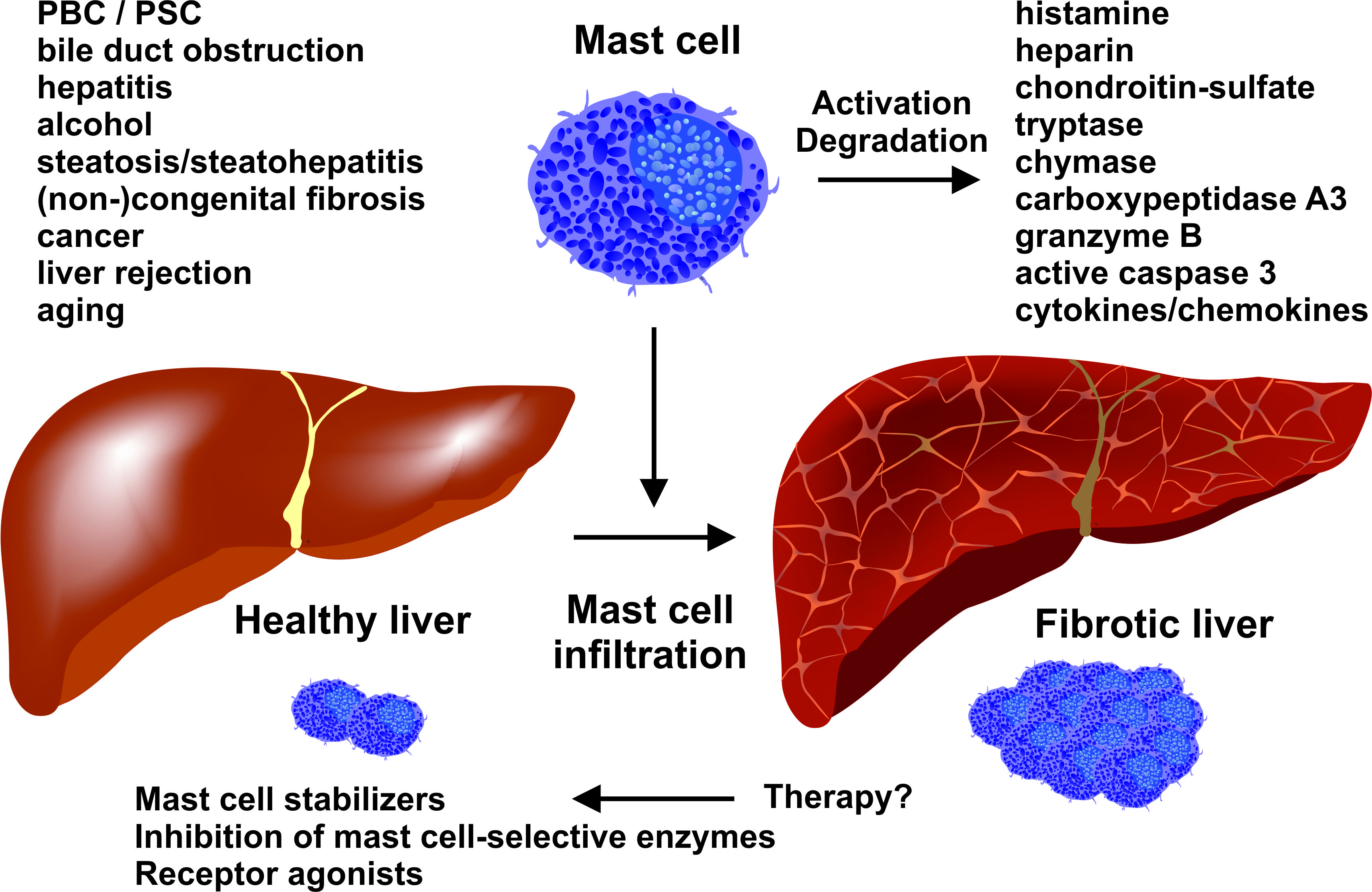
2.3. Mouse Models of Liver Fibrosis: What Can be Learned for Human Disease?
2.3.1. Chemical-Based Injury Models
2.3.2. Surgery-Based Injury Models
2.3.3. Genetic Models
2.3.4. Mouse Models in Translational Research of Human Liver Diseases
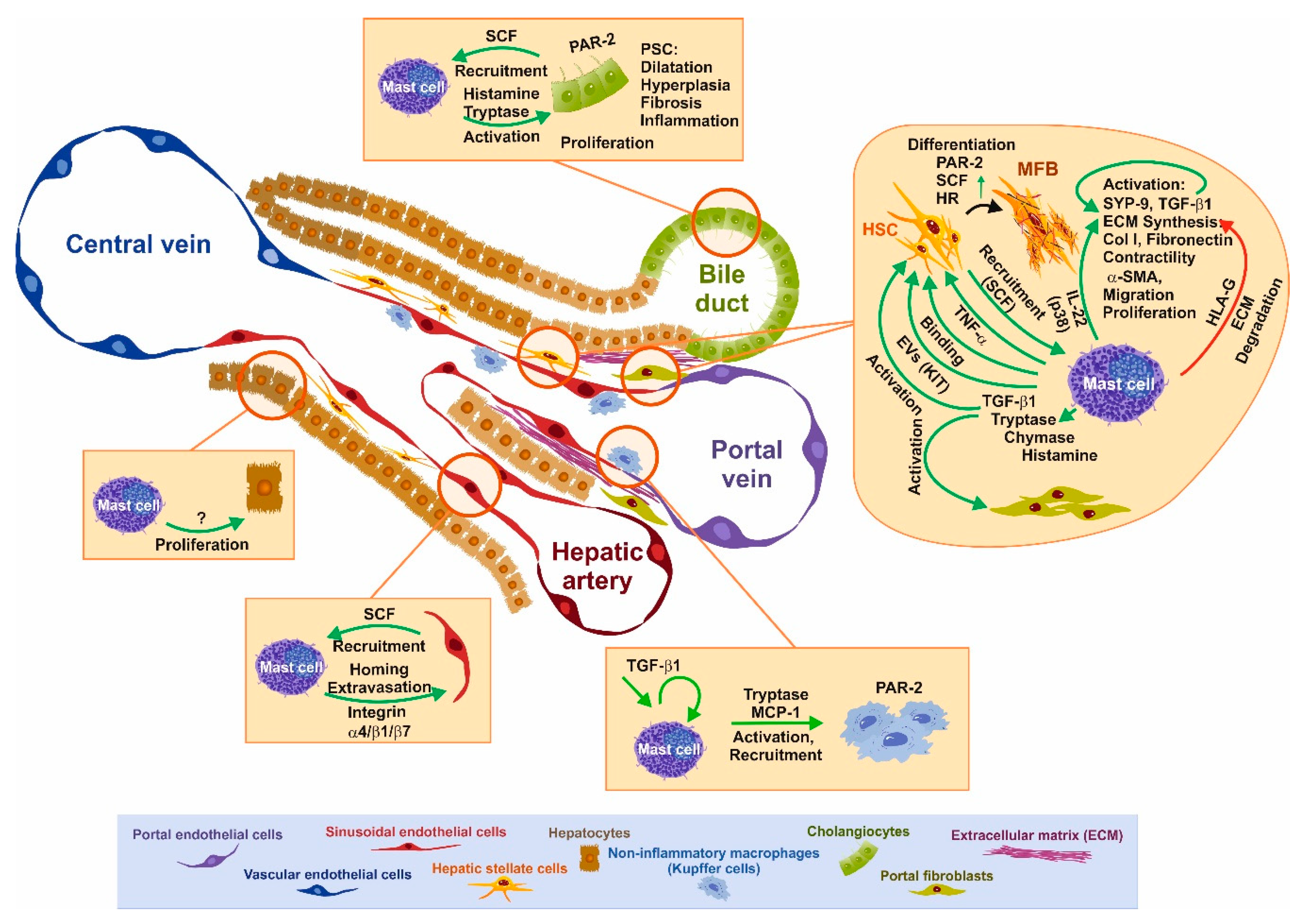
3. Functional Interaction of Mast Cells and Other Cell Types Relevant for Fibrosis
3.1. SCF–KIT Axis as Chemotactic Guidance
3.2. Impact of Mast Cells and Mediators on Liver Disease and Individual Cells
3.3. BDL-Induced Cholestasis in Mast Cell-Depleted KitW-sh/W-sh Mice
3.4. Tryptase/Protease-Activated Receptor-2 Axis
3.5. Chymase as an Interface for the Activation of Several Pro-fibrogenic Pathways
3.6. The Histamine/Histamin Receptor Axis
3.7. TGF-β1
4. Mast Cells in Human Liver Disease
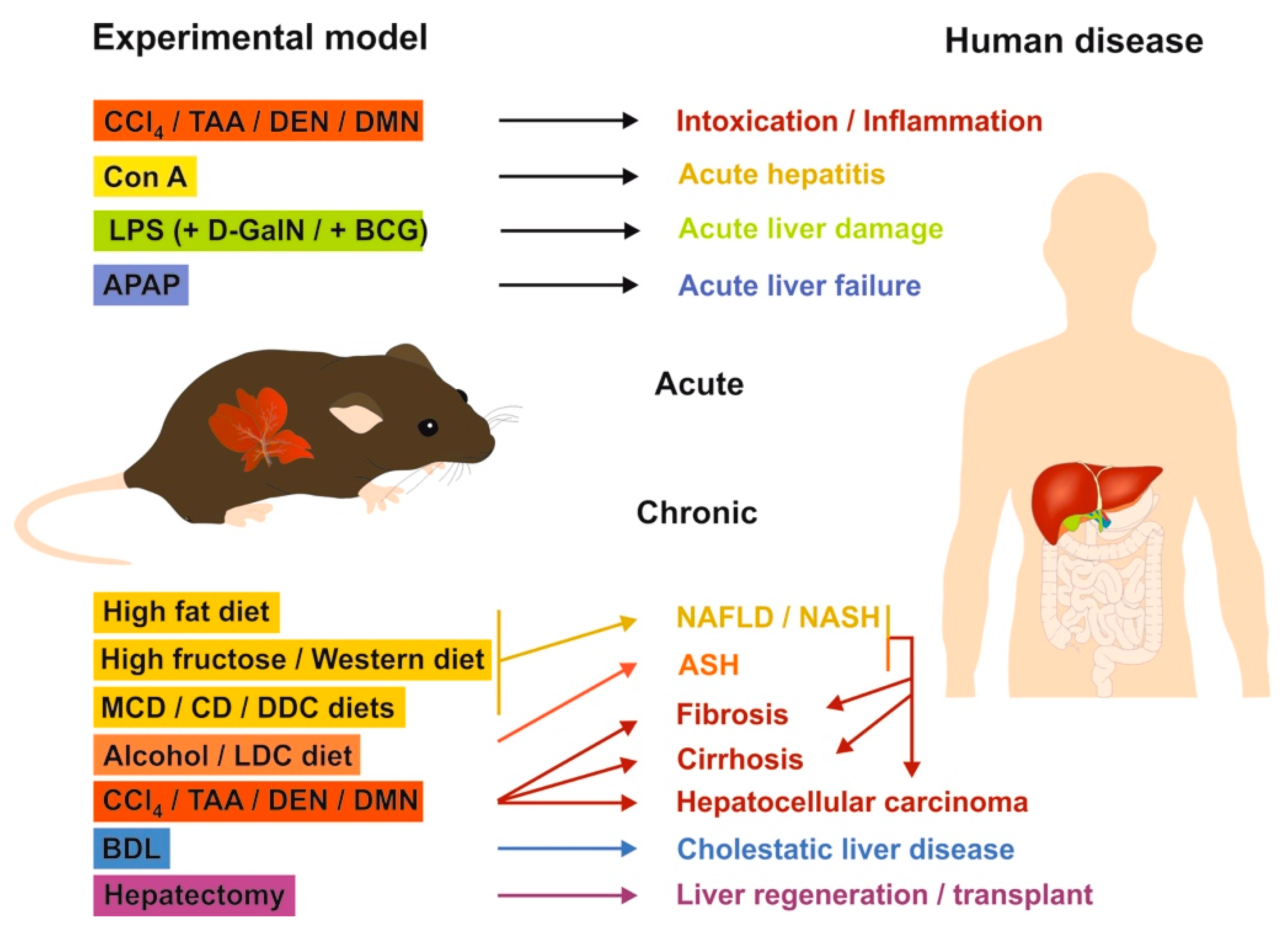
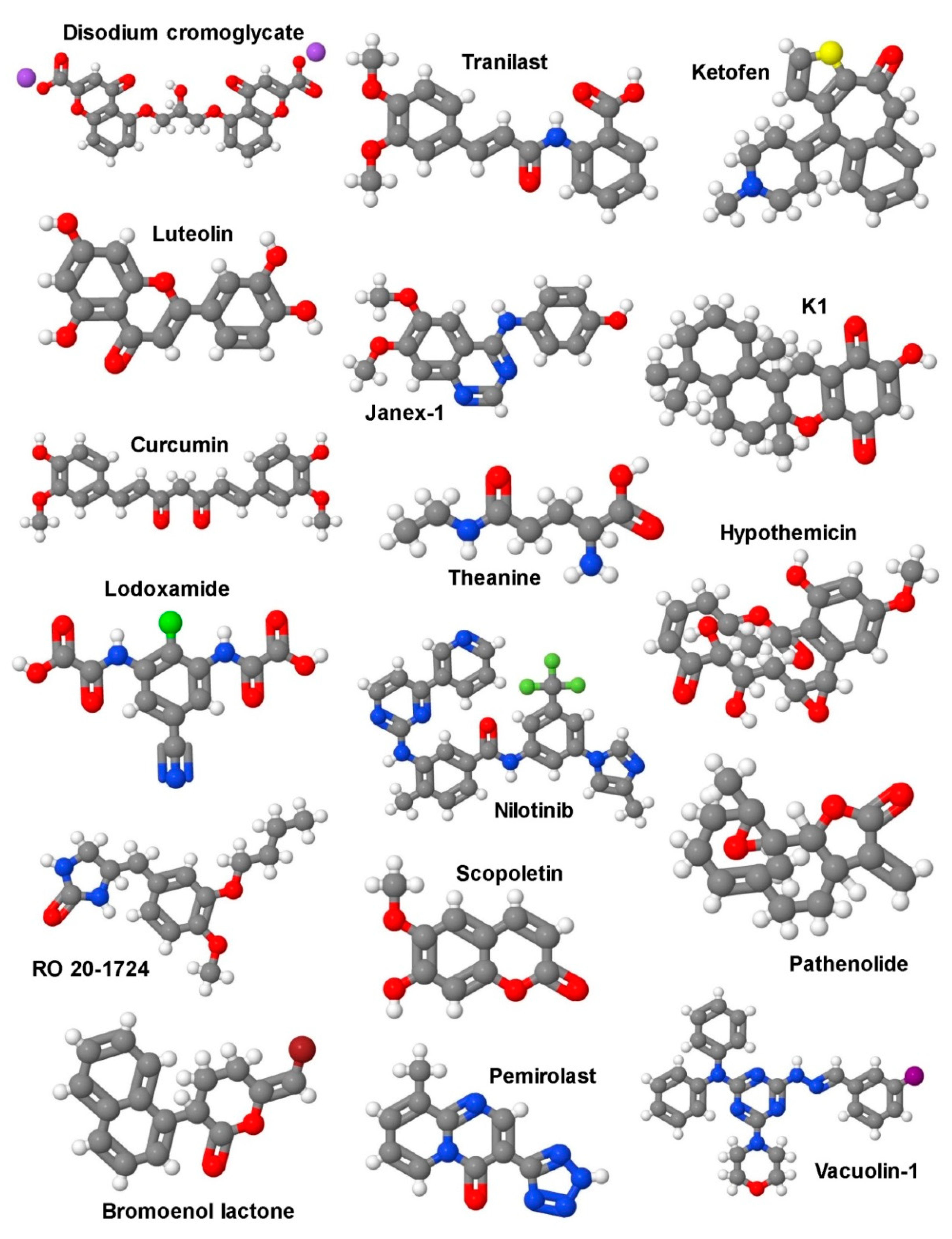
5. Therapeutic Options
5.1. Selective Targeting of Mast Cells in Liver Fibrosis
5.2. Signaling Pathways as Targets for Potential Future Therapies in Liver Fibrosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ribatti, D.; Crivellato, E. Mast cell ontogeny: An historical overview. Immunol. Lett. 2014, 159, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Metz, M.; Maurer, M. Mast cells--key effector cells in immune responses. Trends Immunol. 2007, 28, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Ehara, T.; Shigematsu, H. Mast cells in the kidney. Nephrology (Carlton) 2003, 8, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Farrell, D.J.; Hines, J.E.; Walls, A.F.; Kelly, P.J.; Bennett, M.K.; Burt, A.D. Intrahepatic mast cells in chronic liver diseases. Hepatology 1995, 22, 1175–1181. [Google Scholar] [CrossRef]
- Welle, M. Development, significance, and heterogeneity of mast cells with particular regard to the mast cell-specific proteases chymase and tryptase. J. Leukoc. Biol. 1997, 61, 233–245. [Google Scholar] [CrossRef]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef]
- Kitamura, Y.; Go, S.; Hatanaka, K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood 1978, 52, 447–452. [Google Scholar] [CrossRef]
- Nocka, K.; Buck, J.; Levi, E.; Besmer, P. Candidate ligand for the c-kit transmembrane kinase receptor: KL, a fibroblast derived growth factor stimulates mast cells and erythroid progenitors. EMBO J. 1990, 9, 3287–3294. [Google Scholar] [CrossRef]
- Blank, U.; Ra, C.; Miller, L.; White, K.; Metzger, H.; Kinet, J.P. Complete structure and expression in transfected cells of high affinity IgE receptor. Nature 1989, 337, 187–189. [Google Scholar] [CrossRef]
- Hallgren, J.; Gurish, M.F. Pathways of murine mast cell development and trafficking: Tracking the roots and routes of the mast cell. Immunol. Rev. 2007, 217, 8–18. [Google Scholar] [CrossRef]
- Gurish, M.F.; Tao, H.; Abonia, J.P.; Arya, A.; Friend, D.S.; Parker, C.M.; Austen, K.F. Intestinal mast cell progenitors require CD49dβ7 (α4β7 integrin) for tissue-specific homing. J. Exp. Med. 2001, 194, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Abonia, J.P.; Austen, K.F.; Rollins, B.J.; Joshi, S.K.; Flavell, R.A.; Kuziel, W.A.; Koni, P.A.; Gurish, M.F. Constitutive homing of mast cell progenitors to the intestine depends on autologous expression of the chemokine receptor CXCR2. Blood 2005, 105, 4308–4313. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.S.; Ding, Z.; Hallgren, J. Distinguishing mast cell progenitors from mature mast cells in mice. Stem Cells Dev. 2015, 24, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Ochi, H.; Hirani, W.M.; Yuan, Q.; Friend, D.S.; Austen, K.F.; Boyce, J.A. T helper cell type 2 cytokine-mediated comitogenic responses and CCR3 expression during differentiation of human mast cells in vitro. J. Exp. Med. 1999, 190, 267–280. [Google Scholar] [CrossRef]
- Gentek, R.; Ghigo, C.; Hoeffel, G.; Bulle, M.J.; Msallam, R.; Gautier, G.; Launay, P.; Chen, J.; Ginhoux, F.; Bajenoff, M. Hemogenic endothelial fate mapping reveals dual developmental origin of mast cells. Immunity 2018, 48, 1160–1171. [Google Scholar] [CrossRef]
- Li, Z.; Liu, S.; Xu, J.; Zhang, X.; Han, D.; Liu, J.; Xia, M.; Yi, L.; Shen, Q.; Xu, S.; et al. Adult connective tissue-resident mast cells originate from late erythro-myeloid progenitors. Immunity 2018, 49, 640–653. [Google Scholar] [CrossRef]
- Galli, S.J.; Maurer, M.; Lantz, C.S. Mast cells as sentinels of innate immunity. Cur. Opin. Immunol. 1999, 11, 53–59. [Google Scholar] [CrossRef]
- Galli, S.J.; Nakae, S.; Tsai, M. Mast cells in the development of adaptive immune responses. Nat. Immunol. 2005, 6, 135–142. [Google Scholar] [CrossRef]
- Metz, M.; Piliponsky, A.M.; Chen, C.C.; Lammel, V.; Abrink, M.; Pejler, G.; Tsai, M.; Galli, S.J. Mast cells can enhance resistance to snake and honeybee venoms. Science 2006, 313, 526–530. [Google Scholar] [CrossRef]
- Maurer, M.; Wedemeyer, J.; Metz, M.; Piliponsky, A.M.; Weller, K.; Chatterjea, D.; Clouthier, D.E.; Yanagisawa, M.M.; Tsai, M.; Galli, S.J. Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature 2004, 432, 512–516. [Google Scholar] [CrossRef]
- Schneider, L.A.; Schlenner, S.M.; Feyerabend, T.B.; Wunderlin, M.; Rodewald, H.R. Molecular mechanism of mast cell mediated innate defense against endothelin and snake venom sarafotoxin. J. Exp. Med. 2007, 204, 2629–2639. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-GarciaLuna, J.L.; Chan, D.; Samberg, R.; Abou-Rjeili, M.; Wong, T.H.; Li, A.; Feyerabend, T.B.; Rodewald, H.R.; Henderson, J.E.; Martineau, P.A. Defective bone repair in mast cell-deficient Cpa3Cre/+ mice. PLoS ONE 2017, 12, e0174396. [Google Scholar] [CrossRef] [PubMed]
- Ngkelo, A.; Richart, A.; Kirk, J.A.; Bonnin, P.; Vilar, J.; Lemitre, M.; Marck, P.; Branchereau, M.; Le Gall, S.; Renault, N.; et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J. Exp. Med. 2016, 213, 1353–1374. [Google Scholar] [CrossRef] [PubMed]
- Reitz, M.; Brunn, M.L.; Rodewald, H.R.; Feyerabend, T.B.; Roers, A.; Dudeck, A.; Voehringer, D.; Jonsson, F.; Kuhl, A.A.; Breloer, M. Mucosal mast cells are indispensable for the timely termination of Strongyloides ratti infection. Mucosal Immunol. 2017, 10, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef]
- Schubert, N.; Dudeck, J.; Liu, P.; Karutz, A.; Speier, S.; Maurer, M.; Tuckermann, J.; Dudeck, A. Mast cell promotion of T cell-driven antigen-induced arthritis despite being dispensable for antibody-induced arthritis in which T cells are bypassed. Arthritis Rheumatol. 2015, 67, 903–913. [Google Scholar] [CrossRef]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef]
