Opposite Effects of Moderate and Extreme Cx43 Deficiency in Conditional Cx43-Deficient Mice on Angiotensin II-Induced Cardiac Fibrosis


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Experimental Protocol
2.2. Systolic Cardiac Function by Transthoracic Echocardiography
2.3. Collagen Content and Cardiomyocyte Size
2.4. Immunohistochemistry
2.5. Real Time-PCR
2.6. Total Myocardial Homogenates
2.7. Gelatin Zymography
2.8. Cardiac Fibroblasts Isolation
2.9. Western Blot Analysis
2.10. Statistics
3. Results
3.1. AngII Treatment Induces a Similar Hypertrophic Response in Hearts from Cx43fl/fl and Cx43Cre-ER(T)/fl Mice Independently of Cx43 Levels
3.2. AngII Treatment Differentially Modulates Cardiac Fibrosis in Cx43Cre-ER(T)/fl Mice
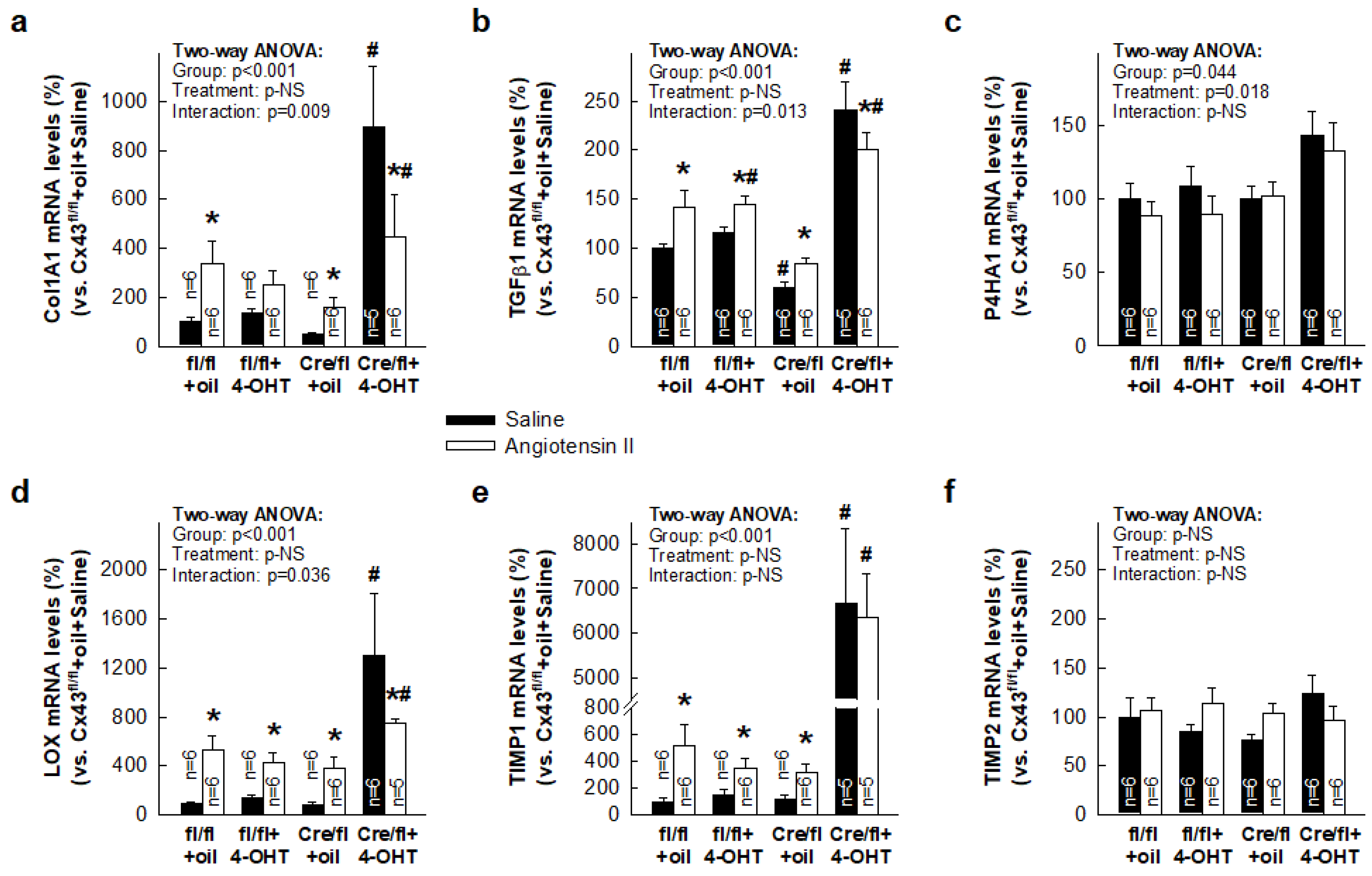
3.3. Paradoxical Overexpression of mRNAs Coding for Proteins Involved in Collagen Turnover in Cx43-Deficient Mice
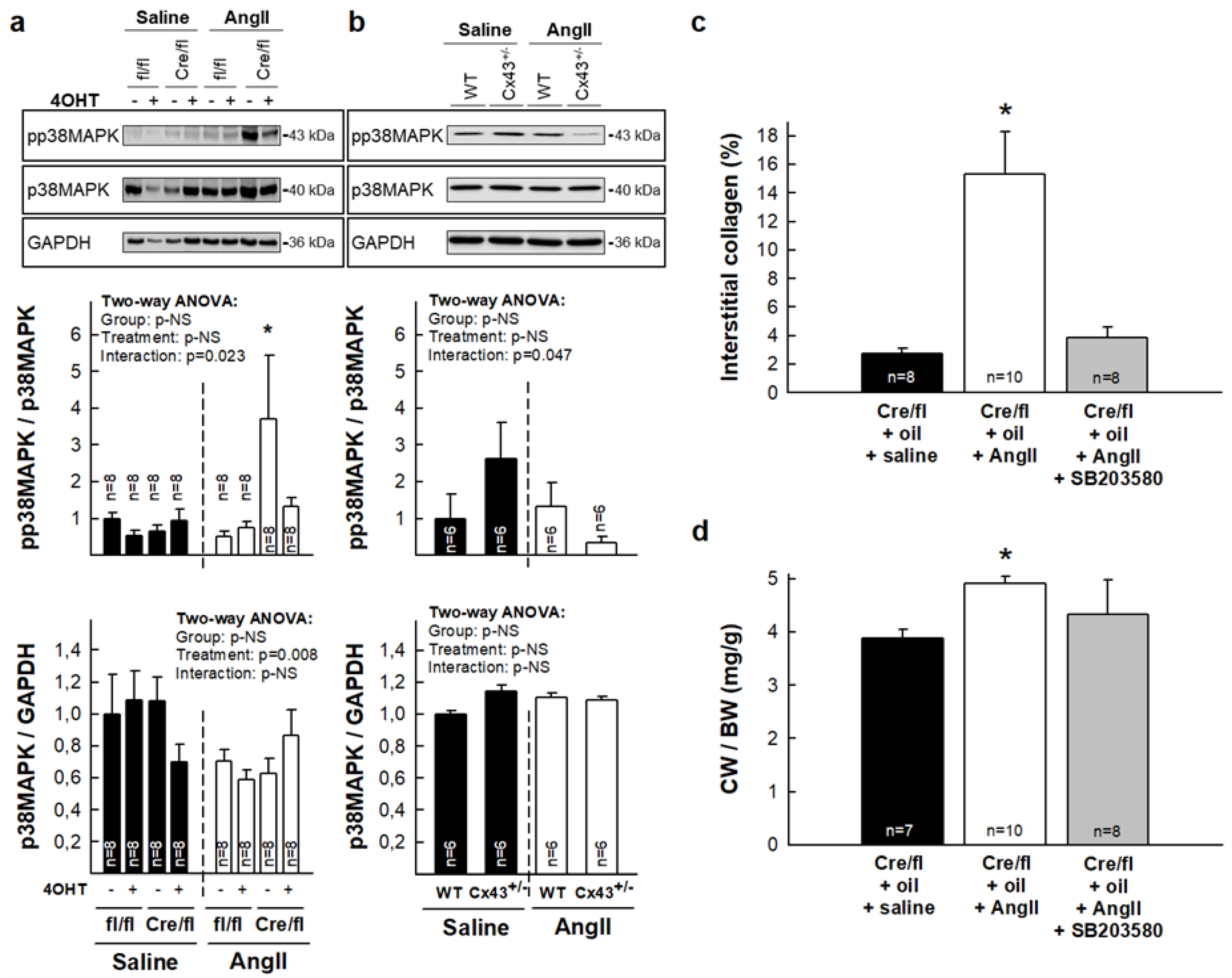
3.4. Enhanced Collagen Deposition in Response to AngII in Hearts from Partially Deficient Cx43Cre-ER(T)/fl Mice Correlates with Increased p38 MAPK Activation
3.5. Normalization of Collagen Content in Hearts from Cx43Cre-ER(T)/fl Mice Injected with 4-OHT After AngII Treatment is Associated with Increased MMP-9 Activity
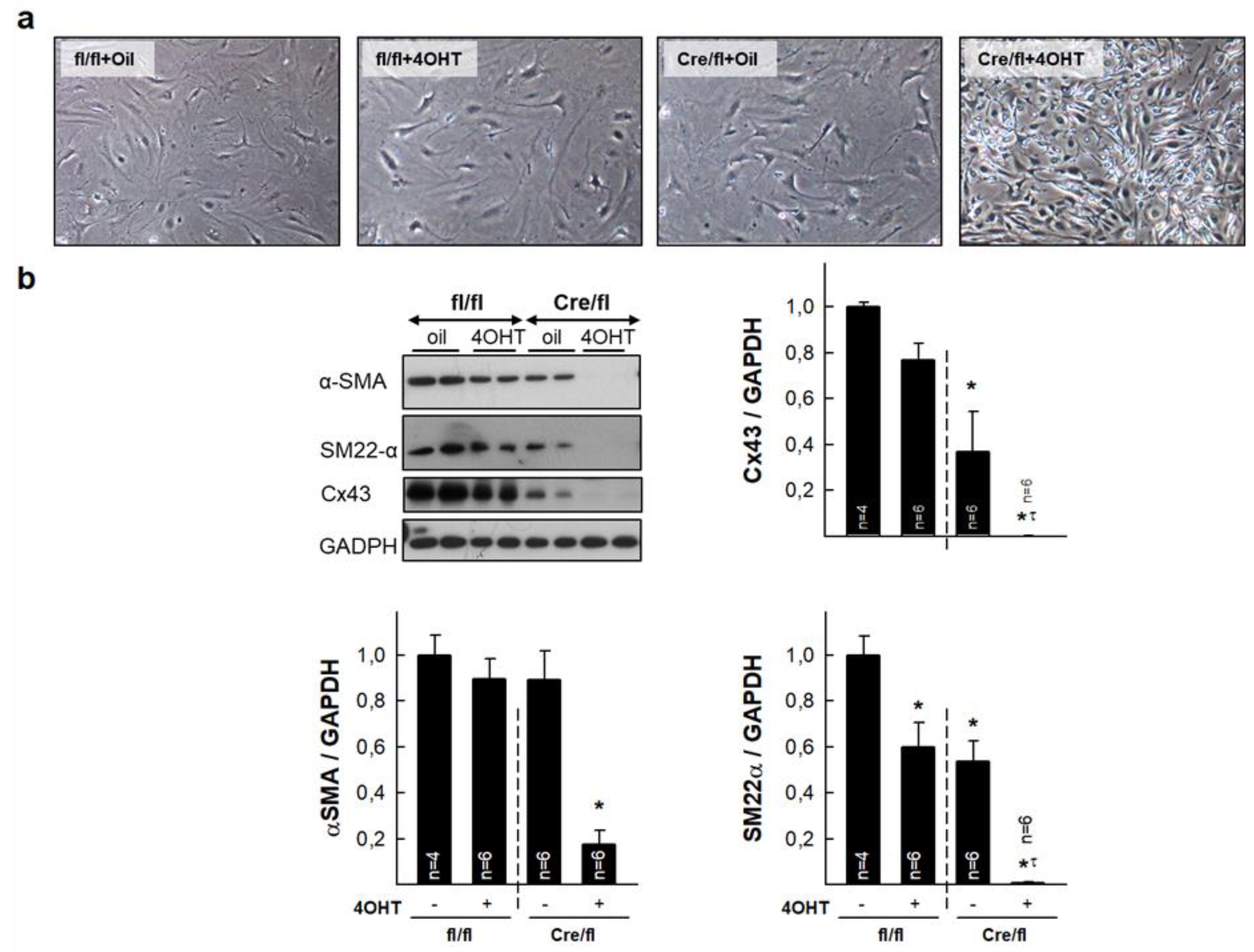
3.6. Isolated Cardiac Fibroblasts from Highly Deficient Cx43Cre-ER(T)/fl Mice Have Altered Phenotype and Differentiation Capacity
3.7. Deletion of Cx43 in Cx43Cre-ER(T)/fl Mice Injected with 4-OHT is Associated with Enhanced Cardiac Expression of Inflammatory Markers
4. Discussion
Ethical Statement
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sosinsky, G.E.; Nicholson, B.J. Structural organization of gap junction channels. Biochim. Biophys. Acta 2005, 1711, 99–125. [Google Scholar] [CrossRef] [PubMed]
- Severs, N.J. The cardiac gap junction and intercalated disc. Int. J. Cardiol. 1990, 26, 137–173. [Google Scholar] [CrossRef]
