
Invertebrate Retinal Progenitors as Regenerative Models in a Microfluidic System

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Drosophila Fly Stocks
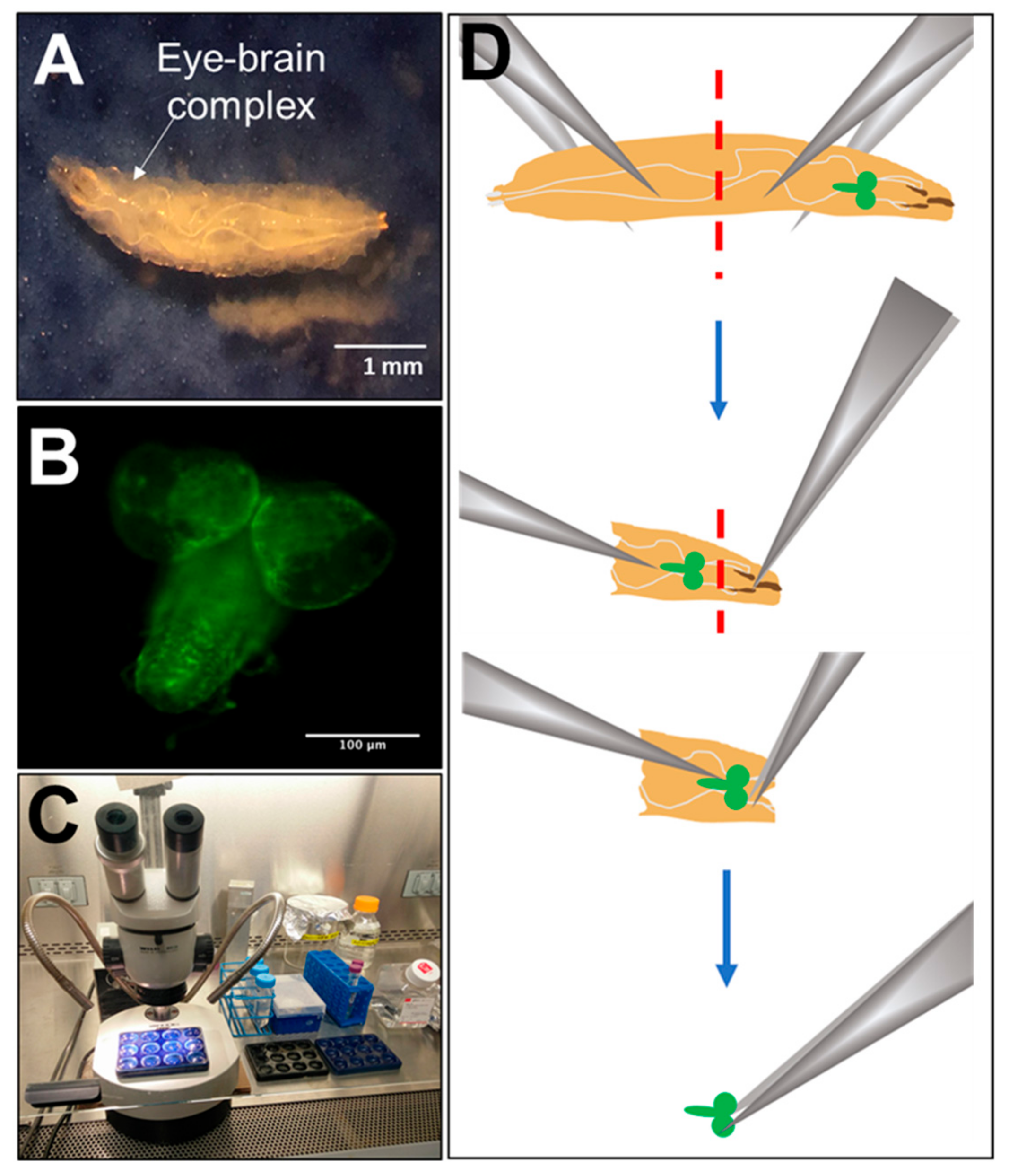
2.2. Dissection, Dissociation and Cell Culture
2.3. Immunocytochemical Assessment of Neuronal and Glial Marker Expression
2.4. Substrate Influence on Cell Survival and Morphology
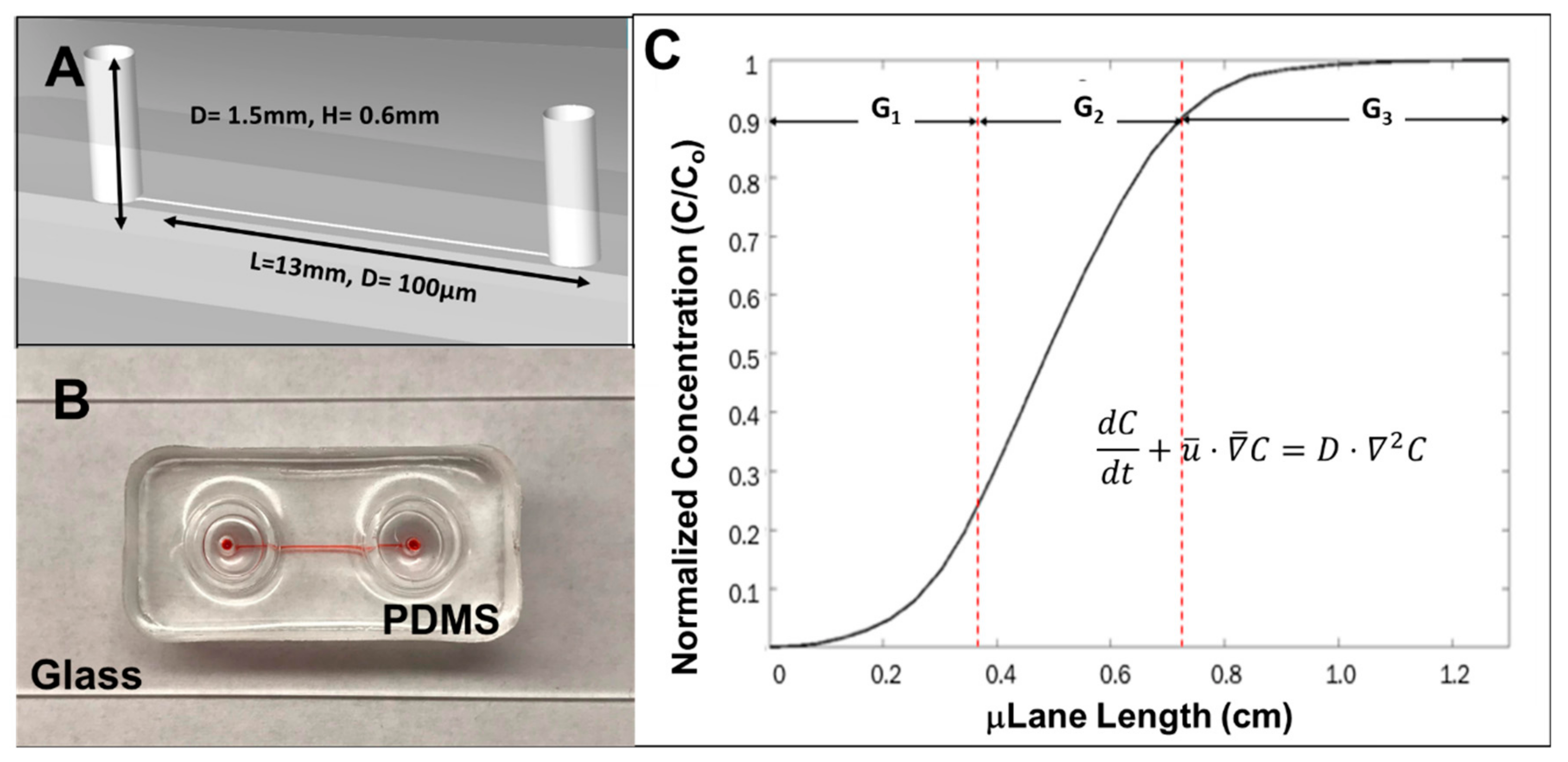
2.5. The μLane Migration Assay
2.6. Microscopy and Imaging
2.7. Data Analysis
2.8. Statistical Tests
3. Results
3.1. Neuron:Glia Distribution
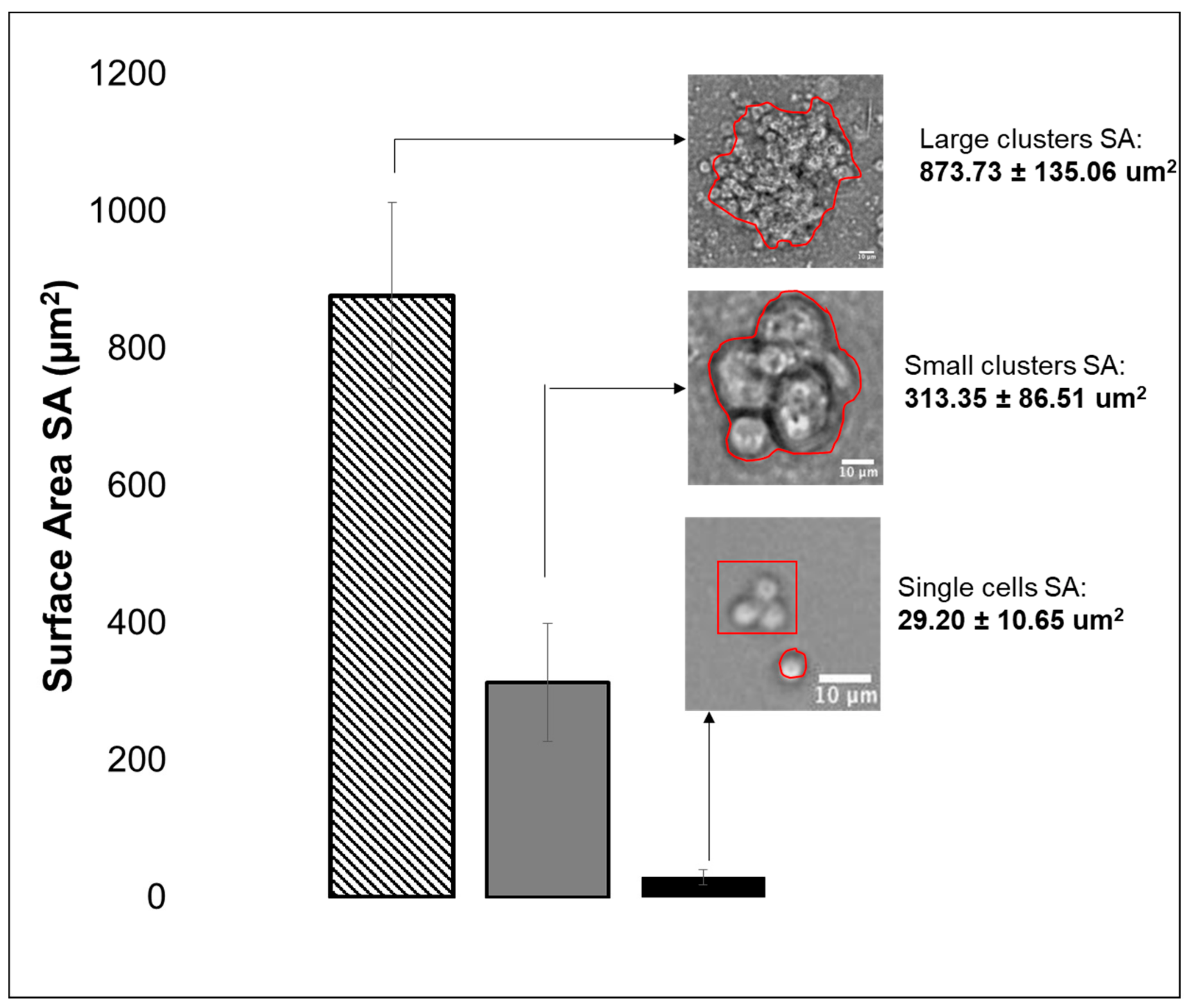
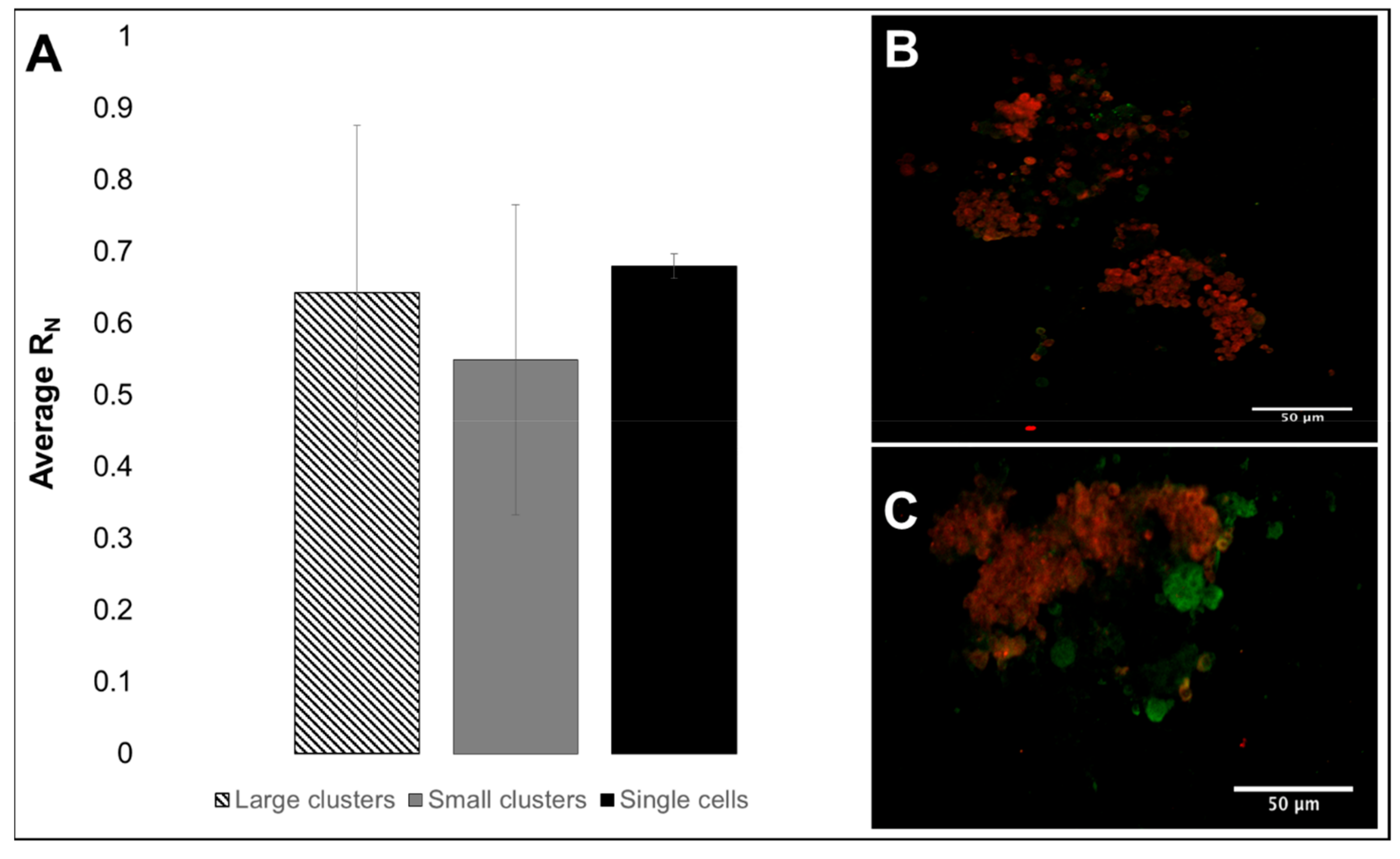
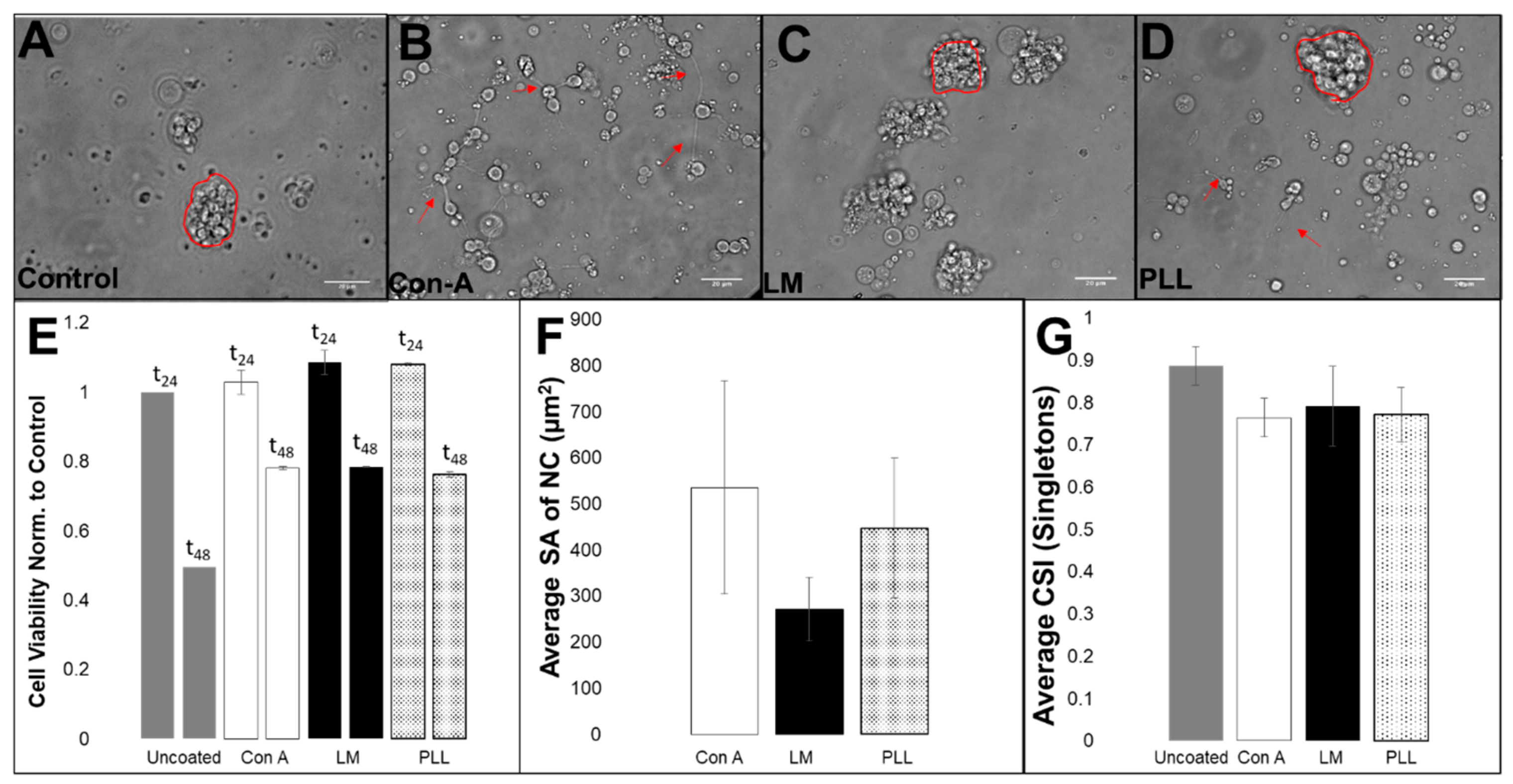
3.2. Viability, Adhesion and Cluster Formation upon Different Substrates
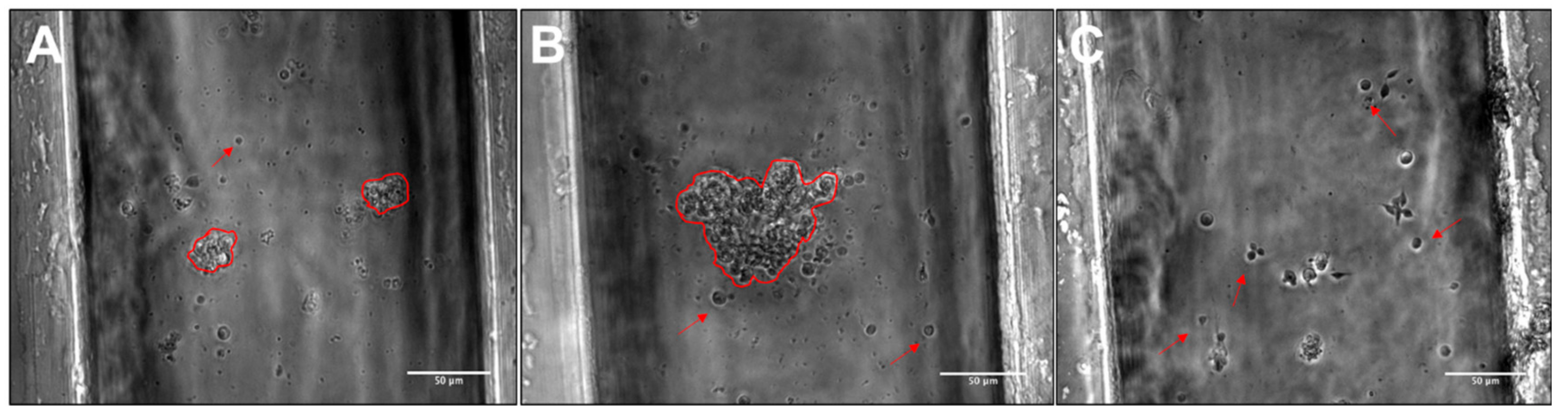
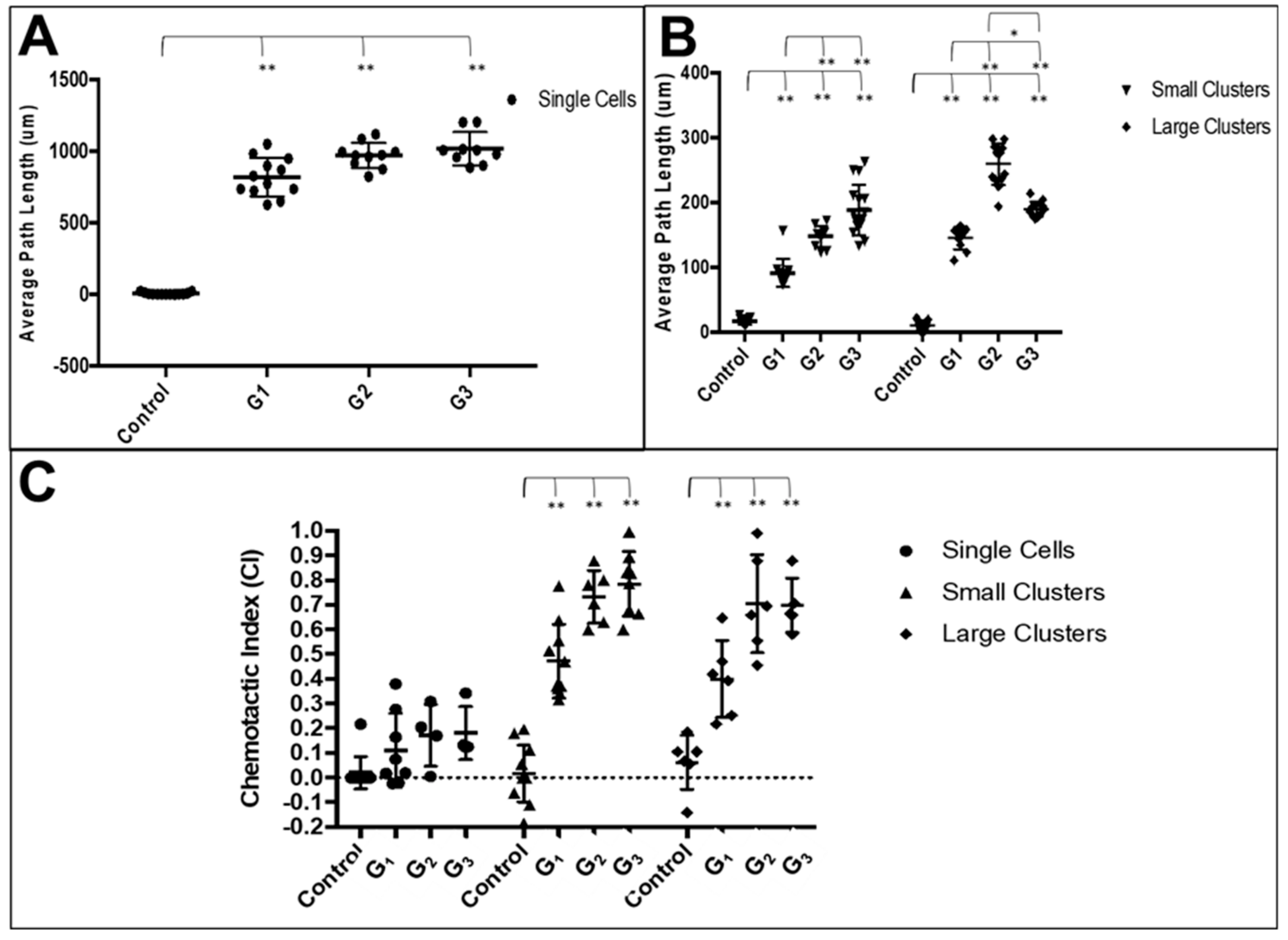
3.3. RPC Chemotactic Migration
4. Discussion
4.1. Invertebrate RPC Models In Vitro
4.2. Ratio of Neuronal:Glial Progenitors
4.3. Collective RPC Interactions with Extracellular Substrates
4.4. Collective RPC Migration
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Scarpa, E.; Mayor, R. Collective cell migration in development. J. Cell Biol. 2016, 212, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Aman, A.; Piotrowski, T. Cell migration during morphogenesis. Dev. Biol. 2010, 341, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Haeger, A.; Wolf, K.; Zegers, M.M.; Friedl, P. Collective cell migration: Guidance principles and hierarchies. Trends Cell Biol. 2015, 25, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Lamba, D.; Karl, M.; Reh, T. Neural regeneration and cell replacement: A view from the eye. Cell Stem Cell 2008, 2, 538–549. [Google Scholar] [CrossRef]
- Macabenta, F.; Stathopoulos, A. Sticking to a plan: Adhesion and signaling control spatial organization of cells within migrating collectives. Curr. Opin. Genet. Dev. 2019, 57, 39–46. [Google Scholar] [CrossRef]
- Norden, C.; Lecaudey, V. Collective cell migration: General themes and new paradigms. Curr. Opin. Genet. Dev. 2019, 57, 54–60. [Google Scholar] [CrossRef]
- Devreotes, P.N.; Bhattacharya, S.; Edwards, M.; PIglesias, A.; Lampert, T.; Miao, Y. Excitable Signal Transduction Networks in Directed Cell Migration. Annu. Rev. Cell Dev. Biol. 2017, 33, 103–125. [Google Scholar] [CrossRef]
- Roca-Cusachs, P.; Sunyer, R.; Trepat, X. Mechanical guidance of cell migration: Lessons from chemotaxis. Curr. Opin. Cell Biol. 2013, 25, 543–549. [Google Scholar] [CrossRef]
- Shellard, A.; Mayor, R. Chemotaxis during neural crest migration. Semin. Cell Dev. Biol. 2016, 55, 111–118. [Google Scholar] [CrossRef]
- Swaney, K.F.; Huang, C.H.; Devreotes, P.N. Eukaryotic chemotaxis: A network of signaling pathways controls motility, directional sensing, and polarity. Annu. Rev. Biophys. 2010, 39, 265–289. [Google Scholar] [CrossRef]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Radice, G.P. Locomotion and cell-substratum contacts of Xenopus epidermal cells in vitro and in situ. J. Cell Sci. 1980, 44, 201–223. [Google Scholar] [PubMed]