- Bradding, P.; Pejler, G. The controversial role of mast cells in fibrosis. Immunol. Rev. 2018, 282, 198–231. [Google Scholar] [CrossRef]
- Dahdah, A.; Gautier, G.; Attout, T.; Fiore, F.; Lebourdais, E.; Msallam, R.; Daeron, M.; Monteiro, R.C.; Benhamou, M.; Charles, N.; et al. Mast cells aggravate sepsis by inhibiting peritoneal macrophage phagocytosis. J. Clin. Investig. 2014, 124, 4577–4589. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; Nilsson, G.; Reiter, A.; Hermine, O.; Sotlar, K.; Sperr, W.R.; Escribano, L.; George, T.I.; et al. Advances in the classification and treatment of mastocytosis: Current status and outlook toward the future. Cancer Res. 2017, 77, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Valent, P. Mast cell activation syndromes: Definition and classification. Allergy 2013, 68, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Bonadonna, P.; Hartmann, K.; Brockow, K.; Niedoszytko, M.; Nedoszytko, B.; Siebenhaar, F.; Sperr, W.R.; Oude Elberink, J.N.G.; et al. Proposed diagnostic algorithm for patients with suspected mast cell activation syndrome. J. Allergy Clin. Immunol. Pract. 2019, 7, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Faroldi, G.; Melo, F.R.; Ronnberg, E.; Grujic, M.; Pejler, G. Active caspase-3 is stored within secretory compartments of viable mast cells. J. Immunol. 2013, 191, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Pardo, J.; Wallich, R.; Ebnet, K.; Iden, S.; Zentgraf, H.; Martin, P.; Ekiciler, A.; Prins, A.; Mullbacher, A.; Huber, M.; et al. Granzyme B is expressed in mouse mast cells in vivo and in vitro and causes delayed cell death independent of perforin. Cell Death Differ. 2007, 14, 1768–1779. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef]
- Zorn, C.N.; Pardo, J.; Martin, P.; Kuhny, M.; Simon, M.M.; Huber, M. Secretory lysosomes of mouse mast cells store and exocytose active caspase-3 in a strictly granzyme B dependent manner. Eur. J. Immunol. 2013, 43, 3209–3218. [Google Scholar] [CrossRef]
- Subramanian, H.; Gupta, K.; Ali, H. Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J. Allergy Clin. Immunol. 2016, 138, 700–710. [Google Scholar] [CrossRef]
- McNeil, B.D.; Pundir, P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015, 519, 237–241. [Google Scholar] [CrossRef]
- Boyce, J.A. Mast cells and eicosanoid mediators: A system of reciprocal paracrine and autocrine regulation. Immunol. Rev. 2007, 217, 168–185. [Google Scholar] [CrossRef]
- Bischoff, S.C. Role of mast cells in allergic and non-allergic immune responses: Comparison of human and murine data. Nat. Rev. Immunol. 2007, 7, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Kalis, C.; Keck, S.; Jiang, Z.; Georgel, P.; Du, X.; Shamel, L.; Sovath, S.; Mudd, S.; Beutler, B.; et al. R-form LPS, the master key to the activation of TLR4/MD2 positive cells. Eur. J. Immunol. 2006, 36, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, N.; Rohde, M.; Geffers, R.; Kroger, A.; Hauser, H.; Weiss, S.; Gekara, N.O. Mast cells elicit proinflammatory but not type I interferon responses upon activation of TLRs by bacteria. Proc. Natl. Acad. Sci. USA 2010, 107, 8748–8753. [Google Scholar] [CrossRef] [PubMed]
- Keck, S.; Muller, I.; Fejer, G.; Savic, I.; Tchaptchet, S.; Nielsen, P.J.; Galanos, C.; Huber, M.; Freudenberg, M.A. Absence of TRIF signaling in lipopolysaccharide-stimulated murine mast cells. J. Immunol. 2011, 186, 5478–5488. [Google Scholar] [CrossRef]
- Duttlinger, R.; Manova, K.; Berrozpe, G.; Chu, T.Y.; DeLeon, V.; Timokhina, I.; Chaganti, R.S.; Zelenetz, A.D.; Bachvarova, R.F.; Besmer, P. The Wsh and Ph mutations affect the c-kit expression profile: C-kit misexpression in embryogenesis impairs melanogenesis in Wsh and Ph mutant mice. Proc. Natl. Acad. Sci. USA 1995, 92, 3754–3758. [Google Scholar] [CrossRef]
- Grimbaldeston, M.A.; Chen, C.C.; Piliponsky, A.M.; Tsai, M.; Tam, S.Y.; Galli, S.J. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 2005, 167, 835–848. [Google Scholar] [CrossRef]
- Feyerabend, T.B.; Weiser, A.; Tietz, A.; Stassen, M.; Harris, N.; Kopf, M.; Radermacher, P.; Moller, P.; Benoist, C.; Mathis, D.; et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 2011, 35, 832–844. [Google Scholar] [CrossRef]
- Scholten, J.; Hartmann, K.; Gerbaulet, A.; Krieg, T.; Muller, W.; Testa, G.; Roers, A. Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res. 2008, 17, 307–315. [Google Scholar] [CrossRef]
- Voehringer, D.; Liang, H.E.; Locksley, R.M. Homeostasis and effector function of lymphopenia-induced “memory-like” T cells in constitutively T cell-depleted mice. J. Immunol. 2008, 180, 4742–4753. [Google Scholar] [CrossRef]
- Dudeck, A.; Dudeck, J.; Scholten, J.; Petzold, A.; Surianarayanan, S.; Kohler, A.; Peschke, K.; Vohringer, D.; Waskow, C.; Krieg, T.; et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 2011, 34, 973–984. [Google Scholar] [CrossRef]
- Steimer, D.A.; Boyd, K.; Takeuchi, O.; Fisher, J.K.; Zambetti, G.P.; Opferman, J.T. Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effector function. Blood 2009, 113, 2805–2815. [Google Scholar] [CrossRef] [PubMed]
- Lilla, J.N.; Chen, C.C.; Mukai, K.; BenBarak, M.J.; Franco, C.B.; Kalesnikoff, J.; Yu, M.; Tsai, M.; Piliponsky, A.M.; Galli, S.J. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood 2011, 118, 6930–6938. [Google Scholar] [CrossRef] [PubMed]
- Reber, L.L.; Marichal, T.; Galli, S.J. New models for analyzing mast cell functions in vivo. Am. J. Pathol. 2012, 33, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef]
- Potey, P.M.; Rossi, A.G.; Lucas, C.D.; Dorward, D.A. Neutrophils in the initiation and resolution of acute pulmonary inflammation: Understanding biological function and therapeutic potential. J. Pathol. 2019, 247, 672–685. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern recognition receptors and the host cell death molecular machinery. Front Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef]
- Gressner, A.M.; Weiskirchen, R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J. Cell Mol. Med. 2006, 10, 76–99. [Google Scholar] [CrossRef]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-β. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Weiskirchen, R. Hepatoprotective and anti-fibrotic agents: It’s time to take the next step. Front. Pharmacol. 2016, 6, 303. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef]