- Kleber, A.G.; Saffitz, J.E. Role of the intercalated disc in cardiac propagation and arrhythmogenesis. Front. Physiol. 2014, 5, 404. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.S.; Nygaard, A.L.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar] [PubMed]
- Ongstad, E.; Kohl, P. Fibroblast-myocyte coupling in the heart: Potential relevance for therapeutic interventions. J. Mol. Cell. Cardiol. 2016, 91, 238–246. [Google Scholar] [CrossRef]
- Lambiase, P.D.; Tinker, A. Connexins in the heart. Cell Tissue Res. 2015, 360, 675–684. [Google Scholar] [CrossRef]
- Sanchez, J.A.; Rodriguez-Sinovas, A.; Fernandez-Sanz, C.; Ruiz-Meana, M.; Garcia-Dorado, D. Effects of a reduction in the number of gap junction channels or in their conductance on ischemia-reperfusion arrhythmias in isolated mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2442–H2453. [Google Scholar] [CrossRef][Green Version]
- Rodriguez-Sinovas, A.; Cabestrero, A.; Lopez, D.; Torre, I.; Morente, M.; Abellan, A.; Miro, E.; Ruiz-Meana, M.; Garcia-Dorado, D. The modulatory effects of connexin 43 on cell death/survival beyond cell coupling. Prog. Biophys. Mol. Biol. 2007, 94, 219–232. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Rodriguez-Sinovas, A.; Ruiz-Meana, M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovasc. Res. 2004, 61, 386–401. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Inserte, J.; Ruiz-Meana, M.; Gonzalez, M.A.; Solares, J.; Julia, M.; Barrabes, J.A.; Soler-Soler, J. Gap junction uncoupler heptanol prevents cell-to-cell progression of hypercontracture and limits necrosis during myocardial reperfusion. Circulation 1997, 96, 3579–3586. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A.; Sanchez, J.A.; Gonzalez-Loyola, A.; Barba, I.; Morente, M.; Aguilar, R.; Agullo, E.; Miro-Casas, E.; Esquerda, N.; Ruiz-Meana, M.; et al. Effects of substitution of Cx43 by Cx32 on myocardial energy metabolism, tolerance to ischemia and preconditioning protection. J. Physiol. 