Mitochondrial Metabolism in Major Neurological Diseases


Abstract
1. General Mitochondrial Function
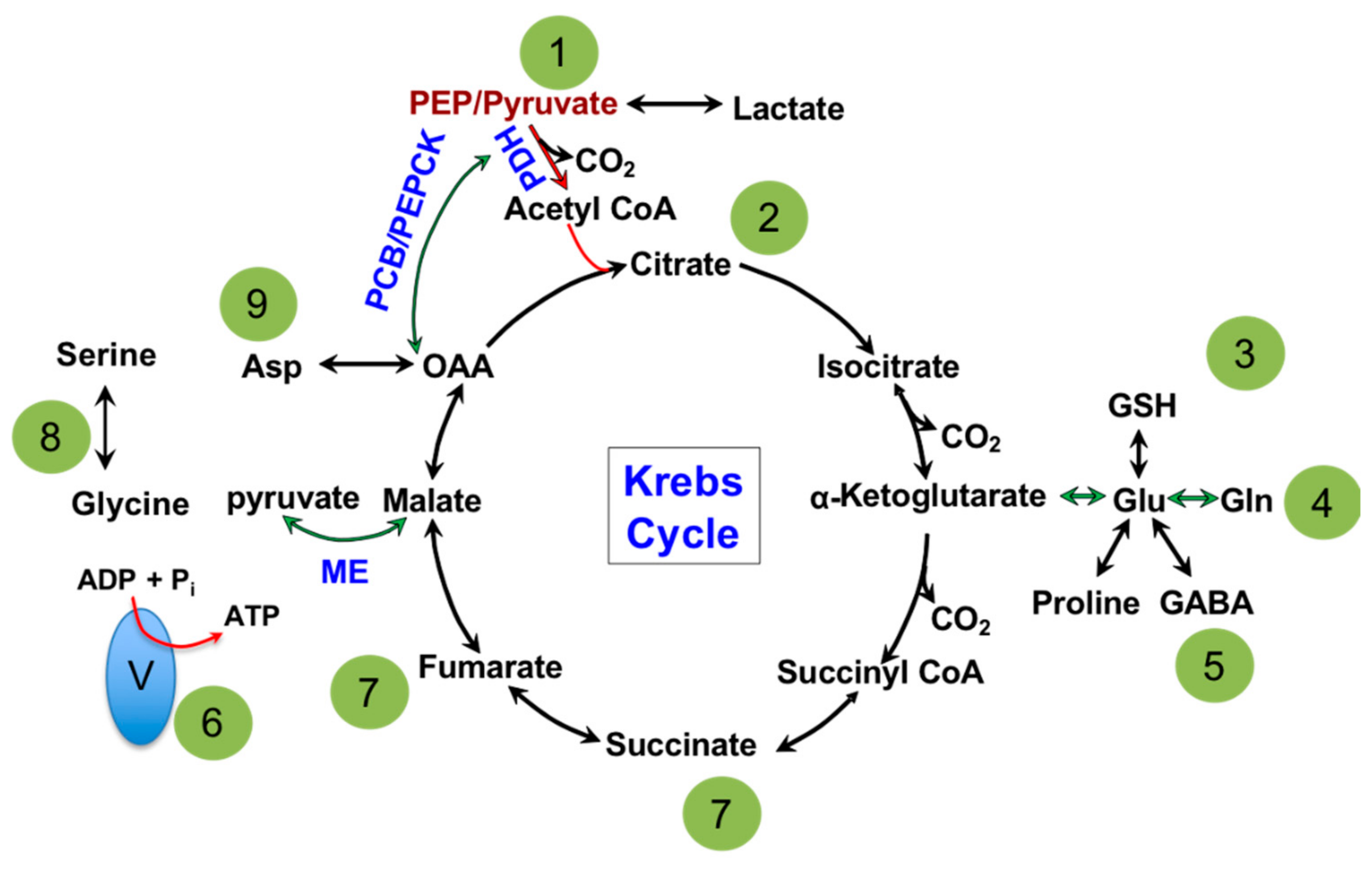
1.1. Mitochondria: Metabolic Hub of a Cell
1.2. Mitochondria and Energy Production
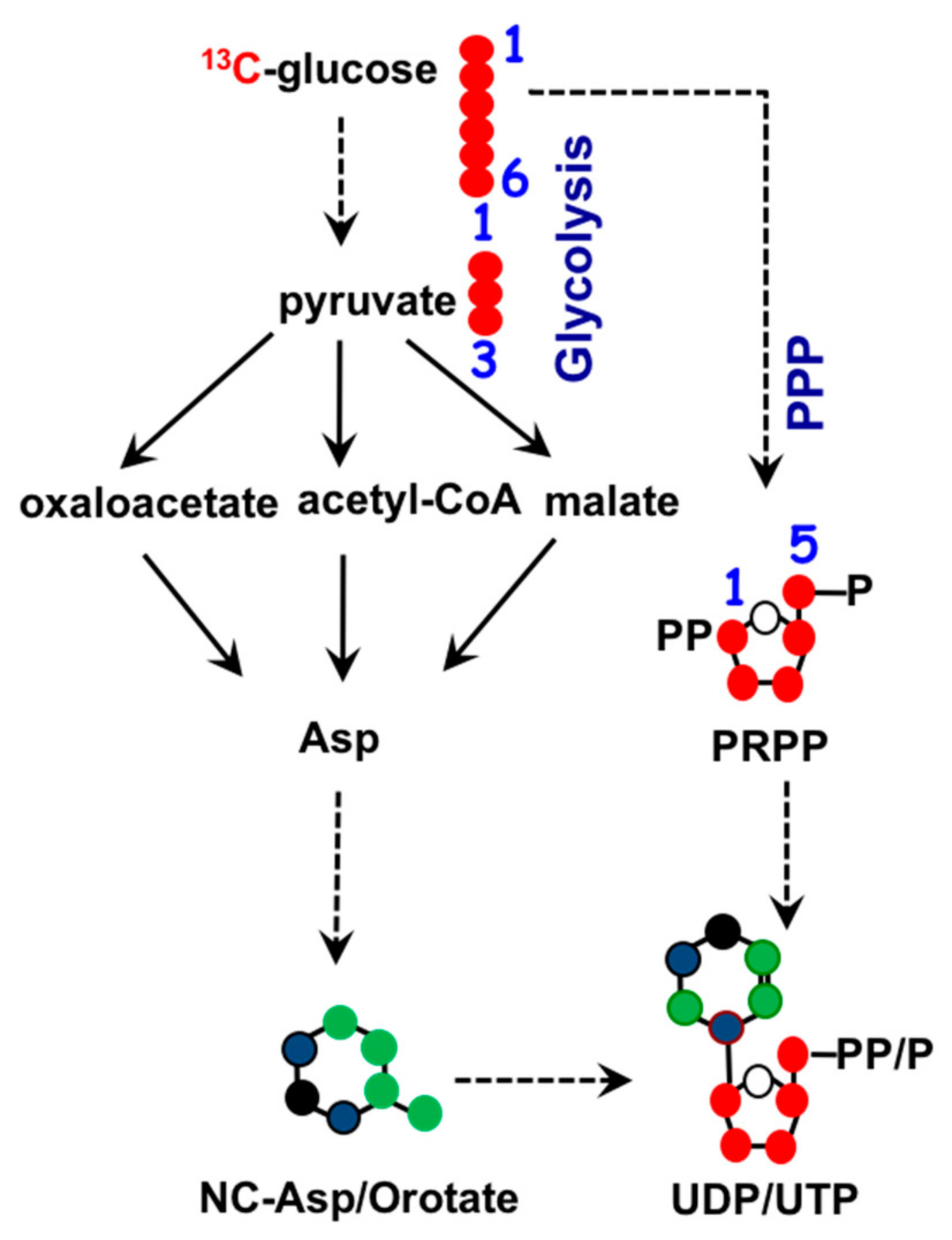
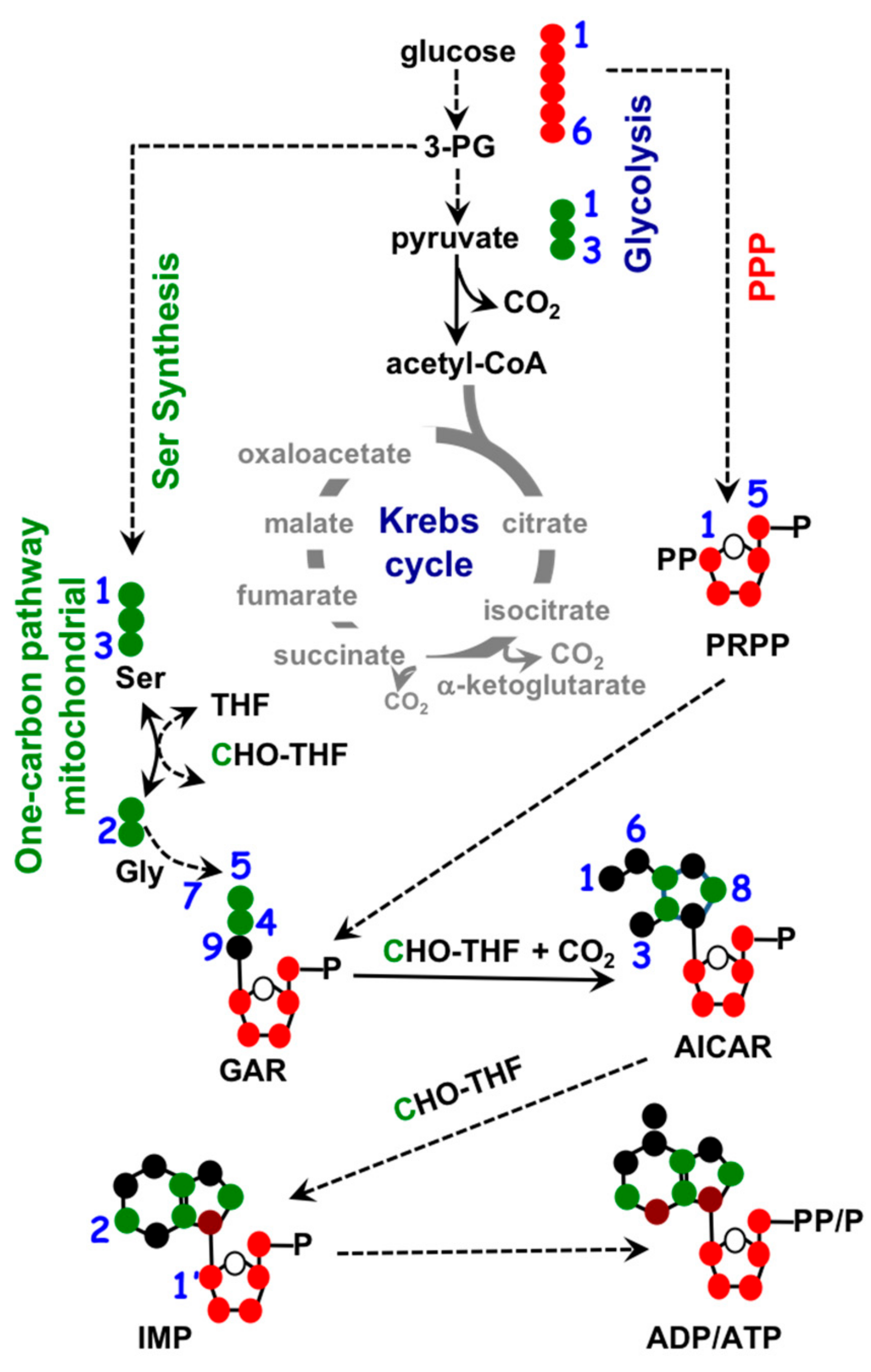
1.3. Mitochondria and Nucleotide Biosynthesis
1.4. Mitochondria and Fatty Acid Metabolism.
1.5. Mitochondria Metabolism and Epigenetics
2. Mitochondria Dysfunction in Alzheimer’s Disease
3. Mitochondrial Disruption Following TBI
4. Role of Mitochondria in Epilepsy
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gray, M.W.; Burger, G.; Lang, B.F. The origin and early evolution of mitochondria. Genome Biol. 2001, 2, reviews1018.1011. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E. The Red Cell. In Hemolytic Anemia in Disorders of Red Cell Metabolism; Beutler, E., Ed.; Springer: Boston, MA, USA, 1978. [Google Scholar]
- Palade, G.E. An electron microscope study of the mitochondrial structure. J. Histochem. Cytochem. 1953, 1, 188–211. [Google Scholar] [CrossRef] [PubMed]
- Moritz, C. Applications of mitochondrial DNA analysis in conservation: A critical review. Mol. Ecol. 1994, 3, 401–411. [Google Scholar] [CrossRef]
- Morgenstern, M.; Stiller, S.B.; Lübbert, P.; Peikert, C.D.; Dannenmaier, S.; Drepper, F.; Weill, U.; Höß, P.; Feuerstein, R.; Gebert, M. Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep. 2017, 19, 2836–2852. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.D.; Thomas, W.K.; Meyer, A.; Edwards, S.V.; Pääbo, S.; Villablanca, F.X.; Wilson, A.C. Dynamics of mitochondrial DNA evolution in animals: Amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. USA 1989, 86, 6196. [Google Scholar] [CrossRef] [PubMed]
- Heijne, G.; Steppuhn, J.; Herrmann, R.G. Domain structure of mitochondrial and chloroplast targeting peptides. Eur. J. Biochem. 1989, 180, 535–545. [Google Scholar] [CrossRef]
- Mesecke, N.; Terziyska, N.; Kozany, C.; Baumann, F.; Neupert, W.; Hell, K.; Herrmann, J.M. A Disulfide Relay System in the Intermembrane Space of Mitochondria that Mediates Protein Import. Cell 2005, 121, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Chacinska, A.; Pfannschmidt, S.; Wiedemann, N.; Kozjak, V.; Sanjuán Szklarz, L.K.; Schulze-Specking, A.; Truscott, K.N.; Guiard, B.; Meisinger, C.; Pfanner, N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO. J. 2004, 23, 3735. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, T.; Schlipf, S.; Sanz, J.; Neubert, K.; Stein, R.; Borner, C. Characterization of the signal that directs Bcl-x7, but not Bcl-2, to the mitochondrial outer membrane. J. Cell Biol. 2003, 160, 53. [Google Scholar] [CrossRef] [PubMed]
- van Gurp, M.; Festjens, N.; van Loo, G.; Saelens, X.; Vandenabeele, P. Mitochondrial intermembrane proteins in cell death. Biochem. Biophys. Res. Commun. 2003, 304, 487–497. [Google Scholar] [CrossRef]
- Nazaret, C.; Heiske, M.; Thurley, K.; Mazat, J.-P. Mitochondrial energetic metabolism: A simplified model of TCA cycle with ATP production. J. Theor. Biol. 2009, 258, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Srere, P.A. The infrastructure of the mitochondrial matrix. Trends Biochem. Sci. 1980, 5, 120–121. [Google Scholar] [CrossRef]
- Hatefi, Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu. Rev. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar] [CrossRef]
- Tattoli, I.; Carneiro, L.A.; Jéhanno, M.; Magalhaes, J.G.; Shu, Y.; Philpott, D.J.; Arnoult, D.; Girardin, S.E. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-κB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, M.; Clément, M.V.; Pervaiz, S.; Brenner, C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 2005, 20, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2004, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, A.S.; Rogers, G.W.; Murphy, A.N. Measuring Mitochondrial Function in Permeabilized Cells Using the Seahorse XF Analyzer or a Clark-Type Oxygen Electrode. Curr. Protocols Toxicol. 2014, 60, 21–25. [Google Scholar] [CrossRef]
- Sun, R.C.; Fan, T.W.M.; Deng, P.; Higashi, R.M.; Lane, A.N.; Le, A.-T.; Scott, T.L.; Sun, Q.; Warmoes, M.O.; Yang, Y. Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat. Commun. 2017, 8, 1646. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Mielke, M.M. Recent advances in the application of metabolomics to Alzheimer’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A.; Johnson, W.A. The role of citric acid in intermediate metabolism in animal tissues. Enzymologia 1937, 4, 148–156. [Google Scholar]
- Chance, B.; Williams, G.R. The respiratory chain and oxidative phosphorylation. Adv. Enzymol. Relat. Sub. Biochem. 1956, 17, 65–134. [Google Scholar]
