ACVR1 Function in Health and Disease




Abstract
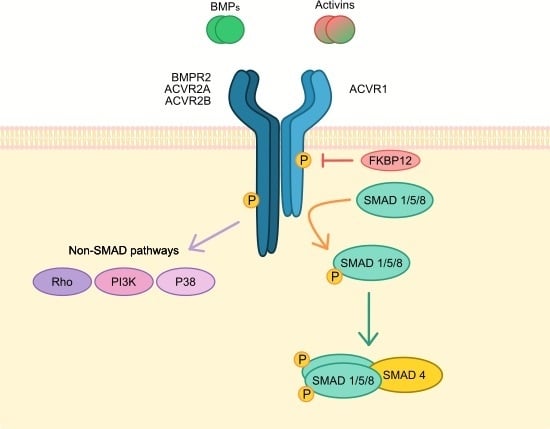
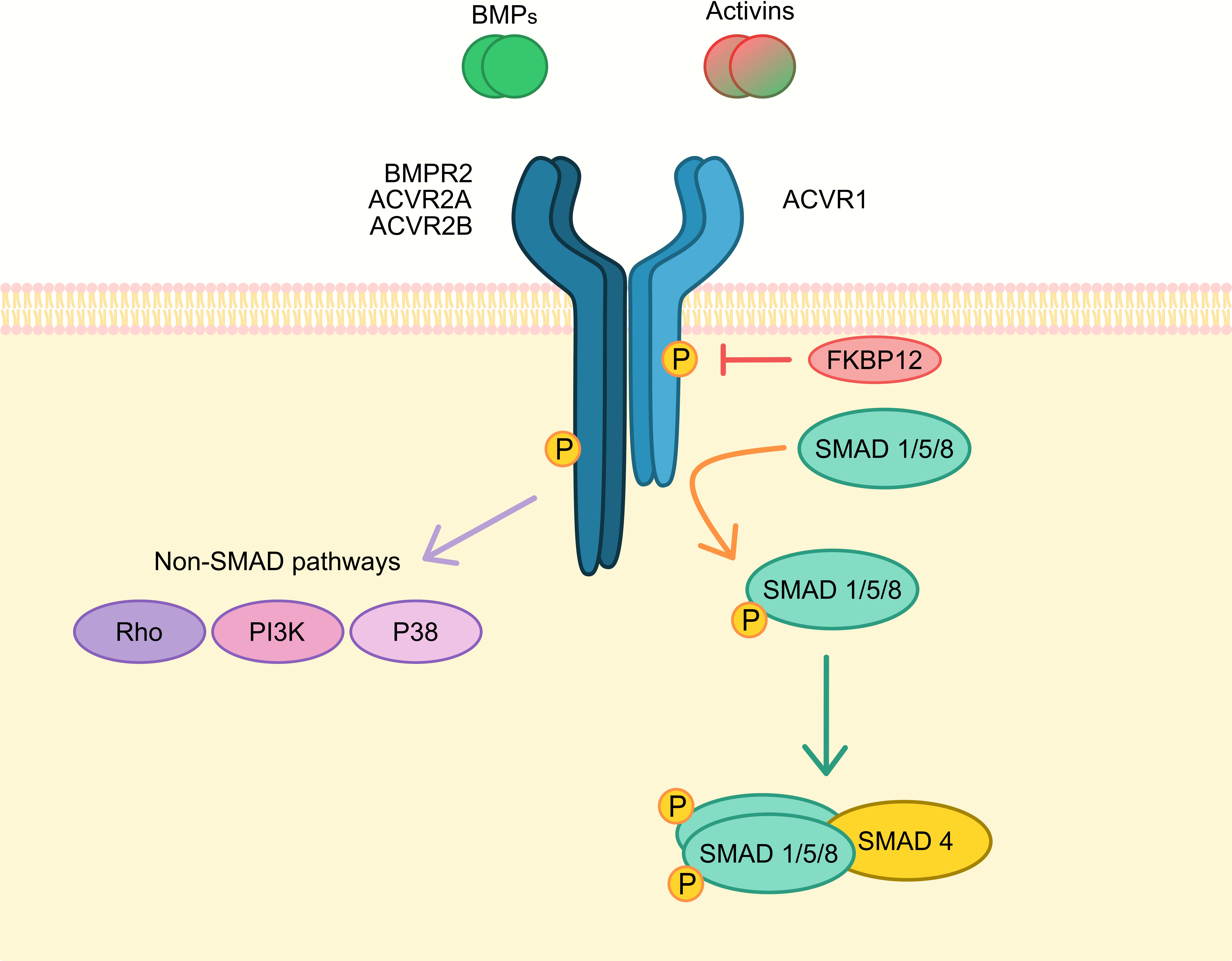
1. ACVR1 Gene and Signalling
2. Fibrodysplasia Ossificans Progressiva
2.1. FOP Clinical Information
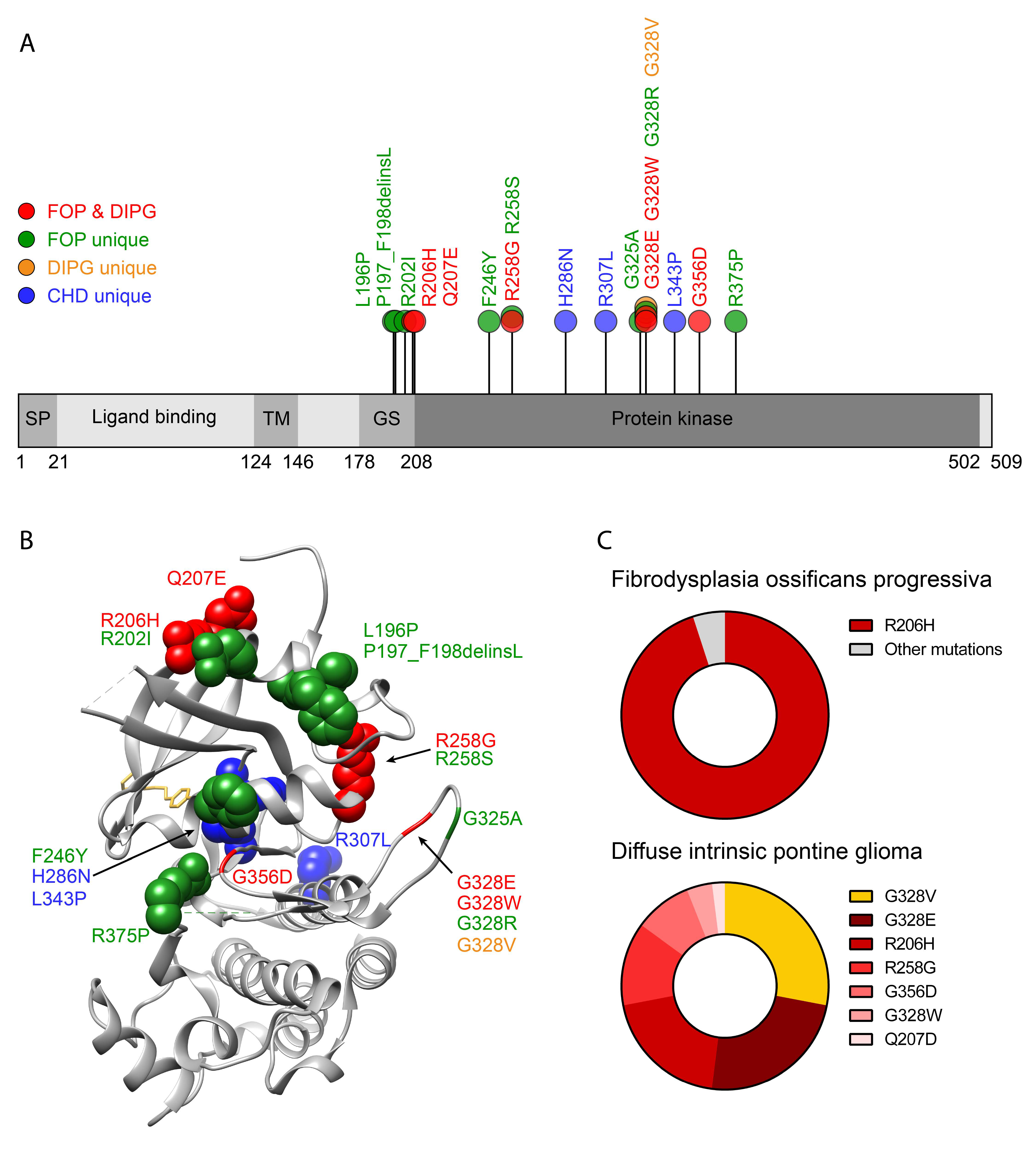
2.2. ACVR1 Genetic Mutations in FOP
2.3. ACVR1 Mutants and Altered BMP Signalling
2.4. Activin A Signalling via ACVR1 Gain of Function Mutants
2.5. ACVR1 Non-Canonical BMP Signalling
3. Bone Regulation and Skeletal Development
4. Cardiovascular Development and Cardiac Diseases
5. Reproductive System
6. ACVR1 in the Central Nervous System
7. BMP Signalling and Pluripotency
8. ACVR1 in Cancer
8.1. Diffuse Intrinsic Pontine Glioma
8.2. Adult Glioblastoma
8.3. Ovarian Cancer
8.4. Endometrial Cancer
8.5. Prostate Cancer
8.6. Multiple Myeloma
8.7. Eye Lens Tumour
8.8. Erythroleukemia
8.9. Head and Neck Squamous Cell Carcinomas
8.10. Dual Role of ACVR1 and BMPs in Cancer Biology
9. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Röijer, E.; Miyazono, K.; Åström, A.K.; Van Kessel, A.G.; Ten Dijke, P.; Stenman, G. Chromosomal localization of three human genes encoding members of the TGF-β superfamily of type I serine/threonine kinase receptors. Mamm. Genome 1998, 9, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Attisano, L.; Cárcamo, J.; Ventura, F.; Weis, F.M.B.; Massagué, J.; Wrana, J.L. Identification of human activin and TGFβ type I receptors that form heteromeric kinase complexes with type II receptors. Cell 1993, 75, 671–680. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Xu, J.; Wang, F.; McKeehan, W.L.; Krummen, L.; Kan, M. A widely expressed transmembrane serine/threonine kinase that does not bind activin, inhibin, transforming growth factor β, or bone morphogenic factor. J. Biol. Chem. 1993, 268, 12719–12723. [Google Scholar]
- Melé, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.G.; Kuriyan, J.; Massagué, J. The TGFβ receptor activation process: An inhibitor- to substrate-binding switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Huse, M.; Chen, Y.G.; Massagué, J.; Kuriyan, J. Crystal structure of the cytoplasmic domain of the type I TGFβ receptor in complex with FKBP12. Cell 1999, 96, 425–436. [Google Scholar] [CrossRef]
- Yadin, D.; Knaus, P.; Mueller, T.D. Structural insights into BMP receptors: Specificity, activation and inhibition. Cytokine Growth Factor Rev. 2016, 27, 13–34. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Cárcamo, J.; Zentella, A.; Doody, J.; Laiho, M.; Wang, X.F.; Massague, J. TGFβ signals through a heteromeric protein kinase receptor complex. Cell 1992, 71, 1003–1014. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Chen, Y.G.; Massagué, J. Smad1 recognition and activation by the ALK1 group of transforming growth factor-β family receptors. J. Biol. Chem. 1999, 274, 3672–3677. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad signaling pathways of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Kaplan, F.S. Inherited human diseases of heterotopic bone formation. Nat. Rev. Rheumatol. 2010, 6, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Chakkalakal, S.A.; Shore, E.M. Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis. Dis. Model. Mech. 2012, 5, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.M.; Evans, D.A. Genetic aspects of fibrodysplasia ossificans progressiva. J. Med. Genet. 1982, 19, 35–39. [Google Scholar] [CrossRef]
- Shore, E.M.; Feldman, G.J.; Xu, M.; Kaplan, F.S. The genetics of fibrodysplasia ossificans progressiva. Clin. Rev. Bone Miner. Metab. 2005, 3, 201–204. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Kobori, J.A.; Orellana, C.; Calvo, I.; Rosello, M.; Martinez, F.; Lopez, B.; Xu, M.; Pignolo, R.J.; Shore, E.M.; et al. Multi-system involvement in a severe variant of fibrodysplasia ossificans progressiva (ACVR1 c.772G>A; R258G): A report of two patients. Am. J. Med. Genet. Part A 2015, 167, 2265–2271. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Suda, R.K.; Kaplan, F.S. The fibrodysplasia ossificans progressiva lesion. Clin. Rev. Bone Miner. Metab. 2005, 3, 195–200. [Google Scholar] [CrossRef]
- Hüning, I.; Gillessen-Kaesbach, G. Fibrodysplasia ossificans progressiva: Clinical course, genetic mutations and genotype-phenotype correlation. Mol. Syndromol. 2014, 5, 201–211. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Bedford-Gay, C.; Liljesthröm, M.; Durbin-Johnson, B.P.; Shore, E.M.; Rocke, D.M.; Kaplan, F.S. The Natural History of Flare-Ups in Fibrodysplasia Ossificans Progressiva (FOP): A Comprehensive Global Assessment. J. Bone Miner. Res. 2016, 31, 650–656. [Google Scholar] [CrossRef]
- Pacifici, M.; Shore, E.M. Common mutations in ALK2/ACVR1, a multi-faceted receptor, have roles in distinct pediatric musculoskeletal and neural orphan disorders. Cytokine Growth Factor Rev. 2016, 27, 93–104. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Zasloff, M.A.; Kitterman, J.A.; Shore, E.M.; Hong, C.C.; Rocke, D.M. Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J. Bone Jt. Surg. Ser. A 2010, 92, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Glaser, D.L.; Shore, E.M.; Deirmengian, G.K.; Gupta, R.; Delai, P.; Morhart, R.; Smith, R.; Le Merrer, M.; Rogers, J.G.; et al. The phenotype of fibrodysplasia ossificans progressiva. Clin. Rev. Bone Miner. Metab. 2005, 3, 183–188. [Google Scholar] [CrossRef]
- Schaffer, A.A.; Kaplan, F.S.; Tracy, M.R.; O’Brien, M.L.; Dormans, J.P.; Shore, E.M.; Harland, R.M.; Kusumi, K. Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel-Feil syndrome: Clues from the BMP signaling pathway. Spine (Phila. Pa. 1976) 2005, 30, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef]