2010, 588, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.A.; Rodriguez-Sinovas, A.; Barba, I.; Miro-Casas, E.; Fernandez-Sanz, C.; Ruiz-Meana, M.; Alburquerque-Bejar, J.J.; Garcia-Dorado, D. Activation of RISK and SAFE pathways is not involved in the effects of Cx43 deficiency on tolerance to ischemia-reperfusion injury and preconditioning protection. Basic Res. Cardiol. 2013, 108, 351. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Coutinho, P.; Frank, S.; Franke, S.; Law, L.Y.; Martin, P.; Green, C.R.; Becker, D.L. Targeting connexin43 expression accelerates the rate of wound repair. Curr. Biol. 2003, 13, 1697–1703. [Google Scholar] [CrossRef]
- Elbadawy, H.M.; Mirabelli, P.; Xeroudaki, M.; Parekh, M.; Bertolin, M.; Breda, C.; Cagini, C.; Ponzin, D.; Lagali, N.; Ferrari, S. Effect of connexin 43 inhibition by the mimetic peptide Gap27 on corneal wound healing, inflammation and neovascularization. Br. J. Pharmacol. 2016, 173, 2880–2893. [Google Scholar] [CrossRef]
- Crespo, Y.S.; da Silva, T.C.; Pereira, I.V.A.; Willebrords, J.; Maes, M.; Sayuri, N.M.; de Castro, I.A.; Leclercq, I.; Romualdo, G.R.; Barbisan, L.F.; et al. TAT-Gap19 and Carbenoxolone Alleviate Liver Fibrosis in Mice. Int. J. Mol. Sci. 2018, 19, 817. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Kovacs, A.; Kanter, E.M.; Yamada, K.A. Reduced expression of Cx43 attenuates ventricular remodeling after myocardial infarction via impaired TGF-beta signaling. Am. J. Physiol. Heart Circ Physiol. 2010, 298, H477–H487. [Google Scholar] [CrossRef]
- Kanno, S.; Kovacs, A.; Yamada, K.A.; Saffitz, J.E. Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J. Am. Coll. Cardiol. 2003, 41, 681–686. [Google Scholar] [CrossRef]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef]