- Doss, G.P.; Agoramoorthy, G.; Chakraborty, C. TNF/TNFR: Drug target for autoimmune diseases and immune-mediated inflammatory diseases. Front. Biosci. (Landmark Ed) 2014, 19, 1028–1040. [Google Scholar] [CrossRef]
- De Bleser, P.J.; Xu, G.; Rombouts, K.; Rogiers, V.; Geerts, A. Glutathione levels discriminate between oxidative stress and transforming growth factor-β signaling in activated rat hepatic stellate cells. J. Biol. Chem. 1999, 274, 33881–33887. [Google Scholar] [CrossRef]
- Manea, S.A.; Constantin, A.; Manda, G.; Sasson, S.; Manea, A. Regulation of Nox enzymes expression in vascular pathophysiology: Focusing on transcription factors and epigenetic mechanisms. Redox Biol. 2015, 5, 358–366. [Google Scholar] [CrossRef]
- Romero, F.; Shah, D.; Duong, M.; Penn, R.B.; Fessler, M.B.; Madenspacher, J.; Stafstrom, W.; Kavuru, M.; Lu, B.; Kallen, C.B.; et al. A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 74–86. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Inagaki, Y.; Rincon-Sanchez, A.R.; Else, C.; Saccomanno, S.; Benedetti, A.; Ramirez, F.; Rojkind, M. Early response of α2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-β independent. Hepatology 2005, 42, 343–352. [Google Scholar] [CrossRef]
- Kubo, H. Extracellular Vesicles in lung disease. Chest 2018, 153, 210–216. [Google Scholar] [CrossRef]
- Momen-Heravi, F.; Bala, S.; Kodys, K.; Szabo, G. Exosomes derived from alcohol-treated hepatocytes horizontally transfer liver specific miRNA-122 and sensitize monocytes to LPS. Sci. Rep. 2015, 5, 9991. [Google Scholar] [CrossRef]
- Heinrich, L.F.; Andersen, D.K.; Cleasby, M.E.; Lawson, C. Long-term high fat feeding of rats results in increased numbers of circulating microvesicles with pro-inflammatory effects on endothelial cells. Br. J. Nutr. 2015, 113, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Cho, Y.E.; Komarow, H.D.; Bandara, G.; Song, B.J.; Olivera, A.; Metcalfe, D.D. Mastocytosis-derived extracellular vesicles exhibit a mast cell signature, transfer KIT to stellate cells, and promote their activation. Proc. Natl. Acad. Sci. USA. 2018, 115, E10692–E10701. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Recent advances in understanding liver fibrosis: Bridging basic science and individualized treatment concepts. F1000Research 2018, 7, 921. [Google Scholar] [CrossRef] [PubMed]
- Campana, L.; Iredale, J.P. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar] [CrossRef]
- Scholten, D.; Trebicka, J.; Liedtke, C.; Weiskirchen, R. The carbon tetrachloride model in mice. Lab Anim. 2015, 49, 4–11. [Google Scholar] [CrossRef]
- Heymann, F.; Hamesch, K.; Weiskirchen, R.; Tacke, F. The concanavalin A model of acute hepatitis in mice. Lab Anim. 2015, 49, 12–20. [Google Scholar] [CrossRef]
- Wallace, M.C.; Hamesch, K.; Lunova, M.; Kim, Y.; Weiskirchen, R.; Strnad, P.; Friedman, S.L. Standard operating procedures in experimental liver research: Thioacetamide model in mice and rats. Lab Anim. 2015, 49, 21–29. [Google Scholar] [CrossRef]
- Mossanen, J.C.; Tacke, F. Acetaminophen-induced acute liver injury in mice. Lab Anim. 2015, 49, 30–36. [Google Scholar] [CrossRef]
- Hamesch, K.; Borkham-Kamphorst, E.; Strnad, P.; Weiskirchen, R. Lipopolysaccharide-induced inflammatory liver injury in mice. Lab Anim. 2015, 49, 37–46. [Google Scholar] [CrossRef]
- Ramadori, P.; Weiskirchen, R.; Trebicka, J.; Streetz, K. Mouse models of metabolic liver injury. Lab Anim. 2015, 49, 47–58. [Google Scholar] [CrossRef]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef]
- Tag, C.G.; Weiskirchen, S.; Hittatiya, K.; Tacke, F.; Tolba, R.H.; Weiskirchen, R. Induction of experimental obstructive cholestasis in mice. Lab Anim. 2015, 49, 70–80. [Google Scholar] [CrossRef]
- Tag, C.G.; Sauer-Lehnen, S.; Weiskirchen, S.; Borkham-Kamphorst, E.; Tolba, R.H.; Tacke, F.; Weiskirchen, R. Bile duct ligation in mice: Induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J. Vis. Exp. 2015. [Google Scholar] [CrossRef]
- Nevzorova, Y.A.; Tolba, R.; Trautwein, C.; Liedtke, C. Partial hepatectomy in mice. Lab Anim. 2015, 49, 81–88. [Google Scholar] [CrossRef]
- Smit, J.J.; Schinkel, A.H.; Oude Elferink, R.P.; Groen, A.K.; Wagenaar, E.; van Deemter, L.; Mol, C.A.; Ottenhoff, R.; Van Der Lugt, N.M.; van Roon, M.A.; et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 1993, 75, 451–462. [Google Scholar] [CrossRef]
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef]
- Gu, X.; Huang, D.; Ci, L.; Shi, J.; Zhang, M.; Yang, H.; Wang, Z.; Sheng, Z.; Sun, R.; Fei, J. Fate tracing of hepatocytes in mouse liver. Sci. Rep. 2017, 7, 16108. [Google Scholar] [CrossRef]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef]
- Metcalfe, D.D. The liver, spleen, and lymph nodes in mastocytosis. J. Investig. Dermatol. 1991, 96, 45S–46S. [Google Scholar] [CrossRef]
- Mican, J.M.; Di Bisceglie, A.M.; Fong, T.L.; Travis, W.D.; Kleiner, D.E.; Baker, B.; Metcalfe, D.D. Hepatic involvement in mastocytosis: Clinicopathologic correlations in 41 cases. Hepatology 1995, 22, 1163–1170. [Google Scholar] [CrossRef]
- Umezu, K.; Yuasa, S.; Sudoh, A. Change of hepatic histamine content during hepatic fibrosis. Biochem. Pharmacol. 1985, 34, 2007–2011. [Google Scholar] [CrossRef] [PubMed]
- Gittlen, S.D.; Schulman, E.C.; Maddrey, W.C. Raised histamine concentrations in chronic cholestatic liver disease. Gut 1990, 31, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.Y.; Wang, D.W.; Xu, Z.H.; Gao, Y.B.; Yang, R.B.; Liu, P.; Wang, Z.P.; Li, Y.P.J. The changes and significance of mast cells in irradiated rat liver. Environ. Pathol. Toxicol. Oncol. 1994, 13, 111–116. [Google Scholar]
- Nakamura, A.; Yamazaki, K.; Suzuki, K.; Sato, S. Increased portal tract infiltration of mast cells and eosinophils in primary biliary cirrhosis. Am. J. Gastroenterol. 1997, 92, 2245–2249. [Google Scholar]
- Rioux, K.P.; Sharkey, K.A.; Wallace, J.L.; Swain, M.G. Hepatic mucosal mast cell hyperplasia in rats with secondary biliary cirrhosis. Hepatology 1996, 23, 888–895. [Google Scholar] [CrossRef]
- Armbrust, T.l.; Batusic, D.; Ringe, B.; Ramadori, G.J. Mast cells distribution in human liver disease and experimental rat liver fibrosis. Indications for mast cell participation in development of liver fibrosis. Hepatology 1997, 26, 1042–1054. [Google Scholar] [CrossRef]
