Involvement of the Chemokine Prokineticin-2 (PROK2) in Alzheimer’s Disease: From Animal Models to the Human Pathology


 ,
,  ,
,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. Surgery and Aβ1–42 Administration
2.4. RNA Extraction and Real-Time PCR
2.5. Immunofluorescence
2.6. In Vivo Blood Assay
2.7. ELISA
2.8. Data Analysis
3. Results
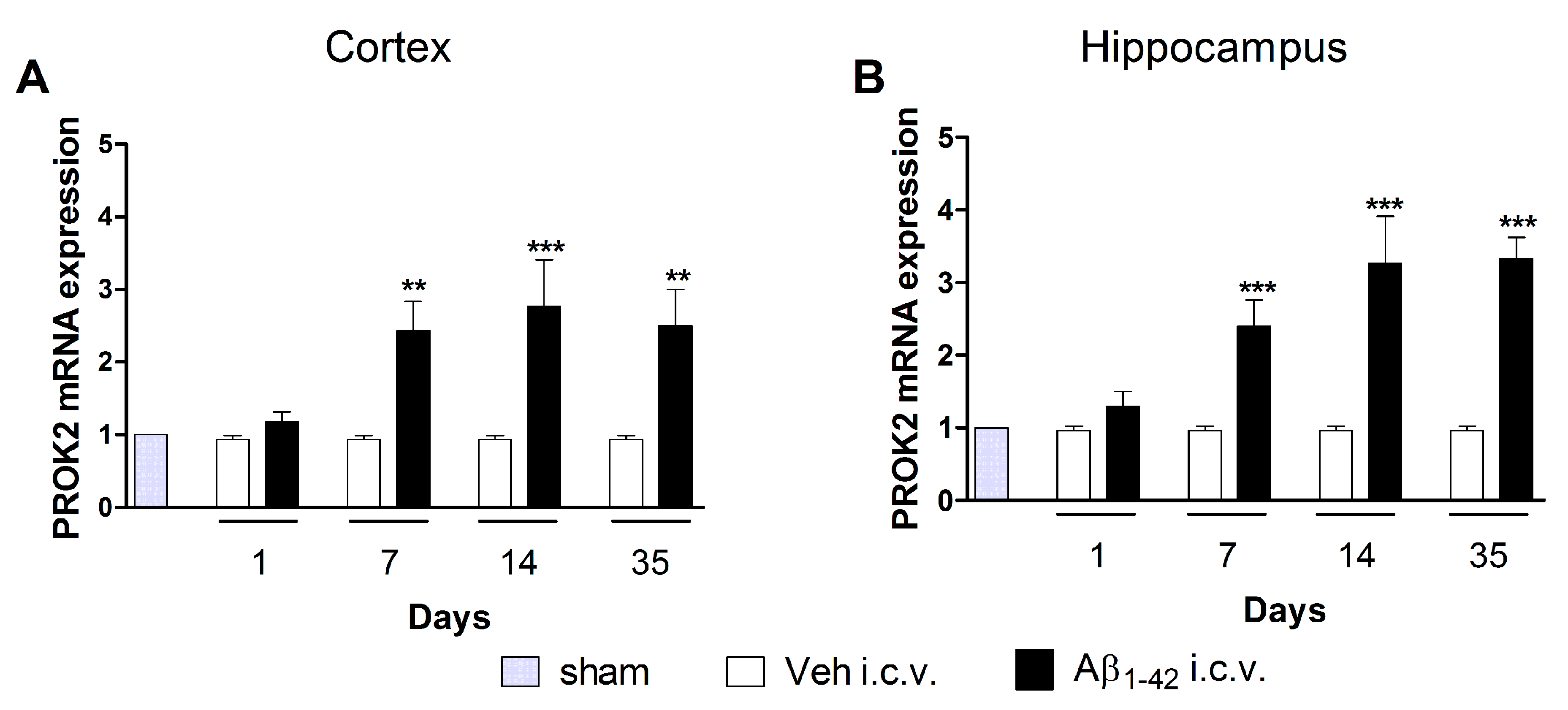
3.1. Upregulation of PROK2 mRNA in Rat Brain Following Aβ Insult
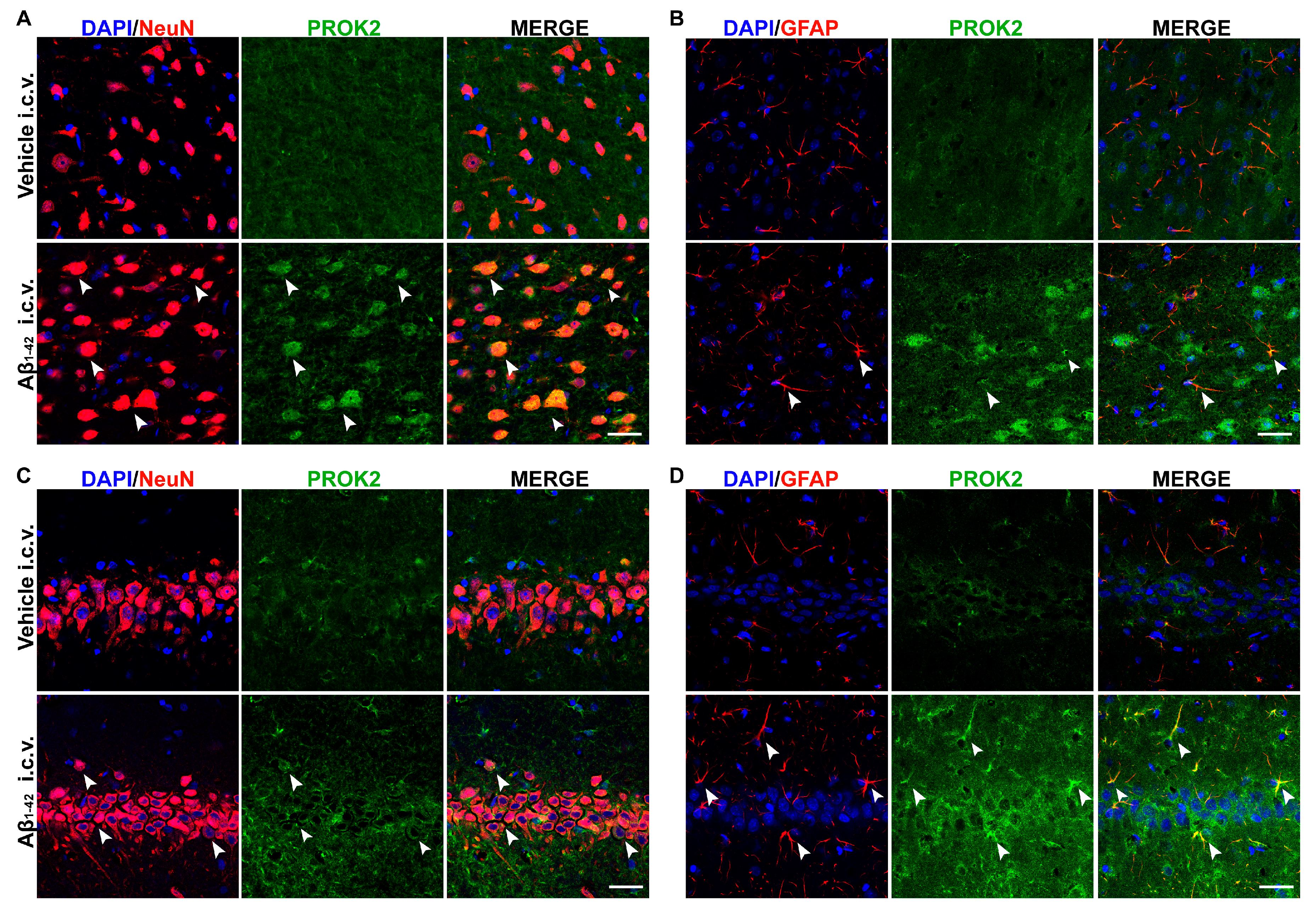
3.2. Localization of PROK2 Protein in Rat Brain Following Aβ Insult
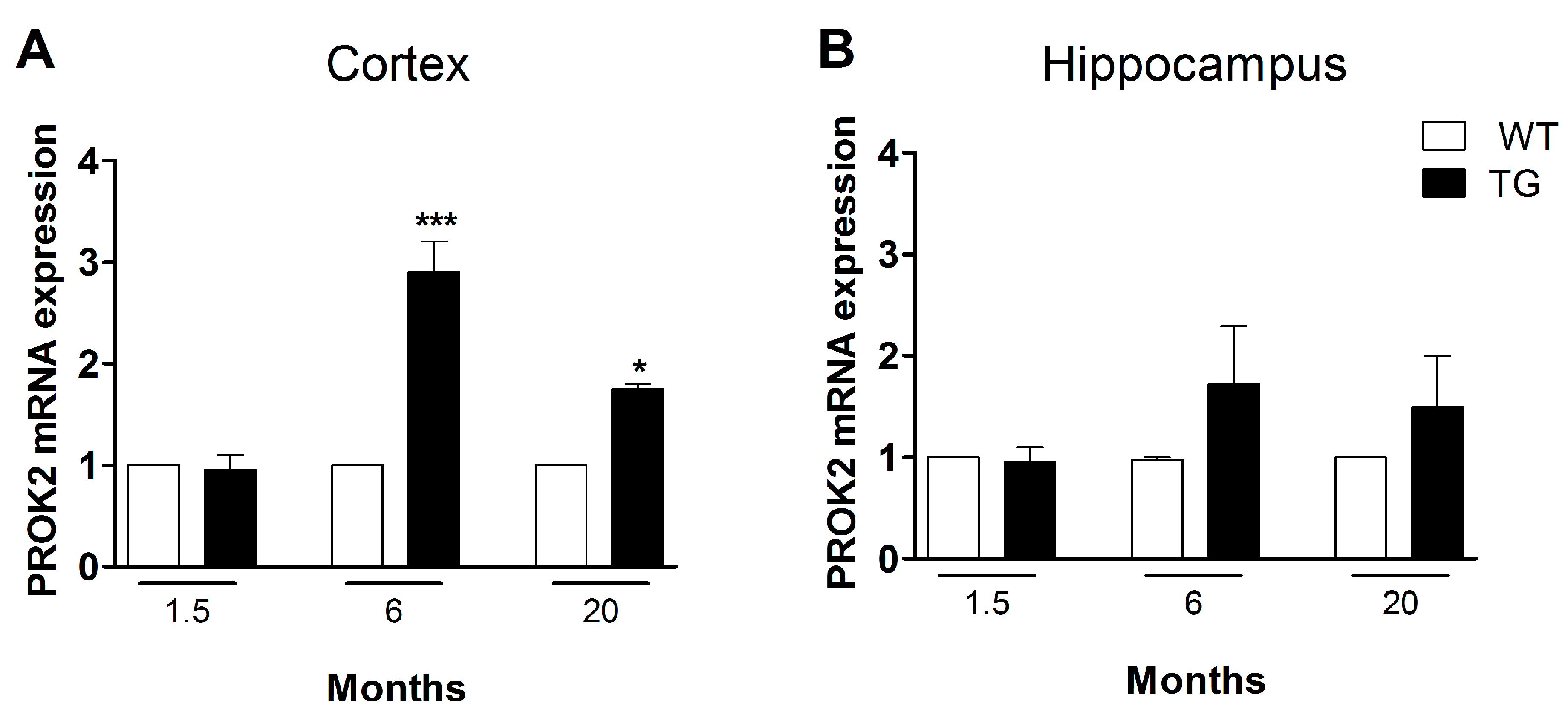
3.3. Upregulation of PROK2 in Tg2576 Brain
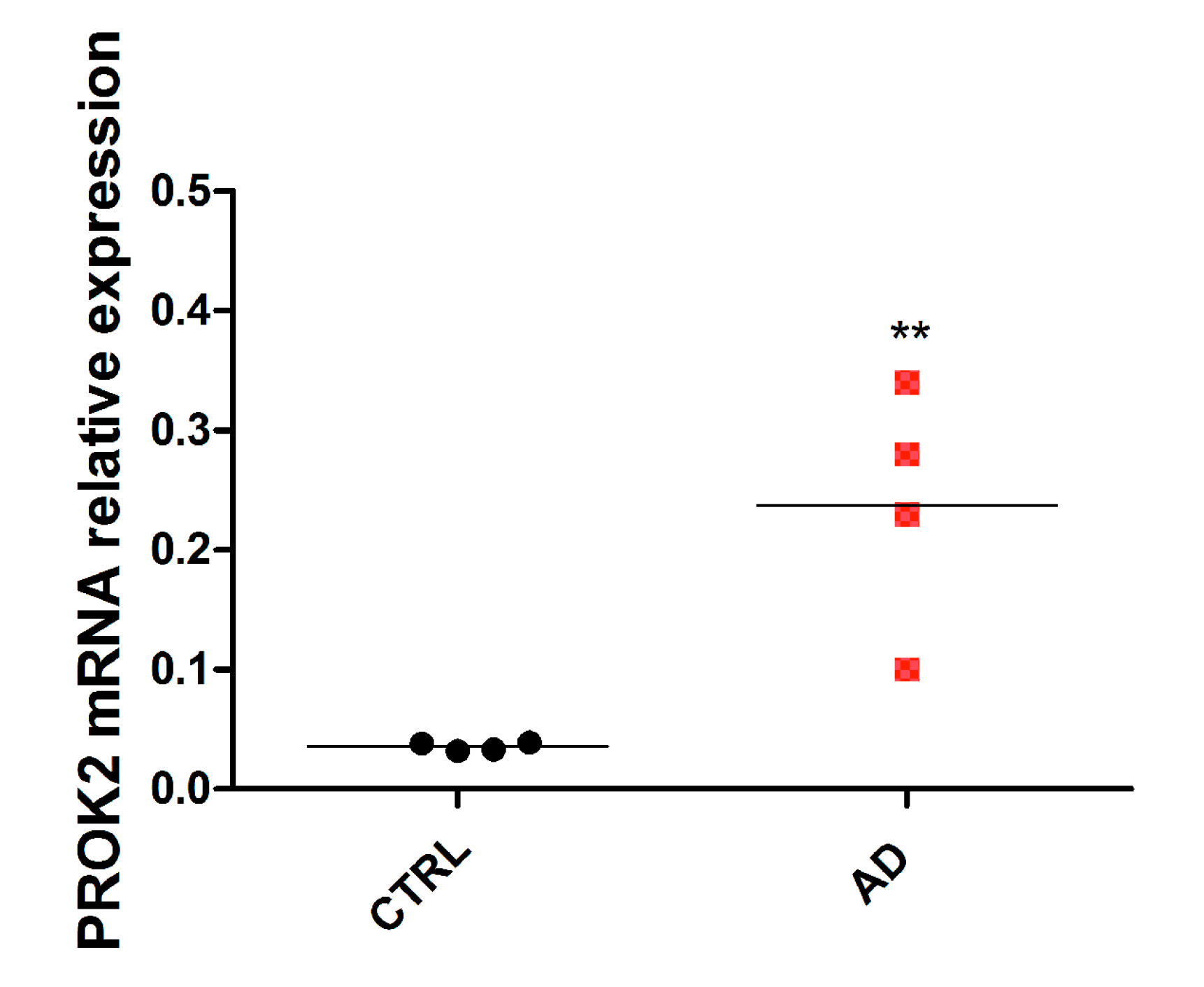
3.4. Upregulation of PROK2 in AD Patients’ Brain
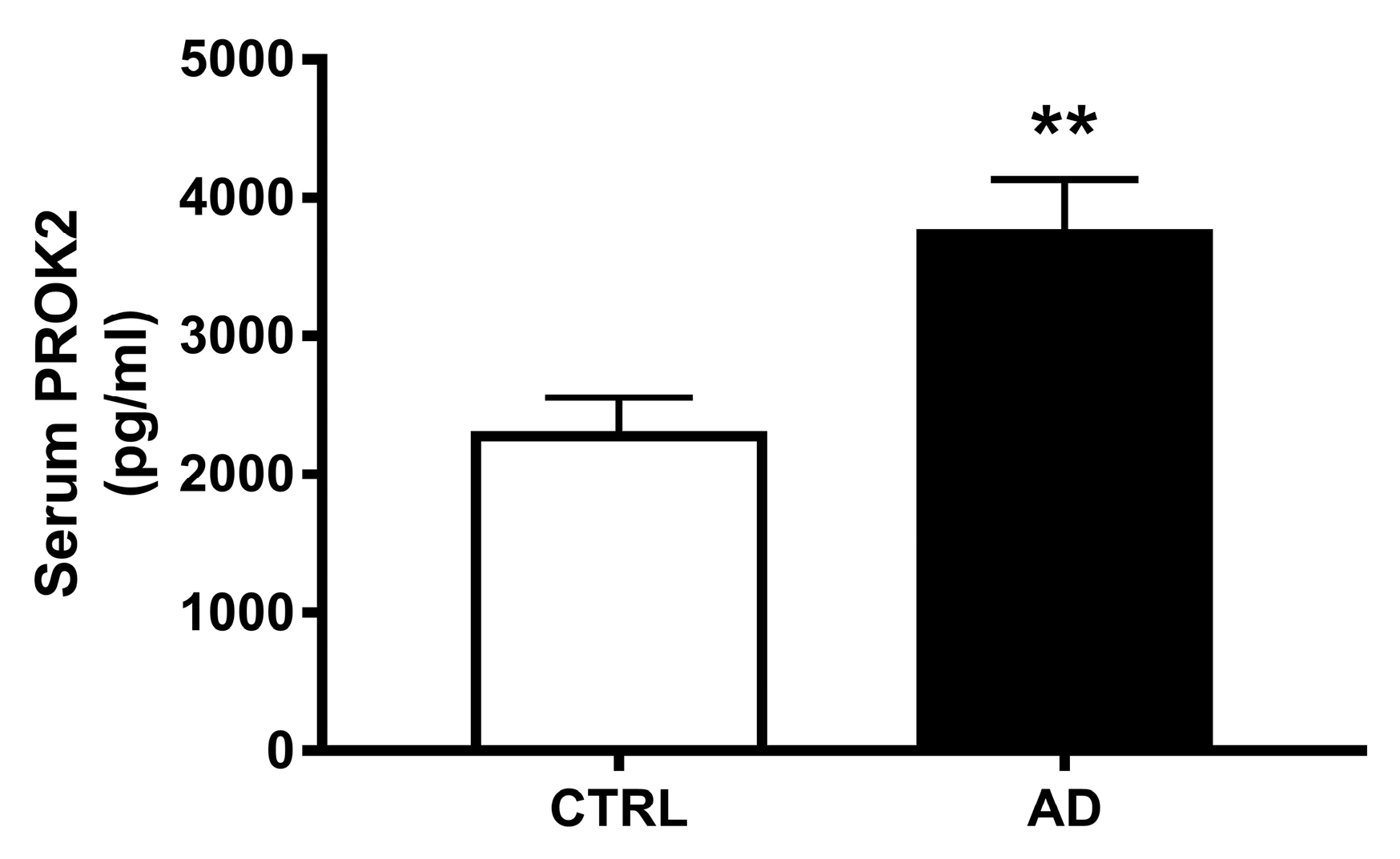
3.5. Increase of PROK2 in AD Patients’ Serum
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.F.; Kodadek, T. The immune system and neuroinflammation as potential sources of blood-based biomarkers for Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. ACS Chem. Neurosci. 2016, 75, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Dá Mesquita, S.; Ferreira, A.C.; Sousa, J.C.; Correia-Neves, M.; Sousa, N.; Marques, F. Insights on the pathophysiology of Alzheimer’s disease: The crosstalk between amyloid pathology, neuroinflammation and the peripheral immune system. Neurosci. Biobehav. Rev. 2016, 68, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Devel. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Darweesh, S.K.L.; Wolters, F.J.; Ikram, M.A.; de Wolf, F.; Bos, D.; Hofman, A. Inflammatory markers and the risk of dementia and Alzheimer’s disease: A meta-analysis. Alzheimers Dement. 2018, 14, 1450–1459. [Google Scholar] [CrossRef]