- Ongstad, E.L.; O’Quinn, M.P.; Ghatnekar, G.S.; Yost, M.J.; Gourdie, R.G. A Connexin43 Mimetic Peptide Promotes Regenerative Healing and Improves Mechanical Properties in Skin and Heart. Adv. Wound Care 2013, 2, 55–62. [Google Scholar] [CrossRef]
- Jansen, J.A.; van Veen, T.A.; de Jong, S.; van der, N.R.; van Stuijvenberg, L.; Driessen, H.; Labzowski, R.; Oefner, C.M.; Bosch, A.A.; Nguyen, T.Q.; et al. Reduced cx43 expression triggers increased fibrosis due to enhanced fibroblast activity. Circ. Arrhythm. Electrophysiol. 2012, 5, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, D.; Theis, M.; Degen, J.; Ott, T.; van Rijen, H.V.; Kirchhoff, S.; Kim, J.S.; de Bakker, J.M.; Willecke, K. Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J. Mol. Cell. Cardiol. 2004, 36, 101–110. [Google Scholar] [CrossRef] [PubMed]
- van Rijen, H.V.; Eckardt, D.; Degen, J.; Theis, M.; Ott, T.; Willecke, K.; Jongsma, H.J.; Opthof, T.; de Bakker, J.M. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 2004, 109, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Galan, M.; Varona, S.; Guadall, A.; Orriols, M.; Navas, M.; Aguilo, S.; de Diego, A.; Navarro, M.A.; Garcia-Dorado, D.; Rodriguez-Sinovas, A.; et al. Lysyl oxidase overexpression accelerates cardiac remodeling and aggravates angiotensin II-induced hypertrophy. FASEB J. 2017, 31, 3787–3799. [Google Scholar] [CrossRef]
- Poncelas, M.; Inserte, J.; Aluja, D.; Hernando, V.; Vilardosa, U.; Garcia-Dorado, D. Delayed, oral pharmacological inhibition of calpains attenuates adverse post-infarction remodelling. Cardiovasc. Res. 2017, 113, 950–961. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, R.; Ferran, B.; Alonso, J.; Marti-Pamies, I.; Aguilo, S.; Calvayrac, O.; Rodriguez, C.; Martinez-Gonzalez, J. NR4A receptors up-regulate the antiproteinase alpha-2 macroglobulin (A2M) and modulate MMP-2 and MMP-9 in vascular smooth muscle cells. Thromb. Haemost. 2015, 113, 1323–1334. [Google Scholar] [CrossRef]