- Olson, H.M.; Nechiporuk, A.V. Using Zebrafish to Study Collective Cell Migration in Development and Disease. Front. Cell Dev. Biol. 2018, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019. ePub October 3, 2019. [Google Scholar] [CrossRef]
- Szabo, A.; Mayor, R. Mechanisms of Neural Crest Migration. Annu. Rev. Genet. 2018, 52, 43–63. [Google Scholar] [CrossRef]
- Rorth, P. Collective guidance of collective cell migration. Trends Cell Biol. 2007, 17, 575–579. [Google Scholar] [CrossRef]
- Ho, M.T.; Teal, C.J.; Shoichet, M.S. A hyaluronan/methylcellulose-based hydrogel for local cell and biomolecule delivery to the central nervous system. Brain Res. Bull. 2019, 148, 46–54. [Google Scholar] [CrossRef]
- Orive, G.; Santos-Vizcaino, E.; Pedraz, J.L.; Hernandez, R.M.; Ramirez, J.E.V.; Dolatshahi-Pirouz, A.; Khademhosseini, A.; Peppas, N.A.; Emerich, D.F. 3D cell-laden polymers to release bioactive products in the eye. Prog. Retin. Eye Res. 2019, 68, 67–82. [Google Scholar] [CrossRef]
- Hunt, N.C.; Hallam, D.; Chichagova, V.; Steel, D.H.; Lako, M. The Application of Biomaterials to Tissue Engineering Neural Retina and Retinal Pigment Epithelium. Adv. Healthc. Mater. 2018, 7, e1800226. [Google Scholar] [CrossRef]
- Warre-Cornish, K.; Barber, A.C.; Sowden, J.C.; Ali, R.R.; Pearson, R.A. Migration, integration and maturation of photoreceptor precursors following transplantation in the mouse retina. Stem Cells Dev. 2014, 23, 941–954. [Google Scholar] [CrossRef]
- Barber, A.C.; Hippert, C.; Duran, Y.; West, E.L.; Bainbridge, J.W.; Warre-Cornish, K.; Luhmann, U.F.; Lakowski, J.; Sowden, J.C.; Ali, R.R.; et al. Repair of the degenerate retina by photoreceptor transplantation. Proc. Natl. Acad. Sci. USA 2013, 110, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Reh, T.A. Photoreceptor Transplantation in Late Stage Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, ORSFg1–ORSFg7. [Google Scholar] [CrossRef] [PubMed]
- Unachukwu, U.J.; Warren, A.; Li, Z.; Mishra, S.; Zhou, J.; Sauane, M.; Lim, H.; Vazquez, M.; Redenti, S. Predicted molecular signaling guiding photoreceptor cell migration following transplantation into damaged retina. Sci. Rep. 2016, 6, 22392. [Google Scholar] [CrossRef] [PubMed]
- Sonnen, K.F.; Merten, C.A. Microfluidics as an Emerging Precision Tool in Developmental Biology. Dev. Cell 2019, 48, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Millet, L.J.; Gillette, M.U. New perspectives on neuronal development via microfluidic environments. Trends Neurosci. 2012, 35, 752–761. [Google Scholar] [CrossRef]
- Liu, Z.; Han, X.; Qin, L. Recent Progress of Microfluidics in Translational Applications. Adv. Healthc. Mater. 2016, 5, 871–888. [Google Scholar] [CrossRef]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef]
- Gupta, N.; Renugopalakrishnan, V.; Liepmann, D.; Paulmurugan, R.; Malhotra, B.D. Cell-based biosensors: Recent trends, challenges and future perspectives. Biosens. Bioelectron. 2019, 141, 111435. [Google Scholar] [CrossRef]
- Lynch, M.; Ramalingam, N. Integrated Fluidic Circuits for Single-Cell Omics and Multi-omics Applications. Adv. Exp. Med. Biol. 2019, 1129, 19–26. [Google Scholar]