- Ozaki, S.; Sato, Y.; Yasoshima, M.; Harada, K.; Nakanuma, Y. Diffuse expression of heparan sulfate proteoglycan and connective tissue growth factor in fibrous septa with many mast cells relate to unresolving hepatic fibrosis of congenital hepatic fibrosis. Liver Int. 2005, 25, 817–828. [Google Scholar] [CrossRef]
- Jarido, V.; Kennedy, L.; Hargrove, L.; Demieville, J.; Thomson, J.; Stephenson, K.; Francis, H. The emerging role of mast cells in liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G89–G101. [Google Scholar] [CrossRef]
- Thomson, J.; Hargrove, L.; Kennedy, L.; Demieville, J.; Francis, H. Cellular crosstalk during cholestatic liver injury. Liver Res. 2017, 1, 26–33. [Google Scholar] [CrossRef]
- Nakano, T.; Lai, C.Y.; Goto, S.; Hsu, L.W.; Kawamoto, S.; Ono, K.; Chen, K.D.; Lin, C.C.; Chiu, K.W.; Wang, C.C.; et al. Immunological and regenerative aspects of hepatic mast cells in liver allograft rejection and tolerance. PLoS ONE 2012, 7, e37202. [Google Scholar] [CrossRef]
- Shen, D.Z. A target role for mast cell in the prevention and therapy of hepatic fibrosis. Med. Hypotheses 2008, 70, 760–764. [Google Scholar] [CrossRef]
- Hargrove, L.; Graf-Eaton, A.; Kennedy, L.; Demieville, J.; Owens, J.; Hodges, K.; Ladd, B.; Francis, H. Isolation and characterization of hepatic mast cells from cholestatic rats. Lab. Investig. 2016, 96, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast cell: A multi-functional master cell. Front. Immunol. 2016, 6, 620. [Google Scholar] [CrossRef]
- Matsunaga, Y.; Terada, T. Mast cell subpopulations in chronic inflammatory hepatobiliary diseases. Liver 2000, 20, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Halova, I.; Draberova, L.; Draber, P. Mast cell chemotaxis—Chemoattractants and signaling pathways. Front. Immunol. 2012, 3, 119. [Google Scholar] [CrossRef] [PubMed]
- Gruber, B.L.; Marchese, M.J.; Kew, R.R. Transforming growth factor-β 1 mediates mast cell chemotaxis. J. Immunol. 1994, 152, 5860–5867. [Google Scholar] [PubMed]
- Olsson, N.; Piek, E.; ten Dijke, P.; Nilsson, G. Human mast cell migration in response to members of the transforming growth factor-β family. J. Leukoc. Biol. 2000, 67, 350–356. [Google Scholar] [CrossRef]
- Meininger, C.J.; Yano, H.; Rottapel, R.; Bernstein, A.; Zsebo, K.M.; Zetter, B.R. The c-kit receptor ligand functions as a mast cell chemoattractant. Blood 1992, 79, 958–963. [Google Scholar] [CrossRef]
- Nilsson, G.; Butterfield, J.H.; Nilsson, K.; Siegbahn, A. Stem cell factor is a chemotactic factor for human mast cells. J. Immunol. 1994, 153, 3717–3723. [Google Scholar]
- Gaça, M.D.; Pickering, J.A.; Arthur, M.J.; Benyon, R.C. Human and rat hepatic stellate cells produce stem cell factor: A possible mechanism for mast cell recruitment in liver fibrosis. J. Hepatol. 1999, 30, 850–858. [Google Scholar] [CrossRef]
- Flanagan, J.G.; Chan, D.C.; Leder, P. Transmembrane form of the kit ligand growth factor is determined by alternative splicing and is missing in the Sld mutant. Cell 1991, 64, 1025–1035. [Google Scholar] [CrossRef]
- Boyce, J.A.; Mellor, E.A.; Perkins, B.; Lim, Y.C.; Luscinskas, F.W. Human mast cell progenitors use α-integrin, VCAM-1, and PSGL-1 E-selectin for adhesive interactions with human vascular endothelium under flow conditions. Blood 2002, 99, 2890–2896. [Google Scholar] [CrossRef] [PubMed]
- Brito, J.M.; Borojevic, R. Liver granulomas in schistosomiasis: Mast cell-dependent induction of SCF expression in hepatic stellate cells is mediated by TNF-α. J. Leukoc. Biol. 1997, 62, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Meadows, V.; Kennedy, L.; Hargrove, L.; Demieville, J.; Meng, F.; Virani, S.; Reinhart, E.; Kyritsi, K.; Invernizzi, P.; Yang, Z.; et al. Downregulation of hepatic stem cell factor by Vivo-Morpholino treatment inhibits mast cell migration and decreases biliary damage/senescence and liver fibrosis in Mdr2−/− mice. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165557. [Google Scholar] [CrossRef] [PubMed]
- Faber, T.W.; Pullen, N.A.; Fernando, J.F.; Kolawole, E.M.; McLeod, J.J.; Taruselli, M.; Williams, K.L.; Rivera, K.O.; Barnstein, B.O.; Conrad, D.H.; et al. ADAM10 is required for SCF-induced mast cell migration. Cell. Immunol. 2014, 290, 80–88. [Google Scholar] [CrossRef]
- Irani, A.A.; Craig, S.S.; Nilsson, G.; Ishizaka, T.; Schwartz, L.B. Characterization of human mast cells developed in vitro from fetal liver cells cocultured with murine 3T3 fibroblasts. Immunology 1992, 77, 136–143. [Google Scholar]
- Irani, A.M.; Nilsson, G.; Miettinen, U.; Craig, S.S.; Ashman, L.K.; Ishizaka, T.; Zsebo, K.M.; Schwartz, L.B. Recombinant human stem cell factor stimulates differentiation of mast cells from dispersed human fetal liver cells. Blood 1992, 80, 3009–3021. [Google Scholar] [CrossRef]
- Du, Z.; Li, Y.; Xia, H.; Irani, A.M.; Schwartz, L.B. Recombinant human granulocyte-macrophage colony-stimulating factor (CSF), but not recombinant human granulocyte CSF, down-regulates the recombinant human stem cell factor-dependent differentiation of human fetal liver-derived mast cells. J. Immunol. 1997, 159, 838–845. [Google Scholar]
- Brito, J.M.; Mermelstein, C.S.; Tempone, A.J.; Borojevic, R. Mast cells can revert dexamethasone-mediated down-regulation of stem cell factor. Eur. J. Pharmacol. 2001, 414, 105–112. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Dahle, M.K.; Agren, J.; Myhre, A.E.; Reinholt, F.P.; Foster, S.J.; Collins, J.L.; Thiemermann, C.; Aasen, A.O.; Wang, J.E. Activation of the liver X receptor protects against hepatic injury in endotoxemia by suppressing Kupffer cell activation. Shock 2006, 25, 141–146. [Google Scholar] [CrossRef]
- Nunomura, S.; Okayama, Y.; Matsumoto, K.; Hashimoto, N.; Endo-Umeda, K.; Terui, T.; Makishima, M.; Ra, C. Activation of LXRs using the synthetic agonist GW3965 represses the production of pro-inflammatory cytokines by murine mast cells. Allergol. Int. 2015, 64, S11–S17. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.M.; Leach, S.; Payne, J.; Mitchell, A.; Dai, Y.; Jackson, G.D. A role for the hepatobiliary system in IgE-mediated intestinal inflammation in the rat. Clin. Exp. Allergy 1999, 29, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, G.; Burnstock, G. A novel cell-to-cell interaction between mast cells and other cell types. Exp. Cell Res. 1983, 147, 1–13. [Google Scholar] [CrossRef]