- Deirmengian, G.K.; Hebela, N.M.; O’Connell, M.; Glaser, D.L.; Shore, E.M.; Kaplan, F.S. Proximal tibial osteochondromas in patients with fibrodysplasia ossificans progressiva. J. Bone Jt. Surg. Ser. A 2008, 90, 366–374. [Google Scholar] [CrossRef]
- Levy, C.E.; Lash, A.T.; Janoff, H.B.; Kaplan, F.S. Conductive Hearing Loss in Individuals with Fibrodysplasia Ossificans Progressiva. Am. J. Audiol. 1999, 8, 29–33. [Google Scholar] [CrossRef]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Brown, M.A.; Kaplan, F.S. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Bocciardi, R.; Bordo, D.; Di Duca, M.; Di Rocco, M.; Ravazzolo, R. Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: Confirmations and advancements. Eur. J. Hum. Genet. 2009, 17, 311–318. [Google Scholar] [CrossRef]
- Guo, H.; Peng, D.; Xu, M.; Xue, J.; Lu, L.; Xu, X.; Liu, Y.; Xiong, Z.; Pan, Q.; Hu, Z.; et al. Report of two FOP cases with 617G > A mutation in the ACVR1 gene from Chinese population. Clin. Dysmorphol. 2010, 19, 206–208. [Google Scholar] [CrossRef]
- Dandara, C.; Scott, C.; Urban, M.; Fieggen, K.; Arendse, R.; Beighton, P. Confirmation of the recurrent ACVR1 617G>A mutation in South Africans with fibrodysplasia ossificans progressiva. South African Med. J. 2012, 102, 631–633. [Google Scholar] [CrossRef][Green Version]
- Morales-Piga, A.; Bachiller-Corral, J.; Trujillo-Tiebas, M.J.; Villaverde-Hueso, A.; Gamir-Gamir, M.L.; Alonso-Ferreira, V.; Vázquez-Díaz, M.; Posada de la Paz, M.; Ayuso-García, C. Fibrodysplasia ossificans progressiva in Spain: Epidemiological, clinical, and genetic aspects. Bone 2012, 51, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Haga, N.; Takikawa, K.; Manabe, N.; Nishimura, G.; Ikegawa, S. The ACVR1 617G>A mutation is also recurrent in three Japanese patients with fibrodysplasia ossificans progressiva. J. Hum. Genet. 2007, 52, 473–475. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, K.; Song, L.; Pang, J.; Ma, H.; Shore, E.M.; Kaplan, F.S.; Wang, P. The phenotype and genotype of fibrodysplasia ossificans progressiva in China: A report of 72 cases. Bone 2013, 57, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.R.; Navarro, M.M.M.; Martins, B.J.A.F.; Coelho, K.E.F.A.; Mello, W.D.; Takata, R.I.; Speck-Martins, C.E. Mutational screening of ACVR1 gene in Brazilian fibrodysplasia ossificans progressiva patients. Clin. Genet. 2010, 77, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Eresen Yazicioǧlu, Ç.; Karatosun, V.; Kizildaǧ, S.; Özsoylu, D.; Kavukçu, S. ACVR1 gene mutations in four Turkish patients diagnosed as fibrodysplasia ossificans progressiva. Gene 2013, 515, 444–446. [Google Scholar] [CrossRef]
- Shukla, A.; Taywade, O.; Stephen, J.; Gupta, D.; Phadke, S.R. Fibrodysplasia ossificans progressiva: Three Indian patients with mutation in the ACVR1 gene. Indian J. Pediatr. 2014, 81, 617–619. [Google Scholar] [CrossRef]
- Tian, S.; Zhu, J.; Lu, Y. Difficult diagnosis and genetic analysis of fibrodysplasia ossificans progressiva: A case report. BMC Med. Genet. 2018, 19. [Google Scholar] [CrossRef]
- Lin, G.T.; Chang, H.W.; Liu, C.S.; Huang, P.J.; Wang, H.C.; Cheng, Y.M. De novo 617G-A nucleotide mutation in the ACVR1 gene in a Taiwanese patient with fibrodysplasia ossificans progressiva. J. Hum. Genet. 2006, 51, 1083–1086. [Google Scholar] [CrossRef]
- Lee, D.Y.; Cho, T.J.; Lee, H.R.; Park, M.S.; Yoo, W.J.; Chung, C.Y.; Choi, I.H. ACVR1 gene mutation in sporadic Korean patients with fibrodysplasia ossificans progressiva. J. Korean Med. Sci. 2009, 24, 433–437. [Google Scholar] [CrossRef]
- Sun, Y.; Xia, W.; Jiang, Y.; Xing, X.; Li, M.; Wang, O.; Zhang, H.; Hu, Y.; Liu, H.; Meng, X.; et al. A recurrent mutation c.617G>A in the ACVR1 gene causes fibrodysplasia ossificans progressiva in two chinese patients. Calcif. Tissue Int. 2009, 84, 361–365. [Google Scholar] [CrossRef]
- Gregson, C.L.; Hollingworth, P.; Williams, M.; Petrie, K.A.; Bullock, A.N.; Brown, M.A.; Tobias, J.H.; Triffitt, J.T. A novel ACVR1 mutation in the glycine/serine-rich domain found in the most benign case of a fibrodysplasia ossificans progressiva variant reported to date. Bone 2011, 48, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, Y.; Katagiri, T.; Ogata, N.; Haga, N. ACVR1 (587T>C) mutation in a variant form of fibrodysplasia ossificans progressiva: Second report. Am. J. Med. Genet. Part A 2014, 164, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Petrie, K.A.; Lee, W.H.; Bullock, A.N.; Pointon, J.J.; Smith, R.; Russell, R.G.G.; Brown, M.A.; Wordsworth, B.P.; Triffitt, J.T. Novel mutations in ACVR1 result in atypical features in two fibrodysplasia ossificans progressiva patients. PLoS ONE 2009, 4, e5005. [Google Scholar] [CrossRef] [PubMed]
- Barnett, C.P.; Dugar, M.; Haan, E.A. Late-onset variant fibrodysplasia ossificans progressiva leading to misdiagnosis of ankylosing spondylitis. Am. J. Med. Genet. Part A 2011, 155, 1492–1495. [Google Scholar] [CrossRef]
- Rafati, M.; Mohamadhashem, F.; Hoseini, A.; Hoseininasab, F.; Ghaffari, S.R. A novel ACVR1 mutation detected by whole exome sequencing in a family with an unusual skeletal dysplasia. Eur. J. Med. Genet. 2016, 59, 330–336. [Google Scholar] [CrossRef]
- Ratbi, I.; Borcciadi, R.; Regragui, A.; Ravazzolo, R.; Sefiani, A. Rarely occurring mutation of ACVR1 gene in Moroccan patient with fibrodysplasia ossificans progressiva. Clin. Rheumatol. 2010, 29, 119–121. [Google Scholar] [CrossRef]
- Whyte, M.P.; Wenkert, D.; Demertzis, J.L.; Dicarlo, E.F.; Westenberg, E.; Mumm, S. Fibrodysplasia ossificans progressiva: Middle-age onset of heterotopic ossification from a unique missense mutation (c.974G > C, p.G325A) in ACVR1. J. Bone Miner. Res. 2012, 27, 729–737. [Google Scholar] [CrossRef]
