Blocking LFA-1 Aggravates Cardiac Inflammation in Experimental Autoimmune Myocarditis


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. EAM Model
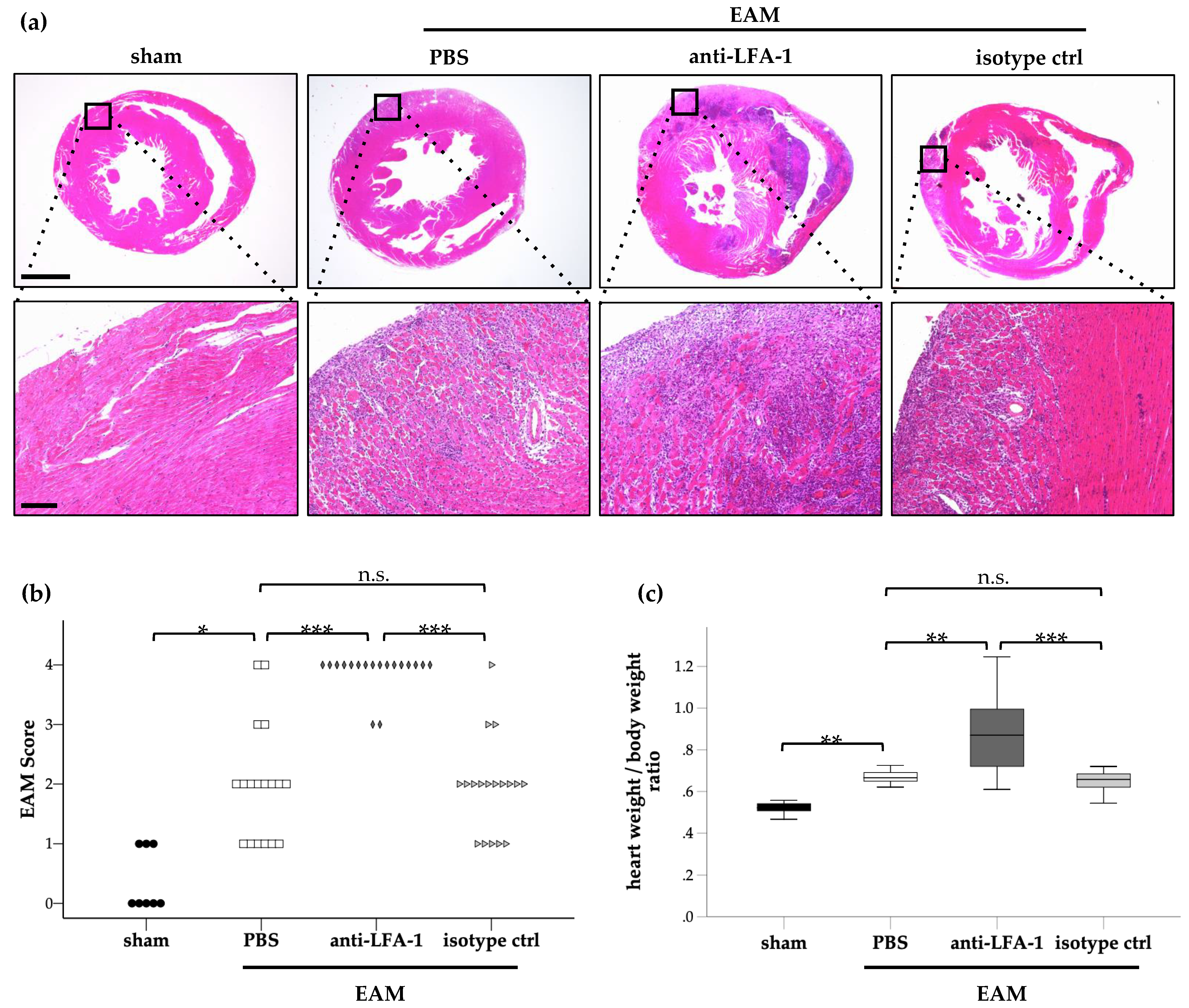
2.3. Histology and Heart Weight/Body Weight Ratio
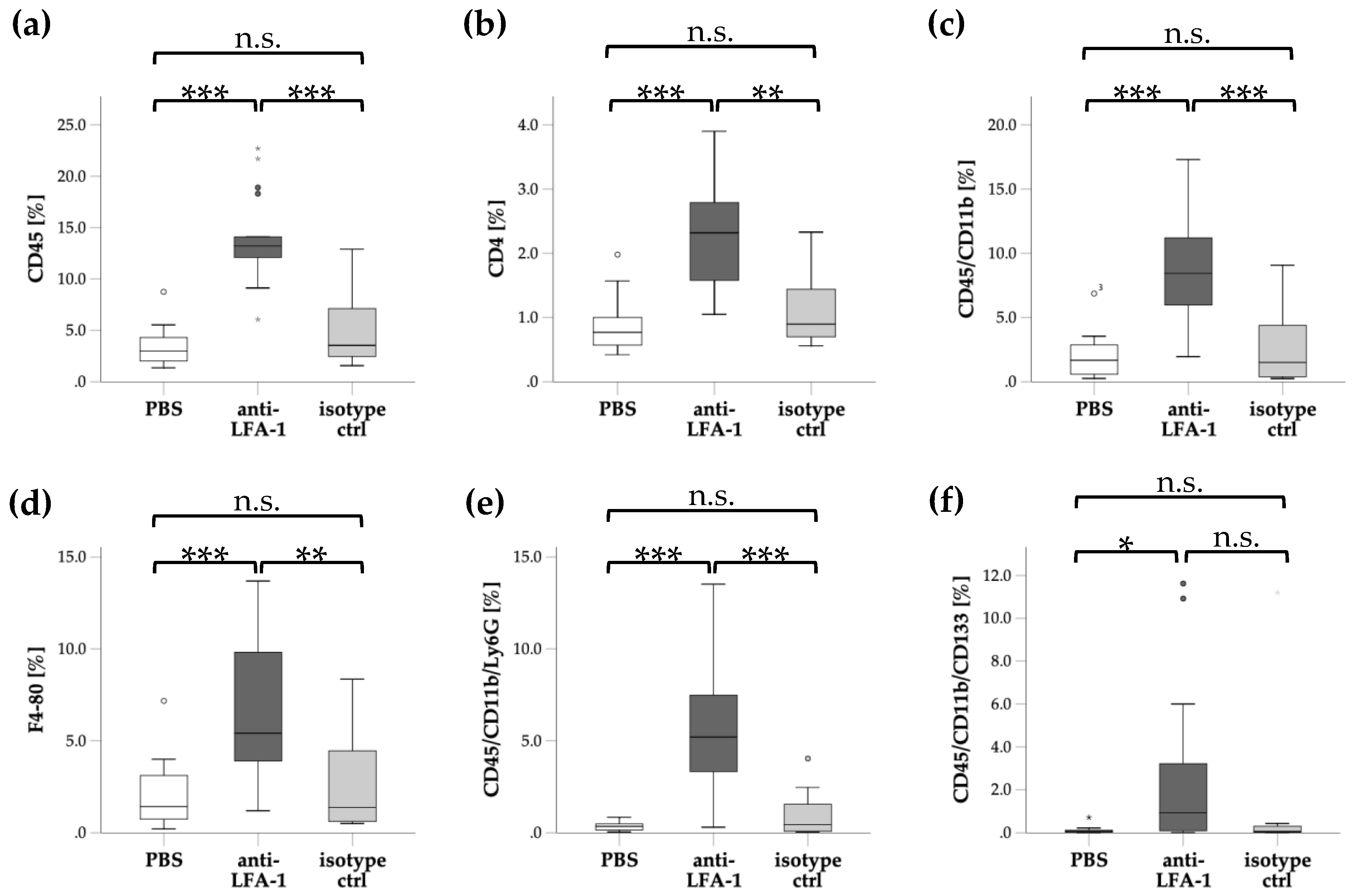
2.4. Flow Cytometry
2.5. Statistical Analysis
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maisch, B.; Richter, A.; Sandmoller, A.; Portig, I.; Pankuweit, S.; Network, B.M.-H.F. Inflammatory dilated cardiomyopathy (DCMI). Herz 2005, 30, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, L.T.; Grabmaier, U.; Uhl, A.; Gess, S.; Boehm, F.; Zehrer, A.; Pick, R.; Salvermoser, M.; Czermak, T.; Pircher, J.; et al. Midkine drives cardiac inflammation by promoting neutrophil trafficking and NETosis in myocarditis. J. Exp. Med. 2019, 216, 350–368. [Google Scholar] [CrossRef] [PubMed]
- Neu, N.; Klieber, R.; Fruhwirth, M.; Berger, P. Cardiac myosin-induced myocarditis as a model of postinfectious autoimmunity. Eur. Heart J. 1991, 12, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, L.T.; Gola, A.; Winkelmann, M.; Jakob, S.M.; Groesser, L.; Borgolte, J.; Pogoda, F.; Pick, R.; Pruenster, M.; Muller-Hocker, J.; et al. The cytokine midkine supports neutrophil trafficking during acute inflammation by promoting adhesion via beta2 integrins (CD11/CD18). Blood 2014, 123, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Kelleher, D. An Introduction to LFA-1/ICAM-1 Interactions in T-Cell Motility. Methods Mol. Biol. 2019, 1930, 1–9. [Google Scholar] [PubMed]
- Lebwohl, M.; Tyring, S.K.; Hamilton, T.K.; Toth, D.; Glazer, S.; Tawfik, N.H.; Walicke, P.; Dummer, W.; Wang, X.; Garovoy, M.R.; et al. A novel targeted T-cell modulator, efalizumab, for plaque psoriasis. N. Engl. J. Med. 2003, 349, 2004–2013. [Google Scholar] [CrossRef] [PubMed]