- Grizzi, F.; Franceschini, B.; Gagliano, N.; Moscheni, C.; Annoni, G.; Vergani, C.; Hermonat, P.L.; Chiriva-Internati, M.; Dioguardi, N. Mast cell density, hepatic stellate cell activation and TGF-β transcripts in the aging Sprague-Dawley rat during early acute liver injury. Toxicol. Pathol. 2003, 31, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, A. 1.; Shibayama, Y. Role of mast cells in hepatic remodeling during cholestasis and its resolution: Relevance to regulation of apoptosis. Exp. Toxicol. Pathol. 2005, 56, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Takahara, Y.; Takahashi, M.; Wagatsuma, H.; Yokoya, F.; Zhang, Q.W.; Yamaguchi, M.; Aburatani, H.; Kawada, N. Gene expression profiles of hepatic cell-type specific marker genes in progression of liver fibrosis. World J. Gastroenterol. 2006, 12, 6473–6499. [Google Scholar] [CrossRef]
- Jeong, W.I.; Lee, C.S.; Park, S.J.; Chung, J.Y.; Jeong, K.S. Kinetics of macrophages, myofibroblasts and mast cells in carbon tetrachloride-induced rat liver cirrhosis. Anticancer Res. 2002, 22, 869–877. [Google Scholar]
- Sugihara, A.; Tsujimura, T.; Fujita, Y.; Nakata, Y.; Terada, N. Evaluation of role of mast cells in the development of liver fibrosis using mast cell-deficient rats and mice. J. Hepatol. 1999, 30, 859–867. [Google Scholar] [CrossRef]
- Amiot, L.; Vu, N.; Drenou, B.; Scrofani, M.; Chalin, A.; Devisme, C.; Samson, M. The anti-fibrotic role of mast cells in the liver is mediated by HLA-G and interaction with hepatic stellate cells. Cytokine 2019, 117, 50–58. [Google Scholar] [CrossRef]
- Amiot, L.; Vu, N.; Rauch, M.; L’Helgoualc’h, A.; Chalmel, F.; Gascan, H.; Turlin, B.; Guyader, D.; Samson, M. Expression of HLA-G by mast cells is associated with hepatitis C virus-induced liver fibrosis. J. Hepatol. 2014, 60, 245–252. [Google Scholar] [CrossRef]
- Miyazawa, S.; Hotta, O.; Doi, N.; Natori, Y.; Nishikawa, K.; Natori, Y. Role of mast cells in the development of renal fibrosis: Use of mast cell-deficient rats. Kidney Int. 2004, 65, 2228–2237. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Ono, K.; Hwang, M.W.; Iwasaki, A.; Okada, M.; Nakatani, K.; Sasayama, S.; Matsumori, A. Evidence for a role of mast cells in the evolution to congestive heart failure. J. Exp. Med. 2002, 195, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Veerappan, A.; O’Connor, N.J.; Brazin, J.; Reid, A.C.; Jung, A.; McGee, D.; Summers, B.; Branch-Elliman, D.; Stiles, B.; Worgall, S.; et al. Mast cells: A pivotal role in pulmonary fibrosis. DNA Cell Biol. 2013, 32, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Shao, Z.; Nazari, M.; Guo, L.; Li, S.H.; Sun, J.; Liu, S.M.; Yuan, H.P.; Weisel, R.D.; Li, R.K. The cardiac repair benefits of inflammation do not persist: Evidence from mast cell implantation. J. Cell Mol. Med. 2015, 19, 2751–2762. [Google Scholar] [CrossRef]
- Huang, Z.G.; Zhai, W.R.; Zhang, Y.E.; Zhang, X.R. Study of heteroserum-induced rat liver fibrosis model and its mechanism. World J. Gastroenterol. 1998, 4, 206–209. [Google Scholar] [CrossRef]
- Akiyoshi, H.; Terada, T. Mast cell, myofibroblast and nerve terminal complexes in carbon tetrachloride-induced cirrhotic rat livers. J. Hepatol. 1998, 29, 112–119. [Google Scholar] [CrossRef]
- Hargrove, L.; Kennedy, L.; Demieville, J.; Jones, H.; Meng, F.; DeMorrow, S.; Karstens, W.; Madeka, T.; Greene JJr Francis, H. Bile duct ligation-induced biliary hyperplasia, hepatic injury, and fibrosis are reduced in mast cell-deficient Kit(W-sh) mice. Hepatology 2017, 65, 1991–2004. [Google Scholar] [CrossRef]
- Gordon, J.R.; Galli, S.J. Promotion of mouse fibroblast collagen gene expression by mast cells stimulated via the FcεRI. Role for mast cell-derived transforming growth factor β and tumor necrosis factor α. J. Exp. Med. 1994, 180, 2027–2037. [Google Scholar] [CrossRef]
- Kendall, J.C.; Li, X.H.; Galli, S.J.; Gordon, J.R. Promotion of mouse fibroblast proliferation by IgE-dependent activation of mouse mast cells: Role for mast cell tumor necrosis factor-α and transforming growth factor-β1. J. Allergy Clin. Immunol. 1997, 99, 113–123. [Google Scholar] [CrossRef]
- Gordon, J.R. TGFβ1 and TNFα secreted by mast cells stimulated via the FcεRI activate fibroblasts for high-level production of monocyte chemoattractant protein-1 (MCP-1). Cell. Immunol. 2000, 201, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Matsumoto, K.; Okumura, S.; Kashiwakura, J.; Oboki, K.; Yokoi, H.; Kambe, N.; Ohta, K.; Okayama, Y. Gene expression profiling of human mast cell subtypes: An in silico study. Allergol. Int. 2006, 55, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.S.; Stevens, R.L.; Lane, W.S.; Carr, M.H.; Austen, K.F.; Serafin, W.E. Different mouse mast cell populations express various combinations of at least six distinct mast cell serine proteases. Proc. Natl. Acad. Sci. USA 1990, 87, 3230–3234. [Google Scholar] [CrossRef]
- Knight, V.; Tchongue, J.; Lourensz, D.; Tipping, P.; Sievert, W. Protease-activated receptor 2 promotes experimental liver fibrosis in mice and activates human hepatic stellate cells. Hepatology 2012, 55, 879–887. [Google Scholar] [CrossRef]
- Gaça, M.D.; Zhou, X.; Benyon, R.C. Regulation of hepatic stellate cell proliferation and collagen synthesis by proteinase-activated receptors. J. Hepatol. 2002, 36, 362–369. [Google Scholar] [CrossRef]
- Lu, J.; Chen, B.; Li, S.; Sun, Q. Tryptase inhibitor APC 366 prevents hepatic fibrosis by inhibiting collagen synthesis induced by tryptase/protease-activated receptor 2 interactions in hepatic stellate cells. Int. Immunopharmacol. 2014, 20, 352–357. [Google Scholar] [CrossRef]
- Komeda, K.; Jin, D.; Takai, S.; Hayashi, M.; Takeshita, A.; Shibayama, Y.; Tanigawa, N.; Miyazaki, M. Significance of chymase-dependent angiotensin II formation in the progression of human liver fibrosis. Hepatol. Res. 2008, 38, 501–510. [Google Scholar] [CrossRef]
- Urata, H.; Kinoshita, A.; Misono, K.S.; Bumpus, F.M.; Husain, A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J. Biol. Chem. 1990, 265, 22348–22357. [Google Scholar]
- Yoshiji, H.; Kuriyama, S.; Yoshii, J.; Ikenaka, Y.; Noguchi, R.; Nakatani, T.; Tsujinoue, H.; Fukui, H. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology 2001, 34, 745–750. [Google Scholar] [CrossRef]
- Ikura, Y.; Ohsawa, M.; Shirai, N.; Sugama, Y.; Fukushima, H.; Suekane, T.; Hirayama, M.; Ehara, S.; Naruko, T.; Ueda, M. Expression of angiotensin II type 1 receptor in human cirrhotic livers: Its relation to fibrosis and portal hypertension. Hepatol. Res. 2005, 32, 107–116. [Google Scholar] [CrossRef]