- Shen, X.N.; Niu, L.D.; Wang, Y.J.; Cao, X.P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.T. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Azizi, G.; Khannazer, N.; Mirshafiey, A. The potential role of chemokines in Alzheimer’s disease pathogenesis. Am. J. Alzheimers Dis. Other Demen. 2014, 29, 415–425. [Google Scholar] [CrossRef]
- Liu, C.; Cui, G.; Zhu, M.; Kang, X.; Guo, H. Neuroinflammation in Alzheimer’s disease: Chemokines produced by astrocytes and chemokine receptors. Int. J. Clin. Exp. Pathol. 2014, 7, 8342–8355. [Google Scholar]
- Domingues, C.; da Cruz, E.; Silva, O.A.B.; Henriques, A.G. Impact of cytokines and chemokines on Alzheimer’s disease neuropathological hallmarks. Curr. Alzheimer Res. 2017, 14, 870–882. [Google Scholar] [CrossRef]
- Le Thuc, O.; Blondeau, N.; Nahon, J.L.; Rovere, C. The complex contribution of chemokines to neuroinflammation: Switching from beneficial to detrimental effects. Ann. N. Y. Acad. Sci. 2015, 1351, 127–140. [Google Scholar] [CrossRef]
- Luster, A.D. Chemokines-chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 1998, 338, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Lim, H.K.; Lee, J.Y.; Kim, D.J.; Park, S.; Lee, C.; Lee, C.U. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2008, 436, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Strobel, S.; Grünblatt, E.; Riederer, P.; Heinsen, H.; Arzberger, T.; Al-Sarraj, S.; Troakes, C.; Ferrer, I.; Monoranu, C.M. Changes in the expression of genes related to neuroinflammation over the course of sporadic Alzheimer’s disease progression: CX3CL1, TREM2, and PPARγ. J. Neural. Transm. 2015, 122, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Iarlori, C.; Feliciani, C.; Gambi, D. Peripheral chemokine receptors, their ligands, cytokines and Alzheimer’s disease. J. Alzheimers Dis. 2008, 14, 147–159. [Google Scholar] [CrossRef]
- Bulati, M.; Buffa, S.; Martorana, A.; Gervasi, F.; Camarda, C.; Azzarello, D.M.; Monastero, R.; Caruso, C.; Colonna-Romano, G. Double negative (IgG+IgD-CD27-) B cells are increased in a cohort of moderate-severe Alzheimer’s disease patients and show a pro-inflammatory trafficking receptor phenotype. J. Alzheimers Dis. 2015, 44, 1241–1251. [Google Scholar] [CrossRef]
- Morgan, A.R.; Touchard, S.; Leckey, C.; O’Hagan, C.; Nevado-Holgado, A.J.; Barkhof, F.; Bertram, L.; Blin, O.; Bos, I.; Dobricic, V.; et al. Annex: NIMA–wellcome trust consortium for neuroimmunology of mood disorders and Alzheimer’s disease. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimers Dement. 2019, 15, 776–787. [Google Scholar] [CrossRef]
- Negri, L.; Lattanzi, R.; Giannini, E.; Melchiorri, P. Bv8/Prokineticin proteins and their receptors. Life Sci. 2007, 81, 1103–1116. [Google Scholar] [CrossRef]
- Negri, L.; Ferrara, N. The prokineticins: Neuromodulators and mediators of inflammation and myeloid cell-dependent angiogenesis. Physiol. Rev. 2018, 98, 1055–1082. [Google Scholar] [CrossRef]