- Kaushik, G.; Leijten, J.; Khademhosseini, A. Concise Review: Organ Engineering: Design, Technology, and Integration. Stem Cells 2017, 35, 51–60. [Google Scholar] [CrossRef]
- Song, M.J.; Bharti, K. Looking into the future: Using induced pluripotent stem cells to build two and three dimensional ocular tissue for cell therapy and disease modeling. Brain Res. 2016, 1638, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Developmentally inspired human ‘organs on chips’. Development 2018, 145, 156125. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Mishra, S.; Pena, J.; Zhou, J.; Redenti, S.; Majeska, R.; Vazquez, M. Collective adhesion and displacement of retinal progenitor cells upon extracellular matrix substrates of transplantable biomaterials. J. Tissue Eng. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Thakur, A.; Redenti, S.; Vazquez, M. A model microfluidics-based system for the human and mouse retina. Biomed. Microdevices 2015, 17, 107. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Bahrami, S.; Mirshekari, H.; Basri, S.M.; Nik, A.B.; Aref, A.R.; Akbari, M.; Hamblin, M.R. Microfluidic systems for stem cell-based neural tissue engineering. Lab Chip 2016, 16, 2551–2571. [Google Scholar] [CrossRef] [PubMed]
- Uzel, S.G.; Amadi, O.C.; Pearl, T.M.; Lee, R.T.; So, P.T.; Kamm, R.D. Simultaneous or Sequential Orthogonal Gradient Formation in a 3D Cell Culture Microfluidic Platform. Small 2016, 12, 612–622. [Google Scholar] [CrossRef]
- Nery, F.C.; Atai, N.A.; da Hora, C.C.; Kim, E.Y.; Hettich, J.; Mempel, T.R.; Breakefield, X.O.; Irimia, D. Microfluidic platform to evaluate migration of cells from patients with DYT1 dystonia. J. Neurosci. Methods 2014, 232, 181–188. [Google Scholar] [CrossRef]
- Reyes, D.R.; Perruccio, E.M.; Becerra, S.P.; Locascio, L.E.; Gaitan, M. Micropatterning neuronal cells on polyelectrolyte multilayers. Langmuir 2004, 20, 8805–8811. [Google Scholar] [CrossRef]
- Fleck, O.; Savin, T. A physical approach to model occlusions in the retinal microvasculature. Eye Lond. 2018, 32, 189–194. [Google Scholar] [CrossRef]
- Terrell, D.; Comander, J. Current Stem-Cell Approaches for the Treatment of Inherited Retinal Degenerations. Semin. Ophthalmol. 2019, 34, 287–292. [Google Scholar] [CrossRef]
- Gagliardi, G.; M’Barek, K.B.; Goureau, O. Photoreceptor cell replacement in macular degeneration and retinitis pigmentosa: A pluripotent stem cell-based approach. Prog. Retin. Eye Res. 2019, 71, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Mishra, S.; Singh, S.; Pei, M.; Gulyas, B.; Padmanabhan, P. Engineering Concepts in Stem Cell Research. Biotechnol. J. 2017, 12, 1700066. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Baranov, P.; Aydin, A.; Abdelgawad, H.; Singh, D.; Niu, W.; Kurisawa, M.; Spector, M.; Young, M.J. In Situ Cross-linking Hydrogel as a Vehicle for Retinal Progenitor Cell Transplantation. Cell Transplant. 2019, 28, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuzzani, O.; Binette, F.; Sternberg, H.; West, M.D.; Nasonkin, I.O. Pluripotent Stem Cells for Retinal Tissue Engineering: Current Status and Future Prospects. Stem Cell Rev. Rep. 2018, 14, 463–483. [Google Scholar] [CrossRef]