- Patel, M.S.; Roche, T.E. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990, 4, 3224–3233. [Google Scholar] [CrossRef] [PubMed]
- Linn, T.C.; Pettit, F.H.; Reed, L.J. α-keto acid dehydrogenase complexes, X. Regulation of the activity of the pyruvate dehydrogenase complex from beef keidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. USA 1969, 62, 234. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.C.H.; Drejer, J.; Hertz, L.; Schousboe, A. Pyruvate Carboxylase Activity in Primary Cultures of Astrocytes and Neurons. J. Neurochem. 1983, 41, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Nordlie, R.C.; Lardy, H.A. Mammalian Liver Phosphoenolpyruvate Carboxykinase Activities. J. Biol. Chem. 1963, 238, 2259–2263. [Google Scholar] [PubMed]
- Hsu, R.Y.; Lardy, H.A. Malic enzyme. In Methods in Enzymology; Cademic Press: Cambridge, MA, USA, 1969; Volume 13, pp. 230–235. [Google Scholar]
- Bradford, H.F.; Ward, H.K. On glutaminase activity in mammalian synaptosomes. Brain Res. 1976, 110, 115–125. [Google Scholar] [CrossRef]
- Frieden, C. Glutamate Dehydrogenase: V. The relation of enzyme structure to the catalytic function. J. Biol. Chem. 1963, 238, 3286–3299. [Google Scholar] [PubMed]
- Yamaya, T.; Oaks, A. Synthesis of glutamate by mitochondria—An anaplerotic function for glutamate dehydrogenase. Physiol. Plant. 1987, 70, 749–756. [Google Scholar] [CrossRef]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The Key Role of Anaplerosis and Cataplerosis for Citric Acid Cycle Function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B. Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Reitzer, L.J.; Wice, B.M.; Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [Google Scholar] [PubMed]
- Lowenstein, J. Ammonia production in muscle and other tissues: The purine nucleotide cycle. Physiol. Rev. 1972, 52, 382–414. [Google Scholar] [CrossRef] [PubMed]
- Jeremiah, S.; Povey, S.; Burley, M.; Kielty, C.; Lee, M.; Spowart, G.; Corney, G.; Cook, P. Mapping studies on human mitochondrial glutamate oxaloacetate transaminase. Ann. Hum. Genet. 1982, 46, 145–152. [Google Scholar] [CrossRef]
- Beardsley, G.; Moroson, B.; Taylor, E.; Moran, R. A new folate antimetabolite, 5, 10-dideaza-5, 6, 7, 8-tetrahydrofolate is a potent inhibitor of de novo purine synthesis. J. Biol. Chem. 1989, 264, 328–333. [Google Scholar] [PubMed]
- Schirch, L. Serine hydroxymethyltransferase. Adv. Enzymol. Relat. Areas Mol. Biol. 1982, 53, 83–112. [Google Scholar] [PubMed]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Drury, E.J.; MacKenzie, R.E. Methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase-formyltetrahydrofolate synthetase. A multifunctional protein from porcine liver. J. Biol. Chem. 1977, 252, 1117–1122. [Google Scholar] [PubMed]
- Appling, D.R. Compartmentation of folate-mediated one-carbon metabolism in eukaryotes. FASEB J. 1991, 5, 2645–2651. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Wakil, S.J. Mechanism of fatty acid synthesis. J. Lipid Res. 1961, 2, 1–24. [Google Scholar]
- Watson, J.A.; Lowenstein, J.M. Citrate and the conversion of carbohydrate into fat fatty acid synthesis by a combination of cytoplasm and mitochondria. J. Biol. Chem. 1970, 245, 5993–6002. [Google Scholar] [PubMed]
- McGarry, J.D.; Leatherman, G.F.; Foster, D.W. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J. Biol. Chem. 1978, 253, 4128–4136. [Google Scholar] [PubMed]
- McGarry, J.D.; Mannaerts, G.; Foster, D.W. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Invest. 1977, 60, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, J.K.; Schonauer, M.S.; Autio, K.J.; Mittelmeier, T.M.; Kastaniotis, A.J.; Dieckmann, C.L. Mitochondrial fatty acid synthesis type II: More than just fatty acids. J. Biol. Chem. 2009, 284, 9011–9015. [Google Scholar] [CrossRef] [PubMed]
- Moris, N.; Pina, C.; Arias, A.M. Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 2016, 17, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009, 10, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-Mediated Changes in Acetyl-CoA and Histone Acetylation Control the Early Differentiation of Embryonic Stem Cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Mews, P.; Donahue, G.; Drake, A.M.; Luczak, V.; Abel, T.; Berger, S.L. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 2017, 546, 381. [Google Scholar] [CrossRef] [PubMed]