- Yin, M.; Wu, L. Effect of mast cell chymase on activation, proliferation and transdifferentiation of hepatic stellate cells. Hepatogastroenterology 2015, 62, 1007–1010. [Google Scholar] [PubMed]
- Kofford, M.W.; Schwartz, L.B.; Schechter, N.M.; Yager, D.R.; Diegelmann, R.F.; Graham, M.F. Cleavage of type I procollagen by human mast cell chymase initiates collagen fibril formation and generates a unique carboxyl-terminal propeptide. J. Biol. Chem. 1997, 272, 7127–7131. [Google Scholar] [CrossRef] [PubMed]
- Takai, S.; Miyazaki, M. A novel therapeutic strategy against vascular disorders with chymase inhibitor. Curr. Vasc. Pharmacol. 2003, 1, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Furubayashi, K.; Takai, S.; Jin, D.; Miyazaki, M.; Katsumata, T.; Inagaki, S.; Kimura, M.; Tanaka, K.; Nishimoto, M.; Fukumoto, H. Chymase activates promatrix metalloproteinase-9 in human abdominal aortic aneurysm. Clin. Chim. Acta 2008, 388, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Longley, B.J.; Tyrrell, L.; Ma, Y.; Williams, D.A.; Halaban, R.; Langley, K.; Lu, H.S.; Schechter, N.M. Chymase cleavage of stem cell factor yields a bioactive, soluble product. Proc. Natl. Acad. Sci. USA 1997, 94, 9017–9021. [Google Scholar] [CrossRef] [PubMed]
- Takai, S.; Jin, D. Chymase Inhibitor as a novel therapeutic agent for Non-alcoholic steatohepatitis. Front. Pharmacol. 2018, 9, 144. [Google Scholar] [CrossRef]
- Johnson, C.; Huynh, V.; Hargrove, L.; Kennedy, L.; Graf-Eaton, A.; Owens, J.; Trzeciakowski, J.P.; Hodges, K.; DeMorrow, S.; Han, Y.; et al. Inhibition of mast cell-derived histamine decreases human cholangiocarcinoma growth and differentiation via c-Kit/Stem cell factor-dependent signaling. Am. J. Pathol. 2016, 186, 123–133. [Google Scholar] [CrossRef]
- Jones, H.; Hargrove, L.; Kennedy, L.; Meng, F.; Graf-Eaton, A.; Owens, J.; Alpini, G.; Johnson, C.; Bernuzzi, F.; Demieville, J.; et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2−/− mice. Hepatology 2016, 64, 1202–1216. [Google Scholar] [CrossRef]
- Choi, J.S.; Kim, J.K.; Yang, Y.J.; Kim, Y.; Kim, P.; Park, S.G.; Cho, E.Y.; Lee, D.H.; Choi, J.W. Identification of cromolyn sodium as an anti-fibrotic agent targeting both hepatocytes and hepatic stellate cells. Pharmacol. Res. 2015, 102, 176–183. [Google Scholar] [CrossRef]
- Kennedy, L.; Hargrove, L.; Demieville, J.; Karstens, W.; Jones, H.; DeMorrow, S.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; Alpini, G.; et al. Blocking H1/H2 histamine receptors inhibits damage/fibrosis in Mdr2−/− mice and human cholangiocarcinoma tumorigenesis. Hepatology 2018, 68, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Boucek, R.J.; Noble, N.L. Histamine, norepinephrine, and bradykinin stimulation of fibroblast growth and modification of serotonin response. Proc. Soc. Exp. Biol. Med. 1973, 144, 929–933. [Google Scholar] [CrossRef]
- Dayton, E.T.; Caulfield, J.P.; Hein, A.; Austen, K.F.; Stevens, R.L. Regulation of the growth rate of mouse fibroblasts by IL-3-activated mouse bone marrow-derived mast cells. J. Immunol. 1989, 142, 4307–4313. [Google Scholar]
- Hatamochi, A.; Fujiwara, K.; Ueki, H. Effects of histamine on collagen synthesis by cultured fibroblasts derived from guinea pig skin. Arch. Dermatol. Res. 1985, 277, 60–64. [Google Scholar] [CrossRef]
- Levi-Schaffer, F.; Rubinchik, E. Activated mast cells are fibrogenic for 3T3 fibroblasts. J. Investig. Dermatol. 1995, 104, 999–1003. [Google Scholar] [CrossRef]
- Thompson, H.L.; Burbelo, P.D.; Gabriel, G.; Yamada, Y.; Metcalfe, D.D. Murine mast cells synthesize basement membrane components. A potential role in early fibrosis. J. Clin. Investig. 1991, 87, 619–623. [Google Scholar] [CrossRef]
- Schon, H.T.; Weiskirchen, R. Immunomodulatory effects of transforming growth factor-β in the liver. Hepatobiliary Surg. Nutr. 2014, 3, 386–406. [Google Scholar] [CrossRef]
- Miller, H.R.; Wright, S.H.; Knight, P.A.; Thornton, E.M. A novel function for transforming growth factor-β1, upregulation of the expression and the IgE-independent extracellular release of a mucosal mast cell granule-specific β-chymase, mouse mast cell protease-1. Blood 1999, 93, 3473–3486. [Google Scholar] [CrossRef]
- Yamazaki, S.; Nakano, N.; Honjo, A.; Hara, M.; Maeda, K.; Nishiyama, C.; Kitaura, J.; Ohtsuka, Y.; Okumura, K.; Ogawa, H.; et al. The transcription factor Ehf is involved in TGF-β-induced suppression of FcεRI and c-Kit expression and FcεRI-mediated activation in mast cells. J. Immunol. 2015, 195, 3427–3435. [Google Scholar] [CrossRef]
- Ndaw, V.S.; Abebayehu, D.; Spence, A.J.; Paez, P.A.; Kolawole, E.M.; Taruselli, M.T.; Caslin, H.L.; Chumanevich, A.P.; Paranjape, A.; Baker, B.; et al. TGF-β1 suppresses IL-33-induced mast cell function. J. Immunol. 2017, 199, 866–873. [Google Scholar] [CrossRef]
- Norozian, F.; Kashyap, M.; Ramirez, C.D.; Patel, N.; Kepley, C.L.; Barnstein, B.O.; Ryan, J.J. TGFβ1 induces mast cell apoptosis. Exp. Hematol. 2006, 34, 579–587. [Google Scholar] [CrossRef]
- Olsson, N.; Piek, E.; Sundström, M.; ten Dijke, P.; Nilsson, G. Transforming growth factor-β-mediated mast cell migration depends on mitogen-activated protein kinase activity. Cell Signal. 2001, 13, 483–490. [Google Scholar] [CrossRef]
- Ramírez-Valadez, K.A.; Vázquez-Victorio, G.; Macías-Silva, M.; González-Espinosa, C. Fyn kinase mediates cortical actin ring depolymerization required for mast cell migration in response to TGF-β in mice. Eur. J. Immunol. 2017, 47, 1305–1316. [Google Scholar] [CrossRef]
- Gebhardt, T.; Lorentz, A.; Detmer, F.; Trautwein, C.; Bektas, H.; Manns, M.P.; Bischoff, S.C. Growth, phenotype, and function of human intestinal mast cells are tightly regulated by transforming growth factor β1. Gut 2005, 54, 928–934. [Google Scholar] [CrossRef][Green Version]
- Kashyap, M.; Bailey, D.P.; Gomez, G.; Rivera, J.; Huff, T.F.; Ryan, J.J. TGF-β1 inhibits late-stage mast cell maturation. Exp. Hematol. 2005, 33, 1281–1291. [Google Scholar] [CrossRef]
- Gomez, G.; Ramirez, C.D.; Rivera, J.; Patel, M.; Norozian, F.; Wright, H.V.; Kashyap, M.V.; Barnstein, B.O.; Fischer-Stenger, K.; Schwartz, L.B.; et al. TGF-β1 inhibits mast cell FcεRI expression. J. Immunol. 2005, 174, 5987–5993. [Google Scholar] [CrossRef]
- Grizzi, F.; Di Caro, G.; Laghi, L.; Hermonat, P.; Mazzola, P.; Nguyen, D.D.; Radhi, S.; Figueroa, J.A.; Cobos, E.; Annoni, G.; et al. Mast cells and the liver aging process. Immun. Ageing 2013, 10, 9. [Google Scholar] [CrossRef]