- Yao, J.; Ko, C.W.; Baranov, P.Y.; Regatieri, C.V.; Redenti, S.; Tucker, B.A.; Mighty, J.; Tao, S.L.; Young, M.J. Enhanced differentiation and delivery of mouse retinal progenitor cells using a micropatterned biodegradable thin-film polycaprolactone scaffold. Tissue Eng. Part A 2015, 21, 1247–1260. [Google Scholar] [CrossRef]
- Stern, J.H.; Tian, Y.; Funderburgh, J.; Pellegrini, G.; Zhang, K.; Goldberg, J.L.; Ali, R.R.; Young, M.; Xie, Y.; Temple, S. Regenerating Eye Tissues to Preserve and Restore Vision. Cell Stem Cell 2018, 22, 834–849. [Google Scholar] [CrossRef]
- Volkner, M.; Kurth, T.; Karl, M.O. The Mouse Retinal Organoid Trisection Recipe: Efficient Generation of 3D Retinal Tissue from Mouse Embryonic Stem Cells. Methods Mol. Biol. 2019, 1834, 119–141. [Google Scholar]
- Fligor, C.M.; Langer, K.B.; Sridhar, A.; Ren, Y.; Shields, P.K.; Edler, M.C.; Ohlemacher, S.K.; Sluch, V.M.; Zack, D.J.; Zhang, C.; et al. Three-Dimensional Retinal Organoids Facilitate the Investigation of Retinal Ganglion Cell Development, Organization and Neurite Outgrowth from Human Pluripotent Stem Cells. Sci. Rep. 2018, 8, 14520. [Google Scholar] [CrossRef]
- Kumar, J.P. The fly eye: Through the looking glass. Dev. Dyn. 2018, 247, 111–123. [Google Scholar] [CrossRef]
- Contreras, E.G.; Sierralta, J.; Oliva, C. Novel Strategies for the Generation of Neuronal Diversity: Lessons From the Fly Visual System. Front. Mol. Neurosci. 2019, 12, 140. [Google Scholar] [CrossRef]
- Gaspar, P.; Almudi, I.; Nunes, M.D.S.; McGregor, A.P. Human eye conditions: Insights from the fly eye. Hum. Genet. 2019, 138, 973–991. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Singh, T.; Farooqi, A.; Venkatesh, T.; Vazquez, M. Controlled microfluidics to examine growth-factor induced migration of neural progenitors in the Drosophila visual system. J. Neurosci. Methods 2016, 262, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Soderstrom, K.O. Lectin binding to the human retina. Anat. Rec. 1988, 220, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Harvey, A.R.; Dyson, S.E. Regrowth of lesioned retinal axons associated with the transplantation of Schwann cells to the brachial region of the rat optic tract. Restor. Neurol. Neurosci. 1991, 2, 233–248. [Google Scholar]
- Guo, Y.; Wang, P.; Ma, J.H.; Cui, Z.; Yu, Q.; Liu, S.; Xue, Y.; Zhu, D.; Cao, J.; Li, Z.; et al. Modeling Retinitis Pigmentosa: Retinal Organoids Generated From the iPSCs of a Patient With the USH2A Mutation Show Early Developmental Abnormalities. Front. Cell Neurosci. 2019, 13, 361. [Google Scholar] [CrossRef]
- Huang, P.; Stern, M.J. FGF signaling in flies and worms: More and more relevant to vertebrate biology. Cytokine Growth Factor Rev. 2005, 16, 151–158. [Google Scholar] [CrossRef]
- Bae, Y.K.; Trisnadi, N.; Kadam, S.; Stathopoulos, A. The role of FGF signaling in guiding coordinate movement of cell groups: Guidance cue and cell adhesion regulator? Cell Adhes. Migr. 2012, 6, 397–403. [Google Scholar] [CrossRef]
- Duffy, J.B. GAL4 system in Drosophila: A fly geneticist’s Swiss army knife. Genesis 2002, 34, 1–15. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Yoshida, H. Drosophila as a Model Organism. Adv. Exp. Med. Biol. 2018, 1076, 1–10. [Google Scholar]