- Moseley, H.N.; Lane, A.N.; Belshoff, A.C.; Higashi, R.M.; Fan, T.W. A novel deconvolution method for modeling UDP-N-acetyl-D-glucosamine biosynthetic pathways based on 13 C mass isotopologue profiles under non-steady-state conditions. BMC Biol. 2011, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, Y.; Bian, C.; Fujiki, R.; Yu, X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 2012, 493, 561. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.-S.; Duggan, L.; Tolga, N.; Belotserkovskya, R.; Lane, W.S.; Shiekhattar, R.; Berger, S.L. Snf1–a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 2001, 293, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins—Emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Gonçalves, E.; Isaac Johnson, T.; Roberto Zecchini, V.; da Costa, A.S.H.; Gaude, E.; Vercauteren Drubbel, A.; Julian Theobald, S.; Abbo, S.; Tran, M.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Soga, T.; Pollard, P.J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Invest. 2013, 123, 3652–3658. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Toro, J.R.; Nickerson, M.L.; Wei, M.-H.; Warren, M.B.; Glenn, G.M.; Turner, M.L.; Stewart, L.; Duray, P.; Tourre, O.; Sharma, N.; et al. Mutations in the Fumarate Hydratase Gene Cause Hereditary Leiomyomatosis and Renal Cell Cancer in Families in North America. Am. J. Hum. Genet. 2003, 73, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s, A. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015, 11, 332. [Google Scholar]
- Ramirez-Bermudez, J. Alzheimer’s disease: Critical notes on the history of a medical concept. Arch. Med. Res. 2012, 43, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xie, N.; Tang, B.; Li, R.; Shen, Y. Alzheimer’s Disease: From Genetic Variants to the Distinct Pathological Mechanisms. Front. Mol. Neurosci. 2017, 10, 319. [Google Scholar] [CrossRef] [PubMed]
- Craft, S. The role of metabolic disorders in Alzheimer disease and vascular dementia: Two roads converged. Arch. Neurol. 2009, 66, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Harrison, F.E. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients 2015, 7, 7332–7357. [Google Scholar] [CrossRef] [PubMed]
- Narayan, K.M.; Boyle, J.P.; Geiss, L.S.; Saaddine, J.B.; Thompson, T.J. Impact of recent increase in incidence on future diabetes burden: U.S., 2005–2050. Diabetes Care 2006, 29, 2114–2116. [Google Scholar] [CrossRef] [PubMed]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.L.; Chase, T.N.; Fedio, P.; Patronas, N.J.; Brooks, R.A.; Di Chiro, G. Alzheimer’s disease: Focal cortical changes shown by positron emission tomography. Neurology 1983, 33, 961–965. [Google Scholar] [CrossRef] [PubMed]
- de Leon, M.J.; Ferris, S.H.; George, A.E.; Christman, D.R.; Fowler, J.S.; Gentes, C.; Reisberg, B.; Gee, B.; Emmerich, M.; Yonekura, Y.; et al. Positron emission tomographic studies of aging and Alzheimer disease. AJNR Am. J. Neuroradiol. 1983, 4, 568–571. [Google Scholar] [PubMed]
- Ferris, S.H.; de Leon, M.J.; Wolf, A.P.; Farkas, T.; Christman, D.R.; Reisberg, B.; Fowler, J.S.; Macgregor, R.; Goldman, A.; George, A.E.; et al. Positron emission tomography in the study of aging and senile dementia. Neurobiol. Aging 1980, 1, 127–131. [Google Scholar] [CrossRef]
- Friedland, R.P.; Budinger, T.F.; Ganz, E.; Yano, Y.; Mathis, C.A.; Koss, B.; Ober, B.A.; Huesman, R.H.; Derenzo, S.E. Regional cerebral metabolic alterations in dementia of the Alzheimer type: Positron emission tomography with [18F]fluorodeoxyglucose. J. Comput. Assist. Tomogr. 1983, 7, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Small, G.W.; Ercoli, L.M.; Silverman, D.H.; Huang, S.C.; Komo, S.; Bookheimer, S.Y.; Lavretsky, H.; Miller, K.; Siddarth, P.; Rasgon, N.L.; et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 6037–6042. [Google Scholar] [CrossRef] [PubMed]
- Laforce, R., Jr.; Rabinovici, G.D. Amyloid imaging in the differential diagnosis of dementia: Review and potential clinical applications. Alzheimers Res. Ther. 2011, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Grady, C.L.; Haxby, J.V.; Schlageter, N.L.; Berg, G.; Rapoport, S.I. Stability of metabolic and neuropsychological asymmetries in dementia of the Alzheimer type. Neurology 1986, 36, 1390–1392. [Google Scholar] [CrossRef] [PubMed]
- Haxby, J.V.; Grady, C.L.; Koss, E.; Horwitz, B.; Heston, L.; Schapiro, M.; Friedland, R.P.; Rapoport, S.I. Longitudinal study of cerebral metabolic asymmetries and associated neuropsychological patterns in early dementia of the Alzheimer type. Arch. Neurol. 1990, 47, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Blass, J.P. Alzheimer’s disease and Alzheimer’s dementia: Distinct but overlapping entities. Neurobiol. Aging 2002, 23, 1077–1084. [Google Scholar] [CrossRef]
- Small, G.W.; Mazziotta, J.C.; Collins, M.T.; Baxter, L.R.; Phelps, M.E.; Mandelkern, M.A.; Kaplan, A.; La Rue, A.; Adamson, C.F.; Chang, L.; et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 1995, 273, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Caselli, R.J.; Yun, L.S.; Chen, K.; Bandy, D.; Minoshima, S.; Thibodeau, S.N.; Osborne, D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl. J. Med. 1996, 334, 752–758. [Google Scholar] [CrossRef] [PubMed]