- Boengler, K.; Dodoni, G.; Rodriguez-Sinovas, A.; Cabestrero, A.; Ruiz-Meana, M.; Gres, P.; Konietzka, I.; Lopez-Iglesias, C.; Garcia-Dorado, D.; Di Lisa, F.; et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Touyz, R.M.; Schiffrin, E.L. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 2000, 52, 639–672. [Google Scholar]
- Kim, S.; Iwao, H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol. Rev. 2000, 52, 11–34. [Google Scholar]
- Tsushima, K.; Osawa, T.; Yanai, H.; Nakajima, A.; Takaoka, A.; Manabe, I.; Ohba, Y.; Imai, Y.; Taniguchi, T.; Nagai, R. IRF3 regulates cardiac fibrosis but not hypertrophy in mice during angiotensin II-induced hypertension. FASEB J. 2011, 25, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef] [PubMed]
- Ghatnekar, G.S.; O’Quinn, M.P.; Jourdan, L.J.; Gurjarpadhye, A.A.; Draughn, R.L.; Gourdie, R.G. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 2009, 4, 205–223. [Google Scholar] [CrossRef]
- Gilmartin, D.J.; Soon, A.; Thrasivoulou, C.; Phillips, A.R.; Jayasinghe, S.N.; Becker, D.L. Sustained Release of Cx43 Antisense Oligodeoxynucleotides from Coated Collagen Scaffolds Promotes Wound Healing. Adv. Healthc. Mater. 2016, 5, 1786–1799. [Google Scholar] [CrossRef]
- Montgomery, J.; Ghatnekar, G.S.; Grek, C.L.; Moyer, K.E.; Gourdie, R.G. Connexin 43-Based Therapeutics for Dermal Wound Healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef]
- Cogliati, B.; Vinken, M.; Silva, T.C.; Araujo, C.M.; Aloia, T.P.; Chaible, L.M.; Mori, C.M.; Dagli, M.L. Connexin 43 deficiency accelerates skin wound healing and extracellular matrix remodeling in mice. J. Dermatol. Sci. 2015, 79, 50–56. [Google Scholar] [CrossRef]