- Gultner, S.; Kuhlmann, T.; Hesse, A.; Weber, J.P.; Riemer, C.; Baier, M.; Hutloff, A. Reduced Treg frequency in LFA-1-deficient mice allows enhanced T effector differentiation and pathology in EAE. Eur. J. Immunol. 2010, 40, 3403–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, J.; Leach, W.; Pippig, S.; Joshi, A.; Wu, B.; House, R.; Beyer, J. Evaluation of a surrogate antibody for preclinical safety testing of an anti-CD11a monoclonal antibody. Regul Toxicol Pharm. 2004, 40, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Kania, G.; Blyszczuk, P.; Valaperti, A.; Dieterle, T.; Leimenstoll, B.; Dirnhofer, S.; Zulewski, H.; Eriksson, U. Prominin-1+/CD133+ bone marrow-derived heart-resident cells suppress experimental autoimmune myocarditis. Cardiovasc. Res. 2008, 80, 236–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, R.B.; Lim, L.H.; Tessier, P.A.; Gavins, F.N.; Mathies, M.; Perretti, M.; Hogg, N. The use of lymphocyte function-associated antigen (LFA)-1-deficient mice to determine the role of LFA-1, Mac-1, and alpha4 integrin in the inflammatory response of neutrophils. J. Exp. Med. 2001, 194, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Frommhold, D.; Kamphues, A.; Dannenberg, S.; Buschmann, K.; Zablotskaya, V.; Tschada, R.; Lange-Sperandio, B.; Nawroth, P.P.; Poeschl, J.; Bierhaus, A.; et al. RAGE and ICAM-1 differentially control leukocyte recruitment during acute inflammation in a stimulus-dependent manner. BMC Immunol. 2011, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Walling, B.L.; Kim, M. LFA-1 in T Cell Migration and Differentiation. Front. Immunol. 