- De Leon, M.J.; Convit, A.; Wolf, O.T.; Tarshish, C.Y.; DeSanti, S.; Rusinek, H.; Tsui, W.; Kandil, E.; Scherer, A.J.; Roche, A.; et al. Prediction of cognitive decline in normal elderly subjects with 2-[18F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc. Natl. Acad. Sci. USA 2001, 98, 10966–10971. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; De Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; de Leon, M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 1990, 40, 1302–1302. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.K. Cytochrome C Oxidase in Alzheimer’s Disease Brain Purification and Characterization. Neurology 1995, 45, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Aβ toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Hauptmann, S.; Scherping, I.; Meinhardt, J.; Rhein, V.; Dröse, S.; Brandt, U.; Fändrich, M.; Müller, W.E.; Götz, J. Oligomeric and fibrillar species of β-amyloid (Aβ42) both impair mitochondrial function in P301L tau transgenic mice. J. Mol. Med. 2008, 86, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Frackowiak, R.S.; Pozzilli, C.; Legg, N.J.; Du Boulay, G.H.; Marshall, J.; Lenzi, G.L.; Jones, T. Regional cerebral oxygen supply and utilization in dementia. A clinical and physiological study with oxygen-15 and positron tomography. Brain 1981, 104, 753–778. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, H.; Ogawa, M.; Yamauchi, H.; Yamaguchi, S.; Kimura, J.; Yonekura, Y.; Konishi, J. Altered cerebral energy metabolism in Alzheimer’s disease: A PET study. J. Nucl. Med. 1994, 35, 1–6. [Google Scholar] [PubMed]
- Sorbi, S.; Bird, E.D.; Blass, J.P. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann. Neurol. 1983, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Starkov, A.; Blass, J.P.; Ratan, R.R.; Beal, M.F. Cause and consequence: Mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A. meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Raber, J.; Huang, Y.; Ashford, J.W. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 2004, 25, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet 2000, 1, 507–537. [Google Scholar] [CrossRef] [PubMed]
- Eichner, J.E.; Dunn, S.T.; Perveen, G.; Thompson, D.M.; Stewart, K.E.; Stroehla, B.C. Apolipoprotein E polymorphism and cardiovascular disease: A HuGE review. Am. J. Epidemiol. 2002, 155, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Bernardo, A.; Walker, D.; Kanegawa, T.; Mahley, R.W.; Huang, Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 2006, 26, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C. Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 2014, 66, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Altmann, A.; Ng, B.; Landau, S.M.; Jagust, W.J.; Greicius, M.D. Alzheimer’s Disease Neuroimaging, I. Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain 2015, 138, 3734–3746. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Caselli, R.J.; Chen, K.; Alexander, G.E.; Bandy, D.; Frost, J. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 3334–3339. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Benzinger, T.L.; Blazey, T.; Jack, C.R., Jr.; Koeppe, R.A.; Su, Y.; Xiong, C.; Raichle, M.E.; Snyder, A.Z.; Ances, B.M.; Bateman, R.J.; et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, E4502–4509. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.K.; Ji, Z.S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, S.B.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem. 2011, 286, 5215–5221. [Google Scholar] [CrossRef] [PubMed]
- James, R.; Searcy, J.L.; Le Bihan, T.; Martin, S.F.; Gliddon, C.M.; Povey, J.; Deighton, R.F.; Kerr, L.E.; McCulloch, J.; Horsburgh, K. Proteomic analysis of mitochondria in APOE transgenic mice and in response to an ischemic challenge. J. Cereb. Blood Flow. Metab. 2012, 32, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Tambini, M.D.; Pera, M.; Kanter, E.; Yang, H.; Guardia-Laguarta, C.; Holtzman, D.; Sulzer, D.; Area-Gomez, E.; Schon, E.A. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016, 17, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Ran Ma, T.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Watanabe, A.; Fujino, T.; Hosono, T.; Michikawa, M. Apolipoprotein E4 (1-272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol. Neurodegener. 2009, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, G.; Bonafe, M.; De Luca, M.; Rose, G.; Varcasia, O.; Bruni, A.; Maletta, R.; Nacmias, B.; Sorbi, S.; Corsonello, F.; et al. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s disease. Hum. Genet. 