- Gucev, Z.; Tasic, V.; Plaseska-Karanfilska, D.; Dimishkovska, M.; Laban, N.; Bozinovski, Z.; Kostovski, M.; Saveski, A.; Polenakovic, M.; Towler, O.W.; et al. Severe digital malformations in a rare variant of fibrodysplasia ossificans progressiva. Am. J. Med. Genet. Part A 2019, 179, 1310–1314. [Google Scholar] [CrossRef]
- Fukuda, T.; Kanomata, K.; Nojima, J.; Kokabu, S.; Akita, M.; Ikebuchi, K.; Jimi, E.; Komori, T.; Maruki, Y.; Matsuoka, M.; et al. A unique mutation of ALK2, G356D, found in a patient with fibrodysplasia ossificans progressiva is a moderately activated BMP type I receptor. Biochem. Biophys. Res. Commun. 2008, 377, 905–909. [Google Scholar] [CrossRef]
- Chaikuad, A.; Alfano, I.; Kerr, G.; Sanvitale, C.E.; Boergermann, J.H.; Triffitt, J.T.; Von Delft, F.; Knapp, S.; Knaus, P.; Bullock, A.N. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem. 2012, 287, 36990–36998. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, Y.; Suzuki, R.; Katagiri, T.; Toguchida, J.; Haga, N. Phenotypic differences of patients with fibrodysplasia ossificans progressive due to p.Arg258Ser variants of ACVR1. Hum. Genome Var. 2015, 2. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shen, Q.; Little, S.C.; Xu, M.; Haupt, J.; Ast, C.; Katagiri, T.; Mundlos, S.; Seemann, P.; Kaplan, F.S.; Mullins, M.C.; et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J. Clin. Investig. 2009, 119, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Van Dinther, M.; Visser, N.; De Gorter, D.J.J.; Doorn, J.; Goumans, M.J.; De Boer, J.; Ten Dijke, P. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J. Bone Miner. Res. 2010, 25, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Song, G.A.; Kim, H.J.; Woo, K.M.; Baek, J.H.; Kim, G.S.; Choi, J.Y.; Ryoo, H.M. Molecular consequences of the ACVR1 R206H mutation of fibrodysplasia ossificans progressiva. J. Biol. Chem. 2010, 285, 22542–22553. [Google Scholar] [CrossRef] [PubMed]
- Billings, P.C.; Fiori, J.L.; Bentwood, J.L.; O’Connell, M.P.; Jiao, X.; Nussbaum, B.; Caron, R.J.; Shore, E.M.; Kaplan, F.S. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 2008, 23, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Kohda, M.; Kanomata, K.; Nojima, J.; Nakamura, A.; Kamizono, J.; Noguchi, Y.; Iwakiri, K.; Kondo, T.; Kurose, J.; et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J. Biol. Chem. 2009, 284, 7149–7156. [Google Scholar] [CrossRef] [PubMed]
- Haupt, J.; Deichsel, A.; Stange, K.; Ast, C.; Bocciardi, R.; Ravazzolo, R.; Di Rocco, M.; Ferrari, P.; Landi, A.; Kaplan, F.S.; et al. ACVR1 p.Q207E causes classic fibrodysplasia ossificans progressiva and is functionally distinct from the engineered constitutively active ACVR1 p.Q207D variant. Hum. Mol. Genet. 2014, 23, 5364–5377. [Google Scholar] [CrossRef]
- Haupt, J.; Xu, M.; Shore, E.M. Variable signaling activity by FOP ACVR1 mutations. Bone 2018, 109, 232–240. [Google Scholar] [CrossRef]
- Machiya, A.; Tsukamoto, S.; Ohte, S.; Kuratani, M.; Fujimoto, M.; Kumagai, K.; Osawa, K.; Suda, N.; Bullock, A.N.; Katagiri, T. Effects of FKBP12 and type II BMP receptors on signal transduction by ALK2 activating mutations associated with genetic disorders. Bone 2018, 111, 101–108. [Google Scholar] [CrossRef]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, B.Y.; Danielson, P.D.; Shah, P.C.; Rockwell, S.; Lechleider, R.J.; Martin, J.; Manganaro, T.; Donahoe, P.K. The immunophilin FKBP12 functions as a common inhibitor of the TGFβ family type I receptors. Cell 1996, 86, 435–444. [Google Scholar] [CrossRef]
- Groppe, J.C.; Wu, J.; Shore, E.M.; Kaplan, F.S. In vitro analyses of the dysregulated R206H ALK2 kinase-FKBP12 interaction associated with heterotopic ossification in FOP. Cells Tissues Organs 2011, 194, 291–295. [Google Scholar]
- Olsen, O.E.; Sankar, M.; Elsaadi, S.; Hella, H.; Buene, G.; Darvekar, S.R.; Misund, K.; Katagiri, T.; Knaus, P.; Holien, T. BMPR2 inhibits activin and BMP signaling via wild-type ALK2. J. Cell Sci. 2018, 131, jcs213512. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.E.; Wader, K.F.; Hella, H.; Mylin, A.K.; Turesson, I.; Nesthus, I.; Waage, A.; Sundan, A.; Holien, T. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun. Signal. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.A.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.A.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Hildebrand, L.; Stange, K.; Deichsel, A.; Gossen, M.; Seemann, P. The Fibrodysplasia Ossificans Progressiva (FOP) mutation p.R206H in ACVR1 confers an altered ligand response. Cell. Signal. 2017, 29, 23–30. [Google Scholar] [CrossRef]
- Barruet, E.; Morales, B.M.; Lwin, W.; White, M.P.; Theodoris, C.V.; Kim, H.; Urrutia, A.; Wong, S.A.; Srivastava, D.; Hsiao, E.C. The ACVR1 R206H mutation found in fibrodysplasia ossificans progressiva increases human induced pluripotent stem cell-derived endothelial cell formation and collagen production through BMP-mediated SMAD1/5/8 signaling. Stem Cell Res. Ther. 2016, 7. [Google Scholar] [CrossRef]
- Antebi, Y.E.; Linton, J.M.; Klumpe, H.; Bintu, B.; Gong, M.; Su, C.; McCardell, R.; Elowitz, M.B. Combinatorial Signal Perception in the BMP Pathway. Cell 2017, 170, 1184–1196.e24. [Google Scholar] [CrossRef]
- Fiori, J.L.; Billings, P.C.; Serrano De La Peña, L.; Kaplan, F.S.; Shore, E.M. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 2006, 21, 902–909. [Google Scholar]
- Rigueur, D.; Brugger, S.; Anbarchian, T.; Kim, J.K.; Lee, Y.; Lyons, K.M. The type i BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J. Bone Miner. Res. 2015, 30, 733–741. [Google Scholar] [CrossRef]