- Lerit, D.A.; Plevock, K.M.; Rusan, N.M. Live imaging of Drosophila larval neuroblasts. J. Vis. Exp. 2014, 89, e51756. [Google Scholar]
- Moraru, M.M.; Egger, B.; Bao, D.B.; Sprecher, S.G. Analysis of cell identity, morphology, apoptosis and mitotic activity in a primary neural cell culture system in Drosophila. Neural Dev. 2012, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.F.; Suzuki, N.; Poo, M.M. Dissociated neurons from normal and mutant Drosophila larval central nervous system in cell culture. J. Neurosci. 1983, 3, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Schneider, I. Cell lines derived from late embryonic stages of Drosophila melanogaster. J. Embryol. Exp. Morphol. 1972, 27, 353–365. [Google Scholar] [PubMed]
- Luhur, A.; Klueg, K.M.; Zelhof, A.C. Generating and working with Drosophila cell cultures: Current challenges and opportunities. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e339. [Google Scholar] [CrossRef]
- Kong, Q.; Able, R.A., Jr.; Dudu, V.; Vazquez, M. A microfluidic device to establish concentration gradients using reagent density differences. J. Biomech. Eng. 2010, 132, 121012. [Google Scholar] [CrossRef]
- Able, R.A., Jr.; Ngnabeuye, C.; Beck, C.; Holland, E.C.; Vazquez, M. Low Concentration Microenvironments Enhance the Migration of Neonatal Cells of Glial Lineage. Cell. Mol Bioeng. 2012, 5, 128–142. [Google Scholar] [CrossRef][Green Version]
- Dudu, V.; Able, R.A., Jr.; Rotari, V.; Kong, Q.; Vazquez, M. Role of Epidermal Growth Factor-Triggered PI3K/Akt Signaling in the Migration of Medulloblastoma-Derived Cells. Cell. Mol. Bioeng. 2012, 5, 413–502. [Google Scholar] [CrossRef]
- Rico-Varela, J.; Singh, T.; McCutcheon, S.; Vazquez, M. EGF as a New Therapeutic Target for Medulloblastoma Metastasis. Cell. Mol. Bioeng. 2015, 8, 553–565. [Google Scholar] [CrossRef]
- Jones, T.A. Skewness and kurtosis as criteria of normality in observed frequency distributions. J. Sediment. Res. 1969, 39, 1622–1627. [Google Scholar] [CrossRef]
- George, D. SPSS for Windows Step by Step: A Simple Study Guide and Reference, 17.0 Update, 10/e; Pearson Education: Bengaluru, India, 2011. [Google Scholar]
- Jarque, C.M.; Bera, A.K. A Test for Normality of Observations and Regression Rezsiduals. International Statistical Review/Revue Internationale de Statistiqu 1987, 55, 163–172. [Google Scholar]
- Reese, B.E. Development of the retina and optic pathway. Vision Res. 2011, 51, 613–632. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Kumar, N.; Velagala, V.; Zartman, J.J. Tools to reverse-engineer multicellular systems: Case studies using the fruit fly. J. Biol. Eng. 2019, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Cagan, R. Principles of Drosophila eye differentiation. Curr. Top. Dev. Biol. 2009, 89, 115–135. [Google Scholar] [PubMed]
- Hsiung, F.; Moses, K. Retinal development in Drosophila: Specifying the first neuron. Hum. Mol. Genet. 2002, 11, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Irvine, K.D. Drosophila as a model for understanding development and disease. Dev. Dyn. 2012, 241, 1–2. [Google Scholar] [CrossRef]