2001, 108, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Maruszak, A.; Safranow, K.; Branicki, W.; Gaweda-Walerych, K.; Pospiech, E.; Gabryelewicz, T.; Canter, J.A.; Barcikowska, M.; Zekanowski, C. The impact of mitochondrial and nuclear DNA variants on late-onset Alzheimer’s disease risk. J. Alzheimers Dis. 2011, 27, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.; Wolf, A.B.; Chavira, B.; Shonebarger, D.; Meckel, J.P.; Leung, L.; Ballina, L.; Ly, S.; Saini, A.; Jones, T.B.; et al. Altered Energy Metabolism Pathways in the Posterior Cingulate in Young Adult Apolipoprotein E varepsilon4 Carriers. J. Alzheimers Dis. 2016, 53, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Valla, J.; Yaari, R.; Wolf, A.B.; Kusne, Y.; Beach, T.G.; Roher, A.E.; Corneveaux, J.J.; Huentelman, M.J.; Caselli, R.J.; Reiman, E.M. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer’s susceptibility gene. J. Alzheimers Dis. 2010, 22, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Koppel, S.J.; Bothwell, R.; Mahnken, J.; Burns, J.M.; Swerdlow, R.H. Platelet cytochrome oxidase and citrate synthase activities in APOE epsilon4 carrier and non-carrier Alzheimer’s disease patients. Redox. Biol. 2017, 12, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Albers, D.S.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. In Advances in Dementia Research; Springer: Berlin, Germany, 2000; pp. 133–154. [Google Scholar]
- Trushina, E.; McMurray, C. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta (BBA)-Bioenerg. 1998, 1366, 211–223. [Google Scholar] [CrossRef]
- Emerit, J.; Edeas, M.; Bricaire, F. Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 2004, 58, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Faul, M.; Coronado, V. Epidemiology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.; Tavazzi, B.; Signoretti, S.; Amorini, A.M.; Belli, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Finocchiaro, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Mitochondrial-related impairment—Part I. Neurosurgery 2007, 61, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Kong, R.H.; Zhang, L.M.; Zhang, J.N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef] [PubMed]
- Bulstrode, H.; Nicoll, J.A.; Hudson, G.; Chinnery, P.F.; Di Pietro, V.; Belli, A. Mitochondrial DNA and traumatic brain injury. Ann. Neurol. 2014, 75, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Mautes, A.E.; Thome, D.; Steudel, W.I.; Nacimiento, A.C.; Yang, Y.; Shohami, E. Changes in regional energy metabolism after closed head injury in the rat. J. Mol. Neurosci. 2001, 16, 33–39. [Google Scholar] [CrossRef]
- Lifshitz, J.; Friberg, H.; Neumar, R.W.; Raghupathi, R.; Welsh, F.A.; Janmey, P.; Saatman, K.E.; Wieloch, T.; Grady, M.S.; McIntosh, T.K. Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: Evidence for differentially sensitive populations in the cortex and hippocampus. J. Cereb. Blood Flow. Metab. 2003, 23, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Keller, J.N.; Mattson, M.P.; Scheff, S.W. Traumatic brain injury alters synaptic homeostasis: Implications for impaired mitochondrial and transport function. J. Neurotrauma. 1998, 15, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.T.; Soltesz, I. Selective depolarization of interneurons in the early posttraumatic dentate gyrus: Involvement of the Na(+)/K.(+)-ATPase. J. Neurophysiol. 2000, 83, 2916–2930. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Becker, D.P.; Tamura, T.; Hovda, D.A. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 1990, 73, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Katayama, Y.; Hovda, D.A.; Yoshino, A.; Becker, D.P. Lactate accumulation following concussive brain injury: The role of ionic fluxes induced by excitatory amino acids. Brain Res. 1995, 674, 196–204. [Google Scholar] [CrossRef]
- Bentzer, P.; Davidsson, H.; Grande, P.O. Microdialysis-based long-term measurements of energy-related metabolites in the rat brain following a fluid percussion trauma. J. Neurotrauma. 2000, 17, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Qian, Y.Z.; Di, X.; Rice, A.; Zhu, J.P.; Bullock, R. Lactate/glucose dynamics after rat fluid percussion brain injury. J. Neurotrauma. 2000, 17, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Hertz, L.; Huang, R.; Sonnewald, U.; Petersen, S.B.; Westergaard, N.; Larsson, O.; Schousboe, A. Utilization of glutamine and of TCA cycle constituents as precursors for transmitter glutamate and GABA. Dev. Neurosci. 1993, 15, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Tapia, R.; Gonzalez, R.M. Glutamine and glutamate as precursors of the releasable pool of GABA in brain cortex slices. Neurosci. Lett. 1978, 10, 165–169. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Peterson, P.L.; Muizelaar, J.P.; Lee, C.P. Amelioration of mitochondrial function by a novel antioxidant U-101033E following traumatic brain injury in rats. J. Neurotrauma. 1997, 14, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Lifshitz, J.; Sullivan, P.G.; Hovda, D.A.; Wieloch, T.; McIntosh, T.K. Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion 2004, 4, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Rabchevsky, A.G.; Waldmeier, P.C.; Springer, J.E. Mitochondrial permeability transition in CNS trauma: Cause or effect of neuronal cell death? J. Neurosci. Res. 2005, 79, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Slater, E.C.; Cleland, K.W. The effect of calcium on the respiratory and phosphorylative activities of heart-muscle sarcosomes. Biochem. J. 1953, 55, 566–590. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.; Nicholls, D.G. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 2003, 278, 19062–19070. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Qian, L.; Zhou, T.; Iadecola, C. Mitochondria are involved in the neurogenic neuroprotection conferred by stimulation of cerebellar fastigial nucleus. J. Neurochem. 2005, 95, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Brustovetsky, T.; Purl, K.J.; Capano, M.; Crompton, M.; Dubinsky, J.M. Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J. Neurosci. 2003, 23, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.D.; Nukala, V.N.; Sullivan, P.G. Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Front. Neuroenergetics 2013, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; LaNoue, K.F.; Cheung, J.Y.; Scaduto, R.C., Jr. Regulation of citric acid cycle by calcium. J. Biol. Chem. 1989, 264, 13430–13439. [Google Scholar] [PubMed]
- Denton, R.M.; Rutter, G.A.; Midgley, P.J.; McCormack, J.G. Effects of Ca2+ on the activities of the calcium-sensitive dehydrogenases within the mitochondria of mammalian tissues. J. Cardiovasc. Pharmacol. 1988, 12 Suppl 5, S69–S72. [Google Scholar] [CrossRef]
- Sharma, P.; Benford, B.; Li, Z.Z.; Ling, G.S. Role of pyruvate dehydrogenase complex in traumatic brain injury and Measurement of pyruvate dehydrogenase enzyme by dipstick test. J. Emerg. Trauma. Shock 2009, 2, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchyan, G.V.; Ucal, M.; Mullebner, A.; Dumitrescu, S.; Kames, M.; Moldzio, R.; Molcanyi, M.; Schaefer, S.; Weidinger, A.; Schaefer, U.; et al. Thiamine preserves mitochondrial function in a rat model of traumatic brain injury, preventing inactivation of the 2-oxoglutarate dehydrogenase complex. Biochim. Biophys. Acta 2018, 1859, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.Y.; Kreipke, C.W.; Speirs, S.L.; Schafer, P.; Schafer, S.; Rafols, J.A. Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci. Lett. 2009, 453, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Bell, J.D.; Siddiq, I.P.; Baker, A.J. An analysis of regional microvascular loss and recovery following two grades of fluid percussion trauma: A role for hypoxia-inducible factors in traumatic brain injury. J. Cereb. Blood Flow. Metab. 2009, 29, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Higashida, T.; Kreipke, C.W.; Rafols, J.A.; Peng, C.; Schafer, S.; Schafer, P.; Ding, J.Y.; Dornbos, D., 3rd; Li, X.; Guthikonda, M.; et al. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J. Neurosurg. 2011, 114, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Golias, T.; Papandreou, I.; Sun, R.; Kumar, B.; Brown, N.V.; Swanson, B.J.; Pai, R.; Jaitin, D.; Le, Q.T.; Teknos, T.N.; et al. Hypoxic repression of pyruvate dehydrogenase activity is necessary for metabolic reprogramming and growth of model tumours. Sci. Rep. 2016, 6, 31146. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Gu, Q.; Peterson, P.L.; Muizelaar, J.P.; Lee, C.P. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J. Neurotrauma. 1997, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Rossi, S.; Stiefel, M.; Doppenberg, E.; Zauner, A.; Bullock, R.; Marmarou, A. CSF and ECF glutamate concentrations in head injured patients. Acta Neurochir. Suppl. 1999, 75, 17–19. [Google Scholar] [PubMed]
- Rose, M.E.; Huerbin, M.B.; Melick, J.; Marion, D.W.; Palmer, A.M.; Schiding, J.K.; Kochanek, P.M.; Graham, S.H. Regulation of interstitial excitatory amino acid concentrations after cortical contusion injury. Brain Res. 2002, 943, 15–22. [Google Scholar] [CrossRef]
- Tavazzi, B.; Signoretti, S.; Lazzarino, G.; Amorini, A.M.; Delfini, R.; Cimatti, M.; Marmarou, A.; Vagnozzi, R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 2005, 56, 582–589. [Google Scholar] [CrossRef] [PubMed]
- CDC. Epilepsy Data and Statistics. Available online: https://www.cdc.gov/epilepsy/data/index.html (accessed on 3 November 2018).
- Gavvala, J.R.; Schuele, S.U. JAMA Patient Page: Epilepsy. JAMA 2016, 316, 2686. [Google Scholar] [CrossRef] [PubMed]