- Qureshi, A.T.; Dey, D.; Sanders, E.M.; Seavey, J.G.; Tomasino, A.M.; Moss, K.; Wheatley, B.; Cholok, D.; Loder, S.; Li, J.; et al. Inhibition of Mammalian Target of Rapamycin Signaling with Rapamycin Prevents Trauma-Induced Heterotopic Ossification. Am. J. Pathol. 2017, 187, 2536–2545. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Zhao, C.; Horigome, K.; Nishio, M.; Okanishi, Y.; Nagata, S.; Komura, S.; Yamada, Y.; Toguchida, J.; Ohta, A.; et al. An mTOR Signaling Modulator Suppressed Heterotopic Ossification of Fibrodysplasia Ossificans Progressiva. Stem Cell Rep. 2018, 11, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Gámez, B.; Rodríguez-Carballo, E.; Graupera, M.; Rosa, J.L.; Ventura, F. Class I PI-3-Kinase Signaling Is Critical for Bone Formation Through Regulation of SMAD1 Activity in Osteoblasts. J. Bone Miner. Res. 2016, 31, 1617–1630. [Google Scholar] [CrossRef]
- Valer, J.A.; Sánchez-de-Diego, C.; Gámez, B.; Mishina, Y.; Rosa, J.L.; Ventura, F. Inhibition of phosphatidylinositol 3-kinase α ( PI 3Kα) prevents heterotopic ossification. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Haupt, J.; Stanley, A.; McLeod, C.M.; Cosgrove, B.D.; Culbert, A.L.; Wang, L.; Mourkioti, F.; Mauck, R.L.; Shore, E.M. ACVR1 R206H FOP mutation alters mechanosensing and tissue stiffness during heterotopic ossification. Mol. Biol. Cell 2018, 30, 17–29. [Google Scholar] [CrossRef]
- Lin, S.; Svoboda, K.K.H.; Feng, J.Q.; Jiang, X. The biological function of type i receptors of bone morphogenetic protein in bone. Bone Res. 2016, 4. [Google Scholar] [CrossRef]
- Zhang, D.; Schwarz, E.M.; Rosier, R.N.; Zuscik, M.J.; Puzas, J.E.; O’Keefe, R.J. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J. Bone Miner. Res. 2003, 18, 1593–1604. [Google Scholar] [CrossRef]
- Yu, P.B.; Deng, D.Y.; Lai, C.S.; Hong, C.C.; Cuny, G.D.; Bouxsein, M.L.; Hong, D.W.; McManus, P.M.; Katagiri, T.; Sachidanandan, C.; et al. BMP type I receptor inhibition reduces heterotopic ossification. Nat. Med. 2008, 14, 1363–1369. [Google Scholar] [CrossRef]
- Kaplan, J.; Kaplan, F.S.; Shore, E.M. Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele-specific targeting. Gene Ther. 2012, 19, 786–790. [Google Scholar] [CrossRef]
- Shi, S.T.; Cai, J.; de Gorter, D.J.J.; Sanchez-Duffhues, G.; Kemaladewi, D.U.; Hoogaars, W.M.H.; Aartsma-Rus, A.; ’t Hoen, P.A.C.; ten Dijke, P. Antisense-Oligonucleotide Mediated Exon Skipping in Activin-Receptor-Like Kinase 2: Inhibiting the Receptor That Is Overactive in Fibrodysplasia Ossificans Progressiva. PLoS ONE 2013, 8, e69096. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Mandair, G.S.; Zhang, H.; Vanrenterghem, G.G.; Ridella, R.; Takahashi, A.; Zhang, Y.; Kohn, D.H.; Morris, M.D.; Mishina, Y.; et al. Bone morphogenetic protein signaling through ACVR1 and BMPR1A negatively regulates bone mass along with alterations in bone composition. J. Struct. Biol. 2018, 201, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, N.; Kaartinen, V.M.; Mishina, Y. Loss-of-function of ACVR1 in osteoblasts increases bone mass and activates canonical Wnt signaling through suppression of Wnt inhibitors SOST and DKK1. Biochem. Biophys. Res. Commun. 2011, 414, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, Q.; Zhao, H.; Hu, Y.; Liu, C.; Yan, G.; Li, D.; Mishina, Y.; Shi, C.; Sun, H. ACVR1 is essential for periodontium development and promotes alveolar bone formation. Arch. Oral Biol. 2018, 95, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, C.; Zhao, H.; Zhou, Y.; Hu, Y.; Yan, G.; Liu, C.; Li, D.; Hao, X.; Mishina, Y.; et al. Distinctive role of ACVR1 in dentin formation: requirement for dentin thickness in molars and prevention of osteodentin formation in incisors of mice. J. Mol. Histol. 2019, 50, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Puerto, M.C.; Iyengar, P.V.; García de Vinuesa, A.; ten Dijke, P.; Sanchez-Duffhues, G. Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 2019, 247, 9–20. [Google Scholar] [CrossRef]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef]
- Wang, J.; Sridurongrit, S.; Dudas, M.; Thomas, P.; Nagy, A.; Schneider, M.D.; Epstein, J.A.; Kaartinen, V. Atrioventricular cushion transformation is mediated by ALK2 in the developing mouse heart. Dev. Biol. 2005, 286, 299–310. [Google Scholar] [CrossRef]
- Thomas, P.S.; Rajderkar, S.; Lane, J.; Mishina, Y.; Kaartinen, V. AcvR1-mediated BMP signaling in second heart field is required for arterial pole development: Implications for myocardial differentiation and regional identity. Dev. Biol. 2014, 390, 191–207. [Google Scholar] [CrossRef][Green Version]
- Kaartinen, V.; Dudas, M.; Nagy, A.; Sridurongrit, S.; Lu, M.M.; Epstein, J.A. Cardiac outflow tract defects in mice lacking ALK2 in neural crestcells. Development 2004, 131, 3481–3490. [Google Scholar] [CrossRef]
- Thomas, P.S.; Sridurongrit, S.; Ruiz-Lozano, P.; Kaartinen, V. Deficient signaling via Alk2 (Acvr1) leads to Bicuspid aortic valve development. PLoS ONE 2012, 7, e35539. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.; Spagnolli, E.; Ernande, L.; Thoonen, R.; Kolodziej, S.A.; Leyton, P.A.; Cheng, J.; Tainsh, R.E.T.; Mayeur, C.; Rhee, D.K.; et al. BMP type I receptor ALK2 is required for angiotensin II-induced cardiac hypertrophy. Am. J. Physiol. Circ. Physiol. 2016, 310, H984–H994. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smith, K.A.; Joziasse, I.C.; Chocron, S.; Van Dinther, M.; Guryev, V.; Verhoeven, M.C.; Rehmann, H.; Van Der Smagt, J.J.; Doevendans, P.A.; Cuppen, E.; et al. Dominant-negative alk2 allele associates with congenital heart defects. Circulation 2009, 119, 3062–3069. [Google Scholar] [CrossRef] [PubMed]