- Cheng, M.Y.; Lee, A.G.; Culbertson, C.; Sun, G.; Talati, R.K.; Manley, N.C.; Li, X.; Zhao, H.; Lyons, D.M.; Zhou, Q.Y.; et al. Prokineticin 2 is an endangering mediator of cerebral ischemic injury. Proc. Natl. Acad. Sci. USA 2012, 109, 5475–5480. [Google Scholar] [CrossRef]
- Landucci, E.; Lattanzi, R.; Gerace, E.; Scartabelli, T.; Balboni, G.; Negri, L.; Pellegrini-Giampietro, D.E. Prokineticins are neuroprotective in models of cerebral ischemia and ischemic tolerance in vitro. Neuropharmacology 2016, 108, 39–48. [Google Scholar] [CrossRef]
- Severini, C.; Lattanzi, R.; Maftei, D.; Marconi, M.; Ciotti, M.T.; Petrocchi Passeri, P.; Florenzano, F.; Del Duca, E.; Caioli, S.; Zona, C.; et al. Bv8/prokineticin 2 is involved in Aβ-induced neurotoxicity. Sci. Rep. 2015, 5, 15301. [Google Scholar] [CrossRef] [PubMed]
- Caioli, S.; Severini, C.; Ciotti, T.; Florenzano, F.; Pimpinella, D.; Petrocchi Passeri, P.; Balboni, G.; Polisca, P.; Lattanzi, R.; Nisticò, R.; et al. Prokineticin system modulation as a new target to counteract the amyloid beta toxicity induced by glutamatergic alterations in an in vitro model of Alzheimer’s disease. Neuropharmacology 2017, 116, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Maftei, D.; Ratano, P.; Fusco, I.; Marconi, V.; Squillace, S.; Negri, L.; Severini, C.; Balboni, G.; Steardo, L.; Bronzuoli, M.R.; et al. The prokineticin receptor antagonist PC1 rescues memory impairment induced by β amyloid administration through the modulation of prokineticin system. Neuropharmacology 2019, 158, 107739. [Google Scholar] [CrossRef] [PubMed]