- Ghatnekar, G.S.; Grek, C.L.; Armstrong, D.G.; Desai, S.C.; Gourdie, R.G. The effect of a connexin43-based Peptide on the healing of chronic venous leg ulcers: A multicenter, randomized trial. J. Investig. Dermatol. 2015, 135, 289–298. [Google Scholar] [CrossRef]
- Bivi, N.; Nelson, M.T.; Faillace, M.E.; Li, J.; Miller, L.M.; Plotkin, L.I. Deletion of Cx43 from osteocytes results in defective bone material properties but does not decrease extrinsic strength in cortical bone. Calcif. Tissue Int. 2012, 91, 215–224. [Google Scholar] [CrossRef]
- Santiago, J.J.; Dangerfield, A.L.; Rattan, S.G.; Bathe, K.L.; Cunnington, R.H.; Raizman, J.E.; Bedosky, K.M.; Freed, D.H.; Kardami, E.; Dixon, I.M. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: Expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev. Dyn. 2010, 239, 1573–1584. [Google Scholar] [CrossRef]
- Fatigati, V.; Murphy, R.A. Actin and tropomyosin variants in smooth muscles. Dependence on tissue type. J. Biol. Chem. 1984, 259, 14383–14388. [Google Scholar] [PubMed]
- Spinale, F.G. Matrix metalloproteinases: Regulation and dysregulation in the failing heart. Circ. Res. 2002, 90, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Metalloproteinase production from macrophages—A perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp. Physiol. 2016, 101, 1327–1337. [Google Scholar] [CrossRef]
- Doble, B.W.; Dang, X.; Ping, P.; Fandrich, R.R.; Nickel, B.E.; Jin, Y.; Cattini, P.A.; Kardami, E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J. Cell Sci. 2004, 117, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Doble, B.W.; Kardami, E. The carboxy-tail of connexin-43 localizes to the nucleus and inhibits cell growth. Mol. Cell. Biochem. 2003, 242, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Salat-Canela, C.; Sese, M.; Peula, C.; Cajal, S.; Aasen, T. Internal translation of the connexin 43 transcript. Cell Commun. Signal. 2014, 12, 31. [Google Scholar] [CrossRef]