- Joziasse, I.C.; Smith, K.A.; Chocron, S.; Van Dinther, M.; Guryev, V.; Van De Smagt, J.J.; Cuppen, E.; Ten Dijke, P.; Mulder, B.J.M.; Maslen, C.L.; et al. ALK2 mutation in a patient with Down’s syndrome and a congenital heart defect. Eur. J. Hum. Genet. 2011, 19, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Li, A.H.; Hanchard, N.A.; Furthner, D.; Fernbach, S.; Azamian, M.; Nicosia, A.; Rosenfeld, J.; Muzny, D.; D’Alessandro, L.C.A.; Morris, S.; et al. Whole exome sequencing in 342 congenital cardiac left sided lesion cases reveals extensive genetic heterogeneity and complex inheritance patterns. Genome Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Kavsak, P.; Abdollah, S.; Wrana, J.L.; Thomsen, G.H. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 1999, 400, 687–693. [Google Scholar] [CrossRef]
- Kussmaul, W.G.; Esmail, A.N.; Sagar, Y.; Ross, J.; Gregory, S.; Kaplan, F.S. Pulmonary and Cardiac Function in Advanced Fibrodysplasia Ossificans Progressiva. Clin. Orthop. Relat. Res. 1998, 346, 104–109. [Google Scholar] [CrossRef]
- Marseglia, L.; D’Angelo, G.; Manti, S.; Manganaro, A.; Calabrò, M.P.; Salpietro, C.; Gitto, E. Fibrodysplasia ossificans progressiva in a newborn with cardiac involvement. Pediatr. Int. 2015, 57, 719–721. [Google Scholar] [CrossRef]
- Knight, P.G.; Glister, C. TGF-β superfamily members and ovarian follicle development. Reproduction 2006, 132, 191–206. [Google Scholar] [CrossRef]
- Shimasaki, S.; Moore, R.K.; Otsuka, F.; Erickson, G.F. The Bone Morphogenetic Protein System in Mammalian Reproduction. Endocr. Rev. 2004, 25, 72–101. [Google Scholar] [CrossRef]
- Visser, J.A.; Olaso, R.; Verhoef-Post, M.; Kramer, P.; Themmen, A.P.N.; Ingraham, H.A. The Serine/Threonine Transmembrane Receptor ALK2 Mediates Müllerian Inhibiting Substance Signaling. Mol. Endocrinol. 2014, 15, 936–945. [Google Scholar]
- Clarke, T.R.; Hoshiya, Y.; Yi, S.E.; Liu, X.; Lyons, K.M.; Donahoe, P.K. Müllerian Inhibiting Substance Signaling Uses a Bone Morphogenetic Protein (BMP)-Like Pathway Mediated by ALK2 and Induces Smad6 Expression. Mol. Endocrinol. 2014, 15, 946–959. [Google Scholar] [CrossRef]
- Gouédard, L.; Chen, Y.G.; Thevenet, L.; Racine, C.; Borie, S.; Lamarre, I.; Josso, N.; Massaguè, J.; Di Clemente, N. Engagement of bone morphogenetic protein type IB receptor and Smad1 signaling by anti-Mullerian hormone and its type II receptor. J. Biol. Chem. 2000, 275, 27973–27978. [Google Scholar] [CrossRef] [PubMed]
- Jamin, S.P.; Arango, N.A.; Mishina, Y.; Hanks, M.C.; Behringer, R.R. Genetic studies of the AMH/MIS signaling pathway for Müllerian duct regression. Mol. Cell. Endocrinol. 2003, 211, 15–19. [Google Scholar]
- Van Houten, E.L.A.F.; Themmen, A.P.N.; Visser, J.A. Hormone anti-müllérienne (AMH): Régulateur et marqueur de la fonction ovarienne. Ann. Endocrinol. (Paris) 2010, 71, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Durlinger, A.L.L.; Gruijters, M.J.G.; Kramer, P.; Karels, B.; Kumar, T.R.; Matzuk, M.M.; Rose, U.M.; De Jong, F.H.; Uilenbroek, J.T.J.; Grootegoed, J.A.; et al. Anti-Müllerian hormone attenuates the effects of FSH on follicle development in the mouse ovary. Endocrinology 2001, 142, 4891–4899. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, F.; Moore, R.K.; Shimasaki, S. Biological Function and Cellular Mechanism of Bone Morphogenetic Protein-6 in the Ovary. J. Biol. Chem. 2001, 276, 32889–32895. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yoon, S.J.; Yoon, T.K.; Cha, K.Y.; Lee, S.H.; Shimasaki, S.; Lee, S.; Lee, K.A. Effects of bone morphogenetic protein-7 (BMP-7) on primordial follicular growth in the mouse ovary. Mol. Reprod. Dev. 2004, 69, 159–163. [Google Scholar] [CrossRef]
- Kevenaar, M.E.; Themmen, A.P.N.; Van Kerkwijk, A.J.; Valkenburg, O.; Uitterlinden, A.G.; De Jong, F.H.; Laven, J.S.E.; Visser, J.A. Variants in the ACVR1 gene are associated with AMH levels in women with polycystic ovary syndrome. Hum. Reprod. 2009, 24, 241–249. [Google Scholar] [CrossRef][Green Version]
- Slattery, M.L.; John, E.M.; Torres-Mejia, G.; Herrick, J.S.; Giuliano, A.R.; Baumgartner, K.B.; Hines, L.M.; Wolff, R.K. Genetic variation in bone morphogenetic proteins and breast cancer risk in hispanic and non-hispanic white women: The breast cancer health disparities study. Int. J. Cancer 2013, 132, 2928–2939. [Google Scholar] [CrossRef]
- Da Silveira, J.C.; Carnevale, E.M.; Winger, Q.A.; Bouma, G.J. Regulation of ACVR1 and ID2 by cell-secreted exosomes during follicle maturation in the mare. Reprod. Biol. Endocrinol. 2014, 12. [Google Scholar] [CrossRef]
- Bai, L.; Chang, H.M.; Cheng, J.C.; Chu, G.; Leung, P.C.K.; Yang, G. ALK2/ALK3-BMPR2/ACVR2A Mediate BMP2-induced downregulation of pentraxin 3 expression in human granulosa-lutein cells. Endocrinology 2017, 158, 3501–3511. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Chang, H.M.; Huang, H.F.; Sheng, J.Z.; Leung, P.C.K. Bone morphogenetic protein 2 regulates cell-cell communication by down-regulating connexin43 expression in luteinized human granulosa cells. Mol. Hum. Reprod. 2017, 23, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Jeong, J.-W.; Wang, J.; Ma, L.; Martin, J.F.; Tsai, S.Y.; Lydon, J.P.; DeMayo, F.J. Bmp2 Is Critical for the Murine Uterine Decidual Response. Mol. Cell. Biol. 2007, 27, 5468–5478. [Google Scholar] [CrossRef] [PubMed]