- Frozza, R.L.; Bernardi, A.; Hoppe, J.B.; Meneghetti, A.B.; Battastini, A.M.; Pohlmann, A.R.; Guterres, S.S.; Salbego, C. Lipid-core nanocapsules improve the effects of resveratrol against Abeta-induced neuroinflammation. J. Biomed. Nanotechnol. 2013, 9, 2086–2104. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 4th ed.; Academic Press, Inc.: San Diego, CA, USA, 1998. [Google Scholar]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Schirinzi, T.; Di Lorenzo, F.; Sancesario, G.M.; Di Lazzaro, G.; Ponzo, V.; Pisani, A.; Mercuri, N.B.; Koch, G.; Martorana, A. Amyloid-mediated cholinergic dysfunction in motor impairment related to Alzheimer’s disease. J. Alzheimers. Dis. 2018, 64, 525–532. [Google Scholar] [CrossRef]
- Rogers, J.; Strohmeyer, R.; Kovelowski, C.J.; Li, R. Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia 2002, 40, 260–269. [Google Scholar] [CrossRef]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef] [PubMed]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in Mild Cognitive Impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Zuena, A.R.; Casolini, P.; Lattanzi, R.; Maftei, D. Chemokines in Alzheimer’s disease: New insights into prokineticins, chemokine-like proteins. Front. Pharmacol. 2019, 10, 622. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, M.; Gearing, A.J.; Miller, K.M. A central role for astrocytes in the inflammatory response to beta-amyloid chemokines, cytokines and reactive oxygen species are produced. J. Neuroimmunol. 1999, 93, 182–193. [Google Scholar] [CrossRef]