2018, 9, 952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothhammer, V.; Heink, S.; Petermann, F.; Srivastava, R.; Claussen, M.C.; Hemmer, B.; Korn, T. Th17 lymphocytes traffic to the central nervous system independently of alpha4 integrin expression during EAE. J. Exp. Med. 2011, 208, 2465–2476. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Eriksson, U.; Lehtonen, J.; Cooper, L.T., Jr. The Quest for New Approaches in Myocarditis and Inflammatory Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2348–2364. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, M.; Mauermann, N.; Marty, R.R.; Dirnhofer, S.; Kurrer, M.O.; Komnenovic, V.; Penninger, J.M.; Eriksson, U. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J. Exp. Med. 2006, 203, 2009–2019. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weckbach, L.T.; Uhl, A.; Boehm, F.; Seitelberger, V.; Huber, B.C.; Kania, G.; Brunner, S.; Grabmaier, U. Blocking LFA-1 Aggravates Cardiac Inflammation in Experimental Autoimmune Myocarditis. Cells 2019, 8, 1267. https://doi.org/10.3390/cells8101267
Weckbach LT, Uhl A, Boehm F, Seitelberger V, Huber BC, Kania G, Brunner S, Grabmaier U. Blocking LFA-1 Aggravates Cardiac Inflammation in Experimental Autoimmune Myocarditis. Cells. 2019; 8(10):1267. https://doi.org/10.3390/cells8101267
Chicago/Turabian StyleWeckbach, Ludwig T., Andreas Uhl, Felicitas Boehm, Valentina Seitelberger, Bruno C. Huber, Gabriela Kania, Stefan Brunner, and Ulrich Grabmaier. 2019. "Blocking LFA-1 Aggravates Cardiac Inflammation in Experimental Autoimmune Myocarditis" Cells 8, no. 10: 1267. https://doi.org/10.3390/cells8101267
APA StyleWeckbach, L. T., Uhl, A., Boehm, F., Seitelberger, V., Huber, B. C., Kania, G., Brunner, S., & Grabmaier, U. (2019). Blocking LFA-1 Aggravates Cardiac Inflammation in Experimental Autoimmune Myocarditis. Cells, 8(10), 1267. https://doi.org/10.3390/cells8101267