- Clementi, C.; Tripurani, S.K.; Large, M.J.; Edson, M.A.; Creighton, C.J.; Hawkins, S.M.; Kovanci, E.; Kaartinen, V.; Lydon, J.P.; Pangas, S.A.; et al. Activin-Like Kinase 2 Functions in Peri-implantation Uterine Signaling in Mice and Humans. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, Y.; Scott, G.; Nagy, A.; Kaartinen, V.; Mishina, Y. BMP type I receptor ALK2 is essential for proper patterning at late gastrulation during mouse embryogenesis. Dev. Dyn. 2007, 236, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Chuva De Sousa Lopes, S.M.; Roelen, B.A.J.; Monteiro, R.M.; Emmens, R.; Lin, H.Y.; Li, E.; Lawson, K.A.; Mummery, C.L. BMP signaling mediated by ALK2 in the visceral endoderm is necessary for the generation of primordial germ cells in the mouse embryo. Genes Dev. 2004, 18, 1838–1849. [Google Scholar] [CrossRef] [PubMed]
- Kitterman, J.A.; Strober, J.B.; Kan, L.; Rocke, D.M.; Cali, A.; Peeper, J.; Snow, J.; Delai, P.L.R.; Morhart, R.; Pignolo, R.J.; et al. Neurological symptoms in individuals with fibrodysplasia ossificans progressiva. J. Neurol. 2012, 259, 2636–2643. [Google Scholar] [CrossRef]
- Bond, A.M.; Bhalala, O.G.; Kessler, J.A. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev. Neurobiol. 2012, 72, 1068–1084. [Google Scholar] [CrossRef]
- Angley, C.; Kumar, M.; Dinsio, K.J.; Hall, A.K.; Siegel, R.E. Signaling by Bone Morphogenetic Proteins and Smad1 Modulates the Postnatal Differentiation of Cerebellar Cells. J. Neurosci. 2003, 23, 260–268. [Google Scholar] [CrossRef]
- Gomes, W.A.; Mehler, M.F.; Kessler, J.A. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev. Biol. 2003, 255, 164–177. [Google Scholar] [CrossRef]
- Samanta, J. Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development 2004, 131, 4131–4142. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.E.; Mehler, M.F.; Mabie, P.C.; Zang, Z.; Santschi, L.; Kessler, J.A. Bone morphogenetic proteins promote astroglial lineage commitment by mammalian subventricular zone progenitor cells. Neuron 1996, 17, 595–606. [Google Scholar] [CrossRef]
- Mehler, M.F.; Mabie, P.C.; Zhu, G.; Gokhan, S.; Kessler, J.A. Developmental changes in progenitor cell responsiveness to gone morphogenetic; Proteins differentially modulate progressive CNS lineage fate. Dev. Neurosci. 2000, 22, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Gobeske, K.T.; Das, S.; Bonaguidi, M.A.; Weiss, C.; Radulovic, J.; Disterhoft, J.F.; Kessler, J.A. BMP signaling mediates effects of exercise on hippocampal neurogenesis and cognition in mice. PLoS ONE 2009, 4, e7506. [Google Scholar] [CrossRef]
- Bertamino, M.; Severino, M.; Schiaffino, M.C.; Garrè, M.L.; Bocciardi, R.; Ravazzolo, R.; Rossi, A.; Di Rocco, M. New insights into central nervous system involvement in FOP: Case report and review of the literature. Am. J. Med. Genet. Part A 2015, 167, 2817–2821. [Google Scholar] [CrossRef]
- Severino, M.; Bertamino, M.; Tortora, D.; Morana, G.; Uccella, S.; Bocciardi, R.; Ravazzolo, R.; Rossi, A.; Di Rocco, M. Novel asymptomatic CNS findings in patients with ACVR1/ALK2 mutations causing fibrodysplasia ossificans progressiva. J. Med. Genet. 2016, 53, 859–864. [Google Scholar] [CrossRef]
- Shiva Kumar, R.; Keerthiraj, B.; Kesavadas, C. Teaching NeuroImages: MRI in fibrodysplasia ossificans progressiva. Neurology 2010, 74. [Google Scholar] [CrossRef]
- Kan, L.; Kitterman, J.A.; Procissi, D.; Chakkalakal, S.; Peng, C.Y.; McGuire, T.L.; Goldsby, R.E.; Pignolo, R.J.; Shore, E.M.; Kaplan, F.S.; et al. CNS demyelination in fibrodysplasia ossificans progressiva. J. Neurol. 2012, 259, 2644–2655. [Google Scholar] [CrossRef]
- Cate, H.S.; Sabo, J.K.; Merlo, D.; Kemper, D.; Aumann, T.D.; Robinson, J.; Merson, T.D.; Emery, B.; Perreau, V.M.; Kilpatrick, T.J. Modulation of bone morphogenic protein signalling alters numbers of astrocytes and oligodendroglia in the subventricular zone during cuprizone-induced demyelination. J. Neurochem. 2010, 115, 11–22. [Google Scholar] [CrossRef]
- Qi, X.; Li, T.-G.; Hao, J.; Hu, J.; Wang, J.; Simmons, H.; Miura, S.; Mishina, Y.; Zhao, G.-Q. BMP4 supports self-renewal of embryonic stem cells by inhibiting mitogen-activated protein kinase pathways. Proc. Natl. Acad. Sci. USA 2004, 101, 6027–6032. [Google Scholar] [CrossRef]
- Ying, Q.L.; Nichols, J.; Chambers, I.; Smith, A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 2003, 115, 281–292. [Google Scholar] [CrossRef]
- Xu, R.H.; Peck, R.M.; Li, D.S.; Feng, X.; Ludwig, T.; Thomson, J.A. Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat. Methods 2005, 2, 185–190. [Google Scholar] [CrossRef]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.K.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional genomics reveals a BMP-Driven mesenchymal-to-Epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Hashizume, Y.; Yamada, Y.; Katayama, T.; Hohjoh, H.; Fusaki, N.; Nakashima, Y.; Furuya, H.; Haga, N.; Takami, Y.; et al. Pathogenic mutation of ALK2 inhibits induced pluripotent stem cell reprogramming and maintenance: Mechanisms of reprogramming and strategy for drug identification. Stem Cells 2012, 30, 2437–2449. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Ikeya, M.; Hino, K.; Horigome, K.; Fukuta, M.; Watanabe, M.; Nagata, S.; Yamamoto, T.; Otsuka, T.; Toguchida, J. New protocol to optimize iPS cells for genome analysis of fibrodysplasia ossificans progressiva. Stem Cells 2015, 33, 1730–1742. [Google Scholar] [CrossRef]
- Cai, J.; Orlova, V.V.; Cai, X.; Eekhoff, E.M.W.; Zhang, K.; Pei, D.; Pan, G.; Mummery, C.L.; Ten Dijke, P. Induced Pluripotent Stem Cells to Model Human Fibrodysplasia Ossificans Progressiva. Stem Cell Rep. 2015, 5, 963–970. [Google Scholar] [CrossRef]
- Kim, B.Y.; Jeong, S.K.; Lee, S.Y.; Lee, S.M.; Gweon, E.J.; Ahn, H.; Kim, J.; Chung, S.K. Concurrent progress of reprogramming and gene correction to overcome therapeutic limitation of mutant ALK2-iPSC. Exp. Mol. Med. 2016, 48. [Google Scholar] [CrossRef]