- Smits, H.A.; Rijsmus, A.; van Loon, J.H.; Wat, J.W.; Verhoef, J.; Boven, L.A.; Nottet, H.S. Amyloid-beta-induced chemokine production in primary human macrophages and astrocytes. J. Neuroimmunol. 2002, 127, 160–168. [Google Scholar] [CrossRef]
- Severini, C.; Passeri, P.P.; Ciotti, M.; Florenzano, F.; Possenti, R.; Zona, C.; Di Matteo, A.; Guglielmotti, A.; Calissano, P.; Pachter, J.; et al. Bindarit, inhibitor of CCL2 synthesis, protects neurons against Amyloid-β -induced toxicity. J. Alzheimers Dis. 2013, 38, 281–293. [Google Scholar] [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Meijer, J.; Kiliaan, A.; Ruitenberg, A.; van Swieten, J.C.; Stijnen, T.; Hofman, A.; Witteman, J.C.M.; Breteler, M.M.B. Inflammatory proteins in plasma and the risk of dementia: The Rotterdam study. Arch. Neurol. 2004, 61, 668–672. [Google Scholar] [CrossRef]
- Motta, M.; Imbesi, R.; Di Rosa, M.; Stivala, F.; Malaguarnera, L. Altered plasma cytokine levels in Alzheimer’s disease: Correlation with the disease progression. Immunol. Lett. 2007, 114, 46–51. [Google Scholar] [CrossRef]
- Galimberti, D.; Fenoglio, C.; Lovati, C.; Venturelli, E.; Guidi, I.; Corrà, B.; Scalabrini, D.; Clerici, F.; Mariani, C.; Bresolin, N.; et al. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1763–1768. [Google Scholar] [CrossRef]
- Galimberti, D.; Schoonenboom., N.; Scheltens, P.; Fenoglio, C.; Bouwman, F.; Venturelli, E.; Guidi, I.; Blankenstein, M.A.; Bresolin, N.; Scarpini, E. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch. Neurol. 2006, 63, 538–543. [Google Scholar] [CrossRef]