- Debray, F.G.; Lambert, M.; Chevalier, I.; Robitaille, Y.; Decarie, J.C.; Shoubridge, E.A.; Robinson, B.H.; Mitchell, G.A. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics 2007, 119, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Khurana, D.; Salganicoff, L.; Melvin, J.; Hobdell, E.; Valencia, I.; Hardison, H.; Marks, H.; Grover, W.; Legido, A. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathies. Epilepsia 2008, 49, 1972. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Davis, R.E.; Williams, M. Mitochondrial function and dysfunction: An update. J. Pharmacol. Exp. Ther. 2012, 342, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef] [PubMed]
- March, P.A. Seizures: Classification, etiologies, and pathophysiology. Clin. Tech. Small Anim. Prac. 1998, 13, 119–131. [Google Scholar] [CrossRef]
- Rahman, S. Mitochondrial disease and epilepsy. Dev. Med. Child Neurol. 2012, 54, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Dermaut, B.; Löfgren, A.; Martin, J.-J.; Van Broeckhoven, C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001, 28, 211. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Luoma, P.; Rantamäki, M.; Al Memar, A.; Kaakkola, S.; Hackman, P.; Krahe, R.; Löfgren, A.; Martin, J.-J.; De Jonghe, P. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology 2004, 63, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Martin, J.; Dermaut, B.; Löfgren, A.; Wibail, A.; Ververken, D.; Tack, P.; Dehaene, I.; Van Zandijcke, M.; Moonen, M. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscular Disorders 2003, 13, 133–142. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Nguyen, K.V. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann. Neurol. 2004, 55, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Hudson, G.; Ferrari, G.; Fütterer, N.; Ahola, S.; Lamantea, E.; Prokisch, H.; Lochmüller, H.; McFarland, R.; Ramesh, V. Phenotypic spectrum associated with mutations of the mitochondrial polymerase γ gene. Brain 2006, 129, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Zeviani, M. 155th ENMC workshop: Polymerase gamma and disorders of mitochondrial DNA synthesis, 21–23 September 2007, Naarden, The Netherlands. Neuromuscular Disorders 2008, 18, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Neeve, V.C.; Samuels, D.C.; Bindoff, L.A.; van den Bosch, B.; Van Goethem, G.; Smeets, H.; Lombès, A.; Jardel, C.; Hirano, M.; DiMauro, S. What is influencing the phenotype of the common homozygous polymerase-γ mutation p. Ala467Thr? Brain 2012, 135, 3614–3626. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Walter, P.B.; Ames, B.N. The role of heme and iron-sulfur clusters in mitochondrial biogenesis, maintenance, and decay with age. Arch. Biochem. Biophys. 2002, 397, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.M.; Janer, A.; Levandovskiy, V.; MacKay, N.; Rouault, T.A.; Tong, W.-H.; Ogilvie, I.; Shoubridge, E.A.; Robinson, B.H. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am. J. Hum. Genet. 2011, 89, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.R.; Friederich, M.W.; Swanson, M.A.; Shaikh, T.; Bhattacharya, K.; Scharer, G.H.; Aicher, J.; Creadon-Swindell, G.; Geiger, E.; MacLean, K.N. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 2013, 137, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Zimmermann, F.A.; Fauth, C.; Bergheim, C.; Meierhofer, D.; Radmayr, D.; Zschocke, J.; Koch, J.; Sperl, W. Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Am. J. Hum. Genet. 2011, 89, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Raas-Rothschild, A.; Rio, M.; Fiermonte, G.; Encha-Razavi, F.; Palmieri, L.; Palmieri, F.; Ben-Neriah, Z.; Kadhom, N.; Vekemans, M. Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am. J. Hum. Genet. 2005, 76, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Li, D.; Gai, X.; McCormick, E.; Place, E.; Lasorsa, F.M.; Otieno, F.G.; Hou, C.; Kim, C.E.; Abdel-Magid, N. AGC1 deficiency causes infantile epilepsy, abnormal myelination, and reduced N.-acetylaspartate. In JIMD Reports, Volume 14; Springer: Berlin, Germany, 2014; pp. 77–85. [Google Scholar]
- Fiermonte, G.; Palmieri, L.; Todisco, S.; Agrimi, G.; Palmieri, F.; Walker, J.E. Identification of the mitochondrial glutamate transporter: Bacterial expression, reconsitution, functional characterization, tissue distribution of two human isoforms. J. Biol. Chem. 2002, 277, 19289–19294. [Google Scholar] [CrossRef] [PubMed]
- Thangaratnarajah, C.; Ruprecht, J.J.; Kunji, E.R. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nature Commun. 2014, 5, 5491. [Google Scholar] [CrossRef] [PubMed]