- Jones, M.K.; Lu, B.; Girman, S.; Wang, S. Cell-based therapeutic strategies for replacement and preservation in retinal degenerative diseases. Prog. Retin. Eye Res. 2017, 58, 1–27. [Google Scholar] [CrossRef]
- Delgado, R.; Delgado, M.G.; Bastin-Heline, L.; Glavic, A.; O’Day, P.M.; Bacigalupo, J. Light-Induced Opening of the TRP Channel in Isolated Membrane Patches Excised from Photosensitive Microvilli from Drosophila Photoreceptors. Neuroscience 2019, 396, 66–72. [Google Scholar] [CrossRef]
- Fan, A.; Tofangchi, A.; de Venecia, M.; Saif, T. A simple microfluidic platform for the partial treatment of insuspendable tissue samples with orientation control. Lab Chip 2018, 18, 735–742. [Google Scholar] [CrossRef]
- Van Giesen, L.; Neagu-Maier, G.L.; Kwon, J.Y.; Sprecher, S.G. A microfluidics-based method for measuring neuronal activity in Drosophila chemosensory neurons. Nat. Protoc. 2016, 11, 2389–2400. [Google Scholar] [CrossRef]
- Mishra, B.; Ghannad-Rezaie, M.; Li, J.; Wang, X.; Hao, Y.; Ye, B.; Chronis, N.; Collins, C.A. Using microfluidics chips for live imaging and study of injury responses in Drosophila larvae. J. Vis. Exp. 2014, 84, e50998. [Google Scholar] [CrossRef]
- Dolph, P.; Nair, A.; Raghu, P. Preparation of dissociated ommatidia from Drosophila. Cold Spring Harb. Protoc. 2011, 2011, pdb–prot5550. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, S.; Unachukwu, U.; Thakur, A.; Majeska, R.; Redenti, S.; Vazquez, M. In vitro formation of neuroclusters in microfluidic devices and cell migration as a function of stromal-derived growth factor 1 gradients. Cell Adhes. Migr. 2017, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Redenti, S.M.; Jiang, C.; Swift, J.S.; Klassen, H.J.; Smith, M.E.; Wnek, G.E.; Young, M.J. The use of progenitor cell/biodegradable MMP2-PLGA polymer constructs to enhance cellular integration and retinal repopulation. Biomaterials 2010, 31, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ballios, B.G.; Cooke, M.J.; van der Kooy, D.; Shoichet, M.S. A hydrogel-based stem cell delivery system to treat retinal degenerative diseases. Biomaterials 2010, 31, 2555–2564. [Google Scholar] [CrossRef] [PubMed]
- Chiba, C.; Sakai, H.; Kaneko, Y.; Saito, T. Concanavalin A Promotes Regeneration of Processes of Isolated Ganglion Cells from the Adult Newt Retina. Zool Sci 1995, 12, 741–747. [Google Scholar] [CrossRef]
- Suzuki, T.; Mandai, M.; Akimoto, M.; Yoshimura, N.; Takahashi, M. The simultaneous treatment of MMP-2 stimulants in retinal transplantation enhances grafted cell migration into the host retina. Stem Cells 2006, 24, 2406–2411. [Google Scholar] [CrossRef]
- Erdiakov, A.K.; Tikhonovich, M.V.; Rzhavina, E.M.; Gavrilova, S.A. The Characteristics of Retina at the Development of Proliferative Vitreoretinopathy in Rats after Intraocular Injection of Concanavalin a and Dispase. Ross. Fiziol. Zhurnal Im. IM Sechenova 2015, 101, 572–585. [Google Scholar]
- Tikhonovich, M.V.; Erdiakov, A.K.; Gavrilova, S.A. Nonsteroid anti-inflammatory therapy suppresses the development of proliferative vitreoretinopathy more effectively than a steroid one. Int. Ophthalmol. 2018, 38, 1365–1378. [Google Scholar] [CrossRef]