- Tolefree, J.A.; Garcia, A.J.; Farrell, J.; Meadows, V.; Kennedy, L.; Hargrove, L.; Demieville, J.; Francis, N. Mirabel, Francis. Alcoholic liver disease and mast cells: What’s your gut got to do with it? Liver Res. 2019, 3, 46–54. [Google Scholar] [CrossRef]
- Finn, D.F.; Walsh, J.J. Twenty-first century mast cell stabilizers. Br. J. Pharmacol. 2013, 170, 23–37. [Google Scholar] [CrossRef]
- Bernstein, I.L.; Siegel, S.C.; Brandon, M.L.; Brown, E.B.; Evans, R.R.; Feinberg, A.R.; Friedlaender, S.; Krumholz, R.A.; Hadley, R.A.; Handelman, N.I.; et al. A controlled study of cromolyn sodium sponsored by the Drug Committee of the American Academy of Allergy. J. Allergy Clin. Immunol. 1972, 50, 235–245. [Google Scholar] [CrossRef]
- Kusner, E.J.; Dubnick, B.; Herzig, D.J. The inhibition by disodium cromoglycate in vitro of anaphylactically induced histamine release from rat peritoneal mast cells. J. Pharmacol. Exp. Ther. 1973, 184, 41–46. [Google Scholar]
- Oka, T.; Kalesnikoff, J.; Starkl, P.; Tsai, M.; Galli, S.J. Evidence questioning cromolyn’s effectiveness and selectivity as a ‘mast cell stabilizer’ in mice. Lab. Investig. 2012, 92, 1472–1482. [Google Scholar] [CrossRef]
- Mori, H.; Kawada, K.; Zhang, P.; Uesugi, Y.; Sakamoto, O.; Koda, A. Bleomycin-induced pulmonary fibrosis in genetically mast cell-deficient WBB6F1-W/Wv mice and mechanism of the suppressive effect of tranilast, an antiallergic drug inhibiting mediator release from mast cells, on fibrosis. Int. Arch. Allergy Appl. Immunol. 1991, 95, 195–201. [Google Scholar] [CrossRef]
- Yan, Y.; Jun, C.; Lu, Y.; Jiangmei, S. Combination of metformin and luteolin synergistically protects carbon tetrachloride-induced hepatotoxicity: Mechanism involves antioxidant, anti-inflammatory, antiapoptotic, and Nrf2/HO-1 signaling pathway. Biofactors 2019, 45, 598–606. [Google Scholar] [CrossRef]
- Abu-Elsaad, N.; El-Karef, A. Protection against nonalcoholic steatohepatitis through targeting IL-18 and IL-1alpha by luteolin. Pharm. Rep. 2019, 71, 688–694. [Google Scholar] [CrossRef]
- He, Y.; Xia, Z.; Yu, D.; Wang, J.; Jin, L.; Huang, D.; Ye, X.; Li, X.; Zhang, B. Hepatoprotective effects and structure-activity relationship of five flavonoids against lipopolysaccharide/d-galactosamine induced acute liver failure in mice. Int. Immunopharmacol. 2019, 68, 171–178. [Google Scholar] [CrossRef]
- Adewale, O.O.; Samuel, E.S.; Manubolu, M.; Pathakoti, K. Curcumin protects sodium nitrite-induced hepatotoxicity in Wistar rats. Toxicol. Rep. 2019, 6, 1006–1011. [Google Scholar] [CrossRef]
- Al-Dossari, M.H.; Fadda, L.M.; Attia, H.A.; Hasan, I.H.; Mahmoud, A.M. Curcumin and selenium prevent lipopolysaccharide/diclofenac-induced liver injury by suppressing inflammation and oxidative stress. Biol. Trace Elem. Res. 2019. [Google Scholar] [CrossRef]
- Pereira, P.J.; Bergner, A.; Macedo-Ribeiro, S.; Huber, R.; Matschiner, G.; Fritz, H.; Sommerhoff, C.P.; Bode, W. Human β-tryptase is a ring-like tetramer with active sites facing a central pore. Nature 1998, 392, 306–311. [Google Scholar] [CrossRef]
- Sommerhoff, C.P.; Bode, W.; Pereira, P.J.; Stubbs, M.T.; Sturzebecher, J.; Piechottka, G.P.; Matschiner, G.; Bergner, A. The structure of the human βII-tryptase tetramer: Fo(u)r better or worse. Proc. Natl. Acad. Sci. USA 1999, 96, 10984–10991. [Google Scholar] [CrossRef]
- Sommerhoff, C.P.; Schaschke, N. Mast cell tryptase β as a target in allergic inflammation: An evolving story. Curr. Pharm. Des. 2007, 13, 313–332. [Google Scholar] [CrossRef]
- Le, Q.T.; Lyons, J.J.; Naranjo, A.N.; Olivera, A.; Lazarus, R.A.; Metcalfe, D.D.; Milner, J.D.; Schwartz, L.B. Impact of naturally forming human α/β-tryptase heterotetramers in the pathogenesis of hereditary α-tryptasemia. J. Exp. Med. 2019, 216, 2348–2361. [Google Scholar] [CrossRef]
- Steinhoff, M.; Buddenkotte, J.; Shpacovitch, V.; Rattenholl, A.; Moormann, C.; Vergnolle, N.; Luger, T.A.; Hollenberg, M.D. Proteinase-activated receptors: Transducers of proteinase-mediated signaling in inflammation and immune response. Endocr. Rev. 2005, 26, 1–43. [Google Scholar] [CrossRef]
- Covic, L.; Gresser, A.L.; Talavera, J.; Swift, S.; Kuliopulos, A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc. Natl. Acad. Sci. USA 2002, 99, 643–648. [Google Scholar] [CrossRef]
- Shearer, A.M.; Rana, R.; Austin, K.; Baleja, J.D.; Nguyen, N.; Bohm, A.; Covic, L.; Kuliopulos, A. Targeting liver fibrosis with a cell-penetrating protease-activated receptor-2 (PAR2) pepducin. J. Biol. Chem. 2016, 291, 23188–23198. [Google Scholar] [CrossRef]
- Humphries, D.E.; Wong, G.W.; Friend, D.S.; Gurish, M.F.; Qiu, W.T.; Huang, C.; Sharpe, A.H.; Stevens, R.L. Heparin is essential for the storage of specific granule proteases in mast cells. Nature 1999, 400, 769–772. [Google Scholar] [CrossRef]
- Fang, K.C.; Raymond, W.W.; Lazarus, S.C.; Caughey, G.H. Dog mastocytoma cells secrete a 92-kD gelatinase activated extracellularly by mast cell chymase. J. Clin. Investig. 1996, 97, 1589–1596. [Google Scholar] [CrossRef]
- Lindstedt, K.A.; Wang, Y.; Shiota, N.; Saarinen, J.; Hyytiainen, M.; Kokkonen, J.O.; Keski-Oja, J.; Kovanen, P.T. Activation of paracrine TGF-β1 signaling upon stimulation and degranulation of rat serosal mast cells: A novel function for chymase. FASEB J. 2001, 15, 1377–1388. [Google Scholar] [CrossRef]
- Masubuchi, S.; Takai, S.; Jin, D.; Tashiro, K.; Komeda, K.; Li, Z.L.; Otsuki, Y.; Okamura, H.; Hayashi, M.; Uchiyama, K. Chymase inhibitor ameliorates hepatic steatosis and fibrosis on established non-alcoholic steatohepatitis in hamsters fed a methionine- and choline-deficient diet. Hepatol. Res. 2013, 43, 970–978. [Google Scholar] [CrossRef]
- Tashiro, K.; Takai, S.; Jin, D.; Yamamoto, H.; Komeda, K.; Hayashi, M.; Tanaka, K.; Tanigawa, N.; Miyazaki, M. Chymase inhibitor prevents the nonalcoholic steatohepatitis in hamsters fed a methionine- and choline-deficient diet. Hepatol. Res. 2010, 40, 514–523. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Jin, D.; Tashiro, K.; Komeda, K.; Masubuchi, S.; Hirokawa, F.; Hayashi, M.; Takai, S.; Uchiyama, K. Chymase inhibitor prevents the development and progression of non-alcoholic steatohepatitis in rats fed a high-fat and high-cholesterol diet. J. Pharmacol. Sci. 2017, 134, 139–146. [Google Scholar] [CrossRef]