- Westin, K.; Buchhave, P.; Nielsen, H.; Minthon, L.; Janciauskiene, S.; Hansson, O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS ONE 2012, 7, e30525. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Yoshikura, N.; Hayashi, Y.; Inuzuka, T. Cerebrospinal fluid C-C motif chemokine ligand 2 correlates with brain atrophy and cognitive impairment in Alzheimer’s disease. J. Alzheimers Dis. 2018, 61, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Alsadany, M.A.; Shehata, H.H.; Mohamad, M.I.; Mahfouz, R.G. Histone deacetylases enzyme, copper, and IL-8 levels in patients with Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen. 2013, 28, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bettcher, B.M.; Fitch, R.; Wynn, M.J.; Lalli, M.A.; Elofson, J.; Jastrzab, L.; Mitic, L.; Miller, Z.A.; Rabinovici, G.D.; Miller, B.L.; et al. MCP-1 and eotaxin-1 selectively and negatively associate with memory in MCI and Alzheimer’s disease dementia phenotypes. Alzheimers Dement. 2016, 3, 91–97. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Gene | Sequence |
|---|---|---|
| Rat | GAPDH | Fw: 5′-GCG AGA TCC CGC TAA CAT CAA ATG G-3′ |
| Rev: 5′-GCC ATC CAC AGT CTT CTG AGT GGC-3′ | ||
| PROK2 | Fw: 5’-TCA TCA CCG GGG CTT GCG-3’ | |
| Rev: 5’-TAA CTT TCC GAG TCA GGG-3’ | ||
| Mouse | GAPDH | Fw: 5′-GCC AAG GCT GTG GGC AAG GT-3′ |
| Rev: 5′-TCT CCA GGC GGC ACG TCA GA-3′ | ||
| PROK2 | Fw: 5′-CTC GGA AAG TTC CAT TTT GG-3′ | |
| Rev: 5′-TTC CGG GCC AAG CAA ATA AAC C-3′ | ||
| Human | β-actin | Fw: 5′-AGA TGA CCC AGA TCA TGT TTG-3′ |