- Ishikawa, M.; Watanabe, H.; Koike, Y.; Hisatomi, O.; Tokunaga, F.; Tonosaki, A. Demonstration by lectin cytochemistry of rod and cone photoreceptors in the lamprey retina. Cell Tissue Res. 1989, 256, 227–232. [Google Scholar] [CrossRef]
- Sasaki, H.; Karasawa, K.; Hironaka, K.; Tahara, K.; Tozuka, Y.; Takeuchi, H. Retinal drug delivery using eyedrop preparations of poly-L-lysine-modified liposomes. Eur. J. Pharm. Biopharm. 2013, 83, 364–369. [Google Scholar] [CrossRef]
- Du, J.; Sun, Y.; Li, F.H.; Du, L.F.; Duan, Y.R. Enhanced delivery of biodegradable mPEG-PLGA-PLL nanoparticles loading Cy3-labelled PDGF-BB siRNA by UTMD to rat retina. J. Biosci. 2017, 42, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Tezcaner, A.; Hicks, D.; Boulmedais, F.; Sahel, J.; Schaaf, P.; Voegel, J.C.; Lavalle, P. Polyelectrolyte multilayer films as substrates for photoreceptor cells. Biomacromolecules 2006, 7, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Inatani, M.; Honjo, M.; Otori, Y.; Oohira, A.; Kido, N.; Tano, Y.; Honda, Y.; Tanihara, H. Inhibitory effects of neurocan and phosphacan on neurite outgrowth from retinal ganglion cells in culture. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1930–1938. [Google Scholar]
- Varshney, S.; Hunter, D.D.; Brunken, W.J. Extracellular Matrix Components Regulate Cellular Polarity and Tissue Structure in the Developing and Mature Retina. J. Ophthalmic Vis. Res. 2015, 10, 329–339. [Google Scholar] [PubMed]
- Assawachananont, J.; Mandai, M.; Okamoto, S.; Yamada, C.; Eiraku, M.; Yonemura, S.; Sasai, Y.; Takahashi, M. Transplantation of embryonic and induced pluripotent stem cell-derived 3D retinal sheets into retinal degenerative mice. Stem Cell Rep. 2014, 2, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, Y.; Shoichet, M.S. The role of endothelial cells in the retinal stem and progenitor cell niche within a 3D engineered hydrogel matrix. Biomaterials 2012, 33, 5198–5205. [Google Scholar] [CrossRef] [PubMed]
- Sheibani, N.; Wang, S.; Darjatmoko, S.R.; Fisk, D.L.; Shahi, P.K.; Pattnaik, B.R.; Sorenson, C.M.; Bhowmick, R.; Volpert, O.V.; Albert, D.M.; et al. Novel anti-angiogenic PEDF-derived small peptides mitigate choroidal neovascularization. Exp. Eye Res. 2019, 188, 107798. [Google Scholar] [CrossRef]
- Napolitano, F.; di Iorio, V.; di Iorio, G.; Melone, M.A.B.; Gianfrancesco, F.; Simonelli, F.; Esposito, T.; Testa, F.; Sampaolo, S. Early posterior vitreous detachment is associated with LAMA5 dominant mutation. Ophthalmic Genet. 2019, 40, 39–42. [Google Scholar] [CrossRef]
- Shen, D.; Wen, R.; Tuo, J.; Bojanowski, C.M.; Chan, C.C. Exacerbation of retinal degeneration and choroidal neovascularization induced by subretinal injection of Matrigel in CCL2/MCP-1-deficient mice. Ophthalmic Res. 2006, 38, 71–73. [Google Scholar] [CrossRef]
- Camley, B.A.; Zimmermann, J.; Levine, H.; Rappel, W.J. Collective Signal Processing in Cluster Chemotaxis: Roles of Adaptation, Amplification, and Co-attraction in Collective Guidance. PLoS Comput. Biol. 2016, 12, e1005008. [Google Scholar] [CrossRef]