- Komeda, K.; Takai, S.; Jin, D.; Tashiro, K.; Hayashi, M.; Tanigawa, N.; Miyazaki, M. Chymase inhibition attenuates tetrachloride-induced liver fibrosis in hamsters. Hepatol. Res. 2010, 40, 832–840. [Google Scholar] [CrossRef]
- Shimizu, S.; Satomura, K.; Aramaki, T.; Katsuta, Y.; Takano, T.; Omoto, Y. Hepatic chymase level in chronic hepatitis: Co-localization of chymase with fibrosis. Hepatol. Res. 2003, 27, 62–66. [Google Scholar] [CrossRef]
- Alegre, F.; Pelegrin, P.; Feldstein, A.E. Inflammasomes in Liver Fibrosis. Semin. Liver Dis. 2017, 37, 119–127. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Mizutani, H.; Schechter, N.; Lazarus, G.; Black, R.A.; Kupper, T.S. Rapid and specific conversion of precursor interleukin 1β (IL-1β) to an active IL-1 species by human mast cell chymase. J. Exp. Med. 1991, 174, 821–825. [Google Scholar] [CrossRef]
- Roy, A.; Ganesh, G.; Sippola, H.; Bolin, S.; Sawesi, O.; Dagalv, A.; Schlenner, S.M.; Feyerabend, T.; Rodewald, H.R.; Kjellén, L.; et al. Mast cell chymase degrades the alarmins heat shock protein 70, biglycan, HMGB1, and interleukin-33 (IL-33) and limits danger-induced inflammation. J. Biol. Chem. 2014, 289, 237–250. [Google Scholar] [CrossRef]
- Marvie, P.; Lisbonne, M.; L’Helgoualc’h, A.; Rauch, M.; Turlin, B.; Preisser, L.; Bourd-Boittin, K.; Theret, N.; Gascan, H.; Piquet-Pellorce, C.; et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J. Cell Mol. Med. 2010, 14, 1726–1739. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, Y.; Yang, M.; Guo, X.; Zhang, M.; Li, H.; Li, J.; Zhao, J. IL-33 treatment attenuated diet-induced hepatic steatosis but aggravated hepatic fibrosis. Oncotarget 2016, 7, 33649–33661. [Google Scholar] [CrossRef]
- McHedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013, 39, 357–371. [Google Scholar] [CrossRef]
- Tan, Z.; Liu, Q.; Jiang, R.; Lv, L.; Shoto, S.S.; Maillet, I.; Quesniaux, V.; Tang, J.; Zhang, W.; Sun, B.; et al. Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell. Mol. Immunol. 2018, 15, 388–398. [Google Scholar] [CrossRef]
- Arriazu, E.; Ge, X.; Leung, T.M.; Magdaleno, F.; Lopategi, A.; Lu, Y.; Kitamura, N.; Urtasun, R.; Theise, N.; Antoine, D.J.; et al. Signalling via the osteopontin and high mobility group box-1 axis drives the fibrogenic response to liver injury. Gut 2017, 66, 1123–1137. [Google Scholar] [CrossRef]
- Ge, X.; Arriazu, E.; Magdaleno, F.; Antoine, D.J.; Dela Cruz, R.; Theise, N.; Nieto, N. High mobility group box-1 drives fibrosis progression signaling via the receptor for advanced glycation end products in mice. Hepatology 2018, 68, 2380–2404. [Google Scholar] [CrossRef]
- Zhou, K.; Xie, G.; Wen, J.; Wang, J.; Pan, W.; Zhou, Y.; Xiao, Y.; Wang, Y.; Jia, W.; Cai, W. Histamine is correlated with liver fibrosis in biliary atresia. Dig. Liver Dis. 2016, 48, 921–926. [Google Scholar] [CrossRef]
- Branco, A.C.C.C.; Yoshikawa, F.S.Y.; Pietrobon, A.J.; Sato, M.N. Role of histamine in modulating the immune response and inflammation. Mediat. Inflamm. 2018, 2018, 9524075. [Google Scholar] [CrossRef]
- Popov, Y.; Patsenker, E.; Fickert, P.; Trauner, M.; Schuppan, D. Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J. Hepatol. 2005, 43, 1045–1054. [Google Scholar] [CrossRef]
- Jordana, M.; Befus, A.D.; Newhouse, M.T.; Bienenstock, J.; Gauldie, J. Effect of histamine on proliferation of normal human adult lung fibroblasts. Thorax 1988, 43, 552–558. [Google Scholar] [CrossRef]
- Gailit, J.; Marchese, M.J.; Kew, R.R.; Gruber, B.L. The differentiation and function of myofibroblasts is regulated by mast cell mediators. J. Investig. Dermatol. 2001, 117, 1113–1119. [Google Scholar] [CrossRef]
- Garbuzenko, E.; Nagler, A.; Pickholtz, D.; Gillery, P.; Reich, R.; Maquart, F.X.; Levi-Schaffer, F. Human mast cells stimulate fibroblast proliferation, collagen synthesis and lattice contraction: A direct role for mast cells in skin fibrosis. Clin. Exp. Allergy 2002, 32, 237–246. [Google Scholar] [CrossRef]
- Lin, L.; Yamagata, K.; Nakayamada, S.; Sawamukai, N.; Yamaoka, K.; Sakata, K.; Nakano, K.; Tanaka, Y. Histamine inhibits differentiation of skin fibroblasts into myofibroblasts. Biochem. Biophys. Res. Commun. 2015, 463, 434–439. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Procedure and Typical Time of Sacrifice | Outcome | Model for Human Liver Disease | Reference |
|---|---|---|---|---|
| CCl4 | Single or repeated i.p. application (1 h–6 wks), regular inhalation or application by gavage (11–15 wks) | Early: inflammation; late: centrilobular liver damage, fibrosis/cirrhosis induced by forming radicals | Intoxication, acute liver damage, fibrosis | [75] |
| LPS LPS/D-GalN LPS/BCG | I.p. injection or application of LPS in drinking water alone or in combination with D-GalN; i.p. injection of LPS/BCG (2–8 h) | Inflammation (LPS), acute hepatic failure (LPS/D-GalN), lethal hepatitis (LPS/BCG) | Acute systemic and hepatic inflammation; lethal hepatitis | [79] |
| DEN DMN | I.p., oral (drinking water, diet, gavage), inhalation, intratracheal, or intragastric instillation (6–50 wks) | Time-dependent liver damage (neutrophilic infiltration, extensive centrilobular hemorrhagic necrosis, bile duct proliferation, fibrosis, and bridging necrosis ending in hepatocarcinogenesis) | Early: intoxication, late: fibrosis, cirrhosis, HCC | [81] |
| BDL | Surgical ligation of the common biliary duct (5 days–4 wks) | Early: liver cell injury, severe inflammation; late: advanced hepatic fibrosis | Early: liver injury and jaundice, late: cholestatic liver diseases | [82,83] |
| Mdr2−/− | Homozygous disruption of the multidrug resistance 2 gene | Significant increase of bilirubin, alkaline phosphatase, aspartate aminotransferase already at age of 6–14 wks | Cholestatic liver injury | [85] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiskirchen, R.; Meurer, S.K.; Liedtke, C.; Huber, M. Mast Cells in Liver Fibrogenesis. Cells 2019, 8, 1429. https://doi.org/10.3390/cells8111429
Weiskirchen R, Meurer SK, Liedtke C, Huber M. Mast Cells in Liver Fibrogenesis. Cells. 2019; 8(11):1429. https://doi.org/10.3390/cells8111429
Chicago/Turabian StyleWeiskirchen, Ralf, Steffen K. Meurer, Christian Liedtke, and Michael Huber. 2019. "Mast Cells in Liver Fibrogenesis" Cells 8, no. 11: 1429. https://doi.org/10.3390/cells8111429
APA StyleWeiskirchen, R., Meurer, S. K., Liedtke, C., & Huber, M. (2019). Mast Cells in Liver Fibrogenesis. Cells, 8(11), 1429. https://doi.org/10.3390/cells8111429