- Smyth, J.W.; Shaw, R.M. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013, 5, 611–618. [Google Scholar] [CrossRef]
- Kardami, E.; Dang, X.; Iacobas, D.A.; Nickel, B.E.; Jeyaraman, M.; Srisakuldee, W.; Makazan, J.; Tanguy, S.; Spray, D.C. The role of connexins in controlling cell growth and gene expression. Prog. Biophys. Mol. Biol. 2007, 94, 245–264. [Google Scholar] [CrossRef]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambon, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valls-Lacalle, L.; Negre-Pujol, C.; Rodríguez, C.; Varona, S.; Valera-Cañellas, A.; Consegal, M.; Martínez-González, J.; Rodríguez-Sinovas, A. Opposite Effects of Moderate and Extreme Cx43 Deficiency in Conditional Cx43-Deficient Mice on Angiotensin II-Induced Cardiac Fibrosis. Cells 2019, 8, 1299. https://doi.org/10.3390/cells8101299
Valls-Lacalle L, Negre-Pujol C, Rodríguez C, Varona S, Valera-Cañellas A, Consegal M, Martínez-González J, Rodríguez-Sinovas A. Opposite Effects of Moderate and Extreme Cx43 Deficiency in Conditional Cx43-Deficient Mice on Angiotensin II-Induced Cardiac Fibrosis. Cells. 2019; 8(10):1299. https://doi.org/10.3390/cells8101299
Chicago/Turabian StyleValls-Lacalle, Laura, Corall Negre-Pujol, Cristina Rodríguez, Saray Varona, Antoni Valera-Cañellas, Marta Consegal, Jose Martínez-González, and Antonio Rodríguez-Sinovas. 2019. "Opposite Effects of Moderate and Extreme Cx43 Deficiency in Conditional Cx43-Deficient Mice on Angiotensin II-Induced Cardiac Fibrosis" Cells 8, no. 10: 1299. https://doi.org/10.3390/cells8101299
APA StyleValls-Lacalle, L., Negre-Pujol, C., Rodríguez, C., Varona, S., Valera-Cañellas, A., Consegal, M., Martínez-González, J., & Rodríguez-Sinovas, A. (2019). Opposite Effects of Moderate and Extreme Cx43 Deficiency in Conditional Cx43-Deficient Mice on Angiotensin II-Induced Cardiac Fibrosis. Cells, 8(10), 1299. https://doi.org/10.3390/cells8101299