| Rev: 5′-TAG ATG GGC ACA GTG TGG-3′ | ||
| PROK2 | Fw: 5′-ATG TGC TGT GCT GTC AGT AT-3′ | |
| Rev: 5′-AAA ATG GAA CTT TAC GAG TCA-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lattanzi, R.; Maftei, D.; Petrella, C.; Pieri, M.; Sancesario, G.; Schirinzi, T.; Bernardini, S.; Barbato, C.; Ralli, M.; Greco, A.; et al. Involvement of the Chemokine Prokineticin-2 (PROK2) in Alzheimer’s Disease: From Animal Models to the Human Pathology. Cells 2019, 8, 1430. https://doi.org/10.3390/cells8111430
Lattanzi R, Maftei D, Petrella C, Pieri M, Sancesario G, Schirinzi T, Bernardini S, Barbato C, Ralli M, Greco A, et al. Involvement of the Chemokine Prokineticin-2 (PROK2) in Alzheimer’s Disease: From Animal Models to the Human Pathology. Cells. 2019; 8(11):1430. https://doi.org/10.3390/cells8111430
Chicago/Turabian StyleLattanzi, Roberta, Daniela Maftei, Carla Petrella, Massimo Pieri, Giulia Sancesario, Tommaso Schirinzi, Sergio Bernardini, Christian Barbato, Massimo Ralli, Antonio Greco, and et al. 2019. "Involvement of the Chemokine Prokineticin-2 (PROK2) in Alzheimer’s Disease: From Animal Models to the Human Pathology" Cells 8, no. 11: 1430. https://doi.org/10.3390/cells8111430
APA StyleLattanzi, R., Maftei, D., Petrella, C., Pieri, M., Sancesario, G., Schirinzi, T., Bernardini, S., Barbato, C., Ralli, M., Greco, A., Possenti, R., Sancesario, G., & Severini, C. (2019). Involvement of the Chemokine Prokineticin-2 (PROK2) in Alzheimer’s Disease: From Animal Models to the Human Pathology. Cells, 8(11), 1430. https://doi.org/10.3390/cells8111430