- Hayashi, Y.; Hsiao, E.C.; Sami, S.; Lancero, M.; Schlieve, C.R.; Nguyen, T.; Yano, K.; Nagahashi, A.; Ikeya, M.; Matsumoto, Y.; et al. BMP-SMAD-ID promotes reprogramming to pluripotency by inhibiting p16/INK4A-dependent senescence. Proc. Natl. Acad. Sci. USA 2016, 113, 13057–13062. [Google Scholar] [CrossRef]
- López-Rovira, T.; Chalaux, E.; Massagué, J.; Rosa, J.L.; Ventura, F. Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J. Biol. Chem. 2002, 277, 3176–3185. [Google Scholar] [CrossRef]
- Alani, R.M.; Young, A.Z.; Shifflett, C.B. Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proc. Natl. Acad. Sci. USA 2001, 98, 7812–7816. [Google Scholar] [CrossRef]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.D.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, G.; Aldape, K. ACVR1 mutations and the genomic landscape of pediatric diffuse glioma. Nat. Genet. 2014, 46, 421–422. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Bartels, U.; Bouffet, E.; Becher, O.; Hawkins, C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol. 2014, 128, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Hawkins, C. Pathology, Molecular Genetics, and Epigenetics of Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.T. Genetics: ACVR1 mutations-A key piece in paediatric diffuse glioma. Nat. Rev. Clin. Oncol. 2014, 11, 300. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.S.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Hoeman, C.M.; Cordero, F.J.; Hu, G.; Misuraca, K.; Romero, M.M.; Cardona, H.J.; Nazarian, J.; Hashizume, R.; McLendon, R.; Yu, P.; et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef]
- Taylor, K.R.; Vinci, M.; Bullock, A.N.; Jones, C. ACVR1 mutations in DIPG: Lessons learned from FOP. Cancer Res. 2014, 74, 4565–4570. [Google Scholar] [CrossRef] [PubMed]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Nikbakht, H.; Panditharatna, E.; Mikael, L.G.; Li, R.; Gayden, T.; Osmond, M.; Ho, C.Y.; Kambhampati, M.; Hwang, E.I.; Faury, D.; et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Jain, P.; Resnick, A.C. Shared ACVR1 mutations in FOP and DIPG: Opportunities and challenges in extending biological and clinical implications across rare diseases. Bone 2018, 109, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Raja, E.; Komuro, A.; Tanabe, R.; Sakai, S.; Ino, Y.; Saito, N.; Todo, T.; Morikawa, M.; Aburatani, H.; Koinuma, D.; et al. Bone morphogenetic protein signaling mediated by ALK-2 and DLX2 regulates apoptosis in glioma-initiating cells. Oncogene 2017, 36, 4963–4974. [Google Scholar] [CrossRef]
- Ghanem, N.; Andrusiak, M.G.; Svoboda, D.; Al Lafi, S.M.; Julian, L.M.; McClellan, K.A.; De Repentigny, Y.; Kothary, R.; Ekker, M.; Blais, A.; et al. The Rb/E2F Pathway Modulates Neurogenesis through Direct Regulation of the Dlx1/Dlx2 Bigene Cluster. J. Neurosci. 2012, 32, 8219–8230. [Google Scholar] [CrossRef]
- Panganiban, G.; Rubenstein, J.L.R. Developmental functions of the Distal-less/Dlx homeobox genes. Development 2002, 129, 4371–4386. [Google Scholar]
- Wang, T.-H.; Chao, A.; Tsai, C.-L.; Chang, C.-L.; Chen, S.-H.; Lee, Y.-S.; Chen, J.-K.; Lin, Y.-J.; Chang, P.-Y.; Wang, C.-J.; et al. Stress-induced Phosphoprotein 1 as a Secreted Biomarker for Human Ovarian Cancer Promotes Cancer Cell Proliferation. Mol. Cell. Proteomics 2010, 9, 1873–1884. [Google Scholar] [CrossRef]
- Tsai, C.L.; Tsai, C.N.; Lin, C.Y.; Chen, H.W.; Lee, Y.S.; Chao, A.; Wang, T.H.; Wang, H.S.; Lai, C.H. Secreted Stress-Induced Phosphoprotein 1 Activates the ALK2-SMAD Signaling Pathways and Promotes Cell Proliferation of Ovarian Cancer Cells. Cell Rep. 2012, 2, 283–293. [Google Scholar] [CrossRef]
- Herrera, B.; Van Dinther, M.; Ten Dijke, P.; Inman, G.J. Autocrine bone morphogenetic protein-9 signals through activin receptor-like kinase-2/Smad1/Smad4 to promote ovarian cancer cell proliferation. Cancer Res. 2009, 69, 9254–9262. [Google Scholar] [CrossRef] [PubMed]
- La Marca, A.; Volpe, A. The Anti-Mullerian hormone and ovarian cancer. Hum. Reprod. Update 2007, 13, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Anttonen, M.; Färkkilä, A.; Tauriala, H.; Kauppinen, M.; MacLaughlin, D.T.; Unkila-Kallio, L.; Bützow, R.; Heikinheimo, M. Anti-Müllerian hormone inhibits growth of AMH type II receptor-positive human ovarian granulosa cell tumor cells by activating apoptosis. Lab. Investig. 2011, 91, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Deng, L.; Xiong, Q.; Su, S.; Gu, J. Anti-müllerian hormone inhibits proliferation and induces apoptosis in epithelial ovarian cancer cells by regulating the cell cycle and decreasing the secretion of stem cell factor. Oncol. Lett. 2018, 16, 3260–3266. [Google Scholar]
- Masiakos, P.T.; MacLaughlin, D.T.; Maheswaran, S.; Teixeira, J.; Shah, P.C.; Kehas, D.J.; Kenneally, M.K.; Ha, T.U.; Fuller, A.F., Jr.; Dombkowski, D.M.; et al. Human ovarian cancer, cell lines, and primary ascites cells express the human Mullerian inhibiting substance (MIS) type II receptor, bind, and are responsive to MIS. Clin. Cancer Res. 1999, 5, 3488–3499. [Google Scholar]
- Stephen, A.E.; Pearsall, L.A.; Christian, B.P.; Donahoe, P.K.; Vacanti, J.P.; MacLaughlin, D.T. Highly purified Müllerian inhibiting substance inhibits human ovarian cancer in vivo. Clin. Cancer Res. 2002, 8, 2640–2646. [Google Scholar]
- Zhao, B.; Pritchard, J.R. Inherited Disease Genetics Improves the Identification of Cancer-Associated Genes. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- COSMIC The Catalogue Of Somatic Mutations In Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 26 September 2019).