- Delplace, V.; Ortin-Martinez, A.; Tsai, E.L.S.; Amin, A.N.; Wallace, V.; Shoichet, M.S. Controlled release strategy designed for intravitreal protein delivery to the retina. J. Control. Release 2019, 293, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.S.; Wong, C.C.; Chan, B.P.; Lo, A.C. Sustained Delivery of Bioactive GDNF from Collagen and Alginate-Based Cell-Encapsulating Gel Promoted Photoreceptor Survival in an Inherited Retinal Degeneration Model. PLoS ONE 2016, 11, e0159342. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.; Mugler, A.; Brennan, M.D.; Lee, S.H.; Huebner, R.J.; Shamir, E.R.; Woo, L.A.; Kim, J.; Amar, P.; Nemenman, I.; et al. Cell-cell communication enhances the capacity of cell ensembles to sense shallow gradients during morphogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E679–E688. [Google Scholar] [CrossRef] [PubMed]
- Dumstrei, K.; Wang, F.; Hartenstein, V. Role of DE-cadherin in neuroblast proliferation, neural morphogenesis, and axon tract formation in Drosophila larval brain development. J. Neurosci. 2003, 23, 3325–3335. [Google Scholar] [CrossRef] [PubMed]
- Hill, E.; Broadbent, I.D.; Chothia, C.; Pettitt, J. Cadherin superfamily proteins in Caenorhabditis elegans and Drosophila melanogaster. J. Mol. Biol. 2001, 305, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.; Hoch, M. Drosophila eye size is determined by Innexin 2-dependent Decapentaplegic signalling. Dev. Biol. 2015, 408, 26–40. [Google Scholar] [CrossRef]
- Lee, C.H.; Herman, T.; Clandinin, T.R.; Lee, R.; Zipursky, S.L. N-cadherin regulates target specificity in the Drosophila visual system. Neuron 2001, 30, 437–450. [Google Scholar] [CrossRef]
- Bauer, R.; Weimbs, A.; Lechner, H.; Hoch, M. DE-cadherin, a core component of the adherens junction complex modifies subcellular localization of the Drosophila gap junction protein innexin2. Cell Commun. Adhes. 2006, 13, 103–114. [Google Scholar] [CrossRef]
- Welzel, G.; Schuster, S. Long-term potentiation in an innexin-based electrical synapse. Sci. Rep. 2018, 8, 12579. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eye-Brain Complexes | Total # of Cells | Average # of Cells per Brain |
|---|---|---|
| N = 15 | 1.4 ± 0.09 × 105 | 9.2 ± 0.8 × 103 |
| N = 30 | 2.9 ± 0.5 × 105 | 9.9 ± 1.7 × 103 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pena, C.D.; Zhang, S.; Majeska, R.; Venkatesh, T.; Vazquez, M. Invertebrate Retinal Progenitors as Regenerative Models in a Microfluidic System. Cells 2019, 8, 1301. https://doi.org/10.3390/cells8101301
Pena CD, Zhang S, Majeska R, Venkatesh T, Vazquez M. Invertebrate Retinal Progenitors as Regenerative Models in a Microfluidic System. Cells. 2019; 8(10):1301. https://doi.org/10.3390/cells8101301
Chicago/Turabian StylePena, Caroline D., Stephanie Zhang, Robert Majeska, Tadmiri Venkatesh, and Maribel Vazquez. 2019. "Invertebrate Retinal Progenitors as Regenerative Models in a Microfluidic System" Cells 8, no. 10: 1301. https://doi.org/10.3390/cells8101301
APA StylePena, C. D., Zhang, S., Majeska, R., Venkatesh, T., & Vazquez, M. (2019). Invertebrate Retinal Progenitors as Regenerative Models in a Microfluidic System. Cells, 8(10), 1301. https://doi.org/10.3390/cells8101301