- Nassiri, F.; Cusimano, M.D.; Scheithauer, B.W.; Rotondo, F.; Fazio, A.; Yousef, G.M.; Syro, L.V.; Kovacs, K.; Lloyd, R.V. Endoglin (CD105): A review of its role in angiogenesis and tumor diagnosis, progression and therapy. Anticancer Res. 2011, 31, 2283–2290. [Google Scholar]
- Craft, C.S.; Romero, D.; Vary, C.P.H.; Bergan, R.C. Endoglin inhibits prostate cancer motility via activation of the ALK2-Smad1 pathway. Oncogene 2007, 26, 7240–7250. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.; Terzic, A.; Conley, B.A.; Craft, C.S.; Jovanovic, B.; Bergan, R.C.; Vary, C.P.H. Endoglin phosphorylation by ALK2 contributes to the regulation of prostate cancer cell migration. Carcinogenesis 2010, 31, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.E.; Wader, K.F.; Misund, K.; Våtsveen, T.K.; Rø, T.B.; Mylin, A.K.; Turesson, I.; Størdal, B.F.; Moen, S.H.; Standal, T.; et al. Bone morphogenetic protein-9 suppresses growth of myeloma cells by signaling through ALK2 but is inhibited by endoglin. Blood Cancer J. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, R.; Dattilo, L.K.; Kaartinen, V.; Deng, C.X.; Umans, L.; Zwijsen, A.; Roberts, A.B.; Bottinger, E.P.; Beebe, D.C. Functions of the type 1 BMP receptor Acvr1 (Alk2) in lens development: Cell proliferation, terminal differentiation, and survival. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4953–4960. [Google Scholar] [CrossRef] [PubMed]
- Wiley, L.A.; Rajagopal, R.; Dattilo, L.K.; Beebe, D.C. The tumor suppressor gene Trp53 protects the mouse lens against posterior subcapsular cataracts and the BMP receptor Acvr1 acts as a tumor suppressor in the lens. Dis. Model. Mech. 2011, 4, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Choi, O.; Pyo, S.; Choi, S.U.; Park, C.H. Identification of novel ALK2 inhibitors and their effect on cancer cells. Biochem. Biophys. Res. Commun. 2017, 492, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, E.P.; Drigo, S.A.; Bérgamo, N.A.; Rosa, F.E.; Bertonha, F.B.; de Abreu, F.B.; Kowalski, L.P.; Rogatto, S.R. Recurrent copy number gains of ACVR1 and corresponding transcript overexpression are associated with survival in head and neck squamous cell carcinomas. Histopathology 2011, 59, 81–89. [Google Scholar] [CrossRef]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Bach, D.H.; Park, H.J.; Lee, S.K. The Dual Role of Bone Morphogenetic Proteins in Cancer. Mol. Ther. Oncolytics 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Zhang, L.; Ye, Y.; Long, X.; Xiao, P.; Ren, X.; Yu, J. BMP signaling and its paradoxical effects in tumorigenesis and dissemination. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Alarmo, E.L.; Kallioniemi, A. Bone morphogenetic proteins in breast cancer: Dual role in tumourigenesis? Endocr. Relat. Cancer 2010, 17. [Google Scholar] [CrossRef] [PubMed]
- Alarmo, E.L.; Pärssinen, J.; Ketolainen, J.M.; Savinainen, K.; Karhu, R.; Kallioniemi, A. BMP7 influences proliferation, migration, and invasion of breast cancer cells. Cancer Lett. 2009, 275, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhong, C.; Frenkel, B.; Reddi, A.H.; Roy-Burman, P. Diverse biological effect and Smad signaling of bone morphogenetic protein 7 in prostate tumor cells. Cancer Res. 2005, 65, 5769–5777. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peng, J.; Yoshioka, Y.; Mandai, M.; Matsumura, N.; Baba, T.; Yamaguchi, K.; Hamanishi, J.; Kharma, B.; Murakami, R.; Abiko, K.; et al. The BMP signaling pathway leads to enhanced proliferation in serous ovarian cancer-A potential therapeutic target. Mol. Carcinog. 2016, 55, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.J.; Kirkbride, K.C.; How, T.; Blobe, G.C. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a Smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis 2009, 30, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chen, A.; He, W.; Li, Z.; Zhang, G.; Liu, Z.; Liu, G.; Liu, X.; He, S.; Xiao, G.; et al. BMP-2 induces EMT and breast cancer stemness through Rb and CD44. Cell Death Discov. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.H.; Zhou, X.M.; Zhang, M.Y.; Shi, L.; Xiao, R.W.; Zeng, L.S.; Yang, X.Z.; Zheng, X.F.S.; Wang, H.Y.; Mai, S.J. BMP2 promotes proliferation and invasion of nasopharyngeal carcinoma cells via mTORC1 pathway. Aging (Albany. NY) 2017, 9, 1326–1340. [Google Scholar] [CrossRef]
- Wang, L.; Park, P.; Zhang, H.; La Marca, F.; Claeson, A.; Valdivia, J.; Lin, C.Y. BMP-2 inhibits the tumorigenicity of cancer stem cells in human osteosarcoma OS99-1 cell line. Cancer Biol. Ther. 2011, 11, 457–463. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Chen, X.; Qiao, M.; Zhang, B.Q.; Wang, N.; Zhang, Z.; Liao, Z.; Zeng, L.; Deng, Y.; Deng, F.; et al. Bone morphogenetic protein 2 inhibits the proliferation and growth of human colorectal cancer cells. Oncol. Rep. 2014, 32, 1013–1020. [Google Scholar] [CrossRef]
- Beck, S.E.; Jung, B.H.; Fiorino, A.; Gomez, J.; Rosario, E.D.; Cabrera, B.L.; Huang, S.C.; Chow, J.Y.C.; Carethers, J.M. Bone morphogenetic protein signaling and growth suppression in colon cancer. Am. J. Physiol. Liver Physiol. 2006, 291, G135–G145. [Google Scholar] [CrossRef]
- Wang, L.; Park, P.; Zhang, H.; La Marca, F.; Claeson, A.; Than, K.; Rahman, S.; Lin, C.Y. BMP-2 inhibits tumor growth of human renal cell carcinoma and induces bone formation. Int. J. Cancer 2012, 131, 1941–1950. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, X.; Wang, H.; Yan, W.; Zhang, Q.; Chang, X. Bone morphogenetic protein 2 inhibits hepatocellular carcinoma growth and migration through downregulation of the PI3K/AKT pathway. Tumor Biol. 2014, 35, 5189–5198. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Function | Hallmark | Role in Cancer | |
|---|---|---|---|---|
| Diffuse intrinsic pontine glioma | Mutant ACVR1 (including R206H) increases cell proliferation in brainstem progenitors [150,152]. | Proliferative signalling | Oncogene | |
| Mutant forms of ACVR1 lead to increased STAT3 signalling promoting mesenchymal profile and EMT [150]. | Epithelial-mesenchymal transition (EMT) | Oncogene | ||
| Glioblastoma | Overexpression of ACVR1 induced apoptosis in glioma-initiating cells and impaired tumour growth [159]. | Regulation of programmed cell death | Tumour suppressor | |
| Suppression of growth | ||||
| Ovarian cancer | ACVR1-STIP1 interaction promotes human ovarian cancer cell proliferation [163]. | Proliferative signalling | Oncogene | |
| Autocrine BMP9 signalling through ACVR1 promotes ovarian cancer cell proliferation [164]. | Proliferative signalling | Oncogene | ||
| AMH signalling reduces granulosa cell tumour growth, increasing apoptosis through type I ACVR1 and BMPR1A/B [166]. | Regulation of programmed cell death | Tumour suppressor | ||
| AMH inhibits cell proliferation of epithelial ovarian cancer cells [167]. | Proliferative signalling | Tumour suppressor | ||
| AMH inhibits human ovarian cancer cell proliferation [168,169]. | Proliferative signalling | Tumour suppressor | ||
| Prostate cancer | ACVR1-Smad1 phosphorylation upon endoglin interaction promotes cell migration [175]. | Invasion and metastasis | Tumour suppressor | |
| ACVR1 phosphorylation of endoglin promotes cell migration [176]. | Invasion and metastasis | Oncogene | ||
| Multiple myeloma | BMP9 signalling through ACVR1 induces apoptosis and inhibits cell growth [177]. | Proliferative signalling | Tumour suppressor | |
| Regulation of programmed cell death | ||||
| Eye lens tumour | ACVR1 signalling inhibits cell proliferation and impairs tumour growth [179]. | Proliferative signalling | Tumour suppressor | |
| Suppression of growth | ||||
| Erythroleukemia | Overexpression of ACVR1 increases cell proliferation in TF-1 cells [180]. | Proliferative signalling | Oncogene |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valer, J.A.; Sánchez-de-Diego, C.; Pimenta-Lopes, C.; Rosa, J.L.; Ventura, F. ACVR1 Function in Health and Disease. Cells 2019, 8, 1366. https://doi.org/10.3390/cells8111366
Valer JA, Sánchez-de-Diego C, Pimenta-Lopes C, Rosa JL, Ventura F. ACVR1 Function in Health and Disease. Cells. 2019; 8(11):1366. https://doi.org/10.3390/cells8111366
Chicago/Turabian StyleValer, José Antonio, Cristina Sánchez-de-Diego, Carolina Pimenta-Lopes, Jose Luis Rosa, and Francesc Ventura. 2019. "ACVR1 Function in Health and Disease" Cells 8, no. 11: 1366. https://doi.org/10.3390/cells8111366
APA StyleValer, J. A., Sánchez-de-Diego, C., Pimenta-Lopes, C., Rosa, J. L., & Ventura, F. (2019). ACVR1 Function in Health and Disease. Cells, 8(11), 1366. https://doi.org/10.3390/cells8111366