- Niciu, M.J.; Kelmendi, B.; Sanacora, G. Overview of glutamatergic neurotransmission in the nervous system. Pharmacol. Biochem. Behav. 2012, 100, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Bénit, P.; Chretien, D.; Kadhom, N.; de Lonlay-Debeney, P.; Cormier-Daire, V.; Cabral, A.; Peudenier, S.; Rustin, P.; Munnich, A.; Rötig, A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I. deficiency. Am. J. Hum. Genet. 2001, 68, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Benit, P.; Slama, A.; Cartault, F.; Giurgea, I.; Chretien, D.; Lebon, S.; Marsac, C.; Munnich, A.; Rötig, A.; Rustin, P. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J. Med. Genet. 2004, 41, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Scacco, S.; Piccoli, C.; Bellomo, F.; Petruzzella, V.; Trentadue, R.; Minuto, M.; Ripoli, M.; Capitanio, N.; Zeviani, M. Dysfunctions of cellular oxidative metabolism in patients with mutations in the NDUFS1 and NDUFS4 genes of complex I. J. Biol. Chem. 2006, 281, 10374–10380. [Google Scholar] [CrossRef] [PubMed]
- Kirby, D.M.; Salemi, R.; Sugiana, C.; Ohtake, A.; Parry, L.; Bell, K.M.; Kirk, E.P.; Boneh, A.; Taylor, R.W.; Dahl, H.-H.M. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I. deficiency. J. Clin. Invest. 2004, 114, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.; Hogan, V.E.; He, L.; Blakely, E.L.; Worgan, L.; Al-Dosary, M.; Saretzki, G.; Alston, C.L.; Morris, A.A.; Clarke, M. The p. M292T NDUFS2 mutation causes complex I-deficient Leigh syndrome in multiple families. Brain 2010, 133, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Berger, I.; Hershkovitz, E.; Shaag, A.; Edvardson, S.; Saada, A.; Elpeleg, O. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann. Neurol. 2008, 63, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Moreira, D.; Ugalde, C.; Smeets, R.; Rodenburg, R.J.; Lopez-Laso, E.; Ruiz-Falco, M.L.; Briones, P.; Martin, M.A.; Smeitink, J.A.; Arenas, J. X-linked NDUFA1 gene mutations associated with mitochondrial encephalomyopathy. Ann. Neurol. 2007, 61, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Loeffen, J.; Smeitink, J.; Triepels, R.; Smeets, R.; Schuelke, M.; Sengers, R.; Trijbels, F.; Hamel, B.; Mullaart, R.; van den Heuvel, L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am. J. Hum. Genet. 1998, 63, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Ruhoy, I.S.; Saneto, R.P. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2014, 7, 221. [Google Scholar] [PubMed]
- Bianciardi, L.; Imperatore, V.; Fernandez-Vizarra, E.; Lopomo, A.; Falabella, M.; Furini, S.; Galluzzi, P.; Grosso, S.; Zeviani, M.; Renieri, A. Exome sequencing coupled with mRNA analysis identifies NDUFAF6 as a Leigh gene. Mol.Genet. Metabol. 2016, 119, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Tucker, E.J.; Compton, A.G.; Kirby, D.M.; Crawford, G.; Burtt, N.P.; Rivas, M.; Guiducci, C.; Bruno, D.L.; Goldberger, O.A. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I. deficiency. Nature Genet. 2010, 42, 851. [Google Scholar] [CrossRef] [PubMed]
- Herzer, M.; Koch, J.; Prokisch, H.; Rodenburg, R.; Rauscher, C.; Radauer, W.; Forstner, R.; Pilz, P.; Rolinski, B.; Freisinger, P. Leigh disease with brainstem involvement in complex I deficiency due to assembly factor NDUFAF2 defect. Europediatrics 2010, 41, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Mimaki, M.; Wang, X.; McKenzie, M.; Thorburn, D.R.; Ryan, M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1817, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Prabhu, S.P.; Lidov, H.G.; Shi, J.; Anselm, I.; Brownstein, C.A.; Bainbridge, M.N.; Beggs, A.H.; Vargas, S.O.; Agrawal, P.B. AIFM1 mutation presenting with fatal encephalomyopathy and mitochondrial disease in an infant. Mol. Case Stud. 2017, 3, a001560. [Google Scholar] [CrossRef] [PubMed]
- Saada, A.; Vogel, R.O.; Hoefs, S.J.; van den Brand, M.A.; Wessels, H.J.; Willems, P.H.; Venselaar, H.; Shaag, A.; Barghuti, F.; Reish, O. Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I. assembly protein, cause fatal neonatal mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Sugiana, C.; Pagliarini, D.J.; McKenzie, M.; Kirby, D.M.; Salemi, R.; Abu-Amero, K.K.; Dahl, H.-H.M.; Hutchison, W.M.; Vascotto, K.A.; Smith, S.M. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am. J. Hum. Genet. 2008, 83, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Wortmann, S.B.; Smeets, R.J.; Venselaar, H.; Antoine, M.; Visser, G.; Ben-Omran, T.; Van Den Heuvel, L.P.; Timmers, H.J.; Smeitink, J.A. SDHA mutations causing a multisystem mitochondrial disease: Novel mutations and genetic overlap with hereditary tumors. Eur. J. Hum. Genet. 2015, 23, 202. [Google Scholar] [CrossRef] [PubMed]
- Tucker, E.J.; Wanschers, B.F.; Szklarczyk, R.; Mountford, H.S.; Wijeyeratne, X.W.; van den Brand, M.A.; Leenders, A.M.; Rodenburg, R.J.; Reljić, B.; Compton, A.G. Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet. 2013, 9, e1004034. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Garcia, S.; Padgett, K.R.; Moraes, C.T. A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum. Mol. Genet. 2012, 21, 5066–5077. [Google Scholar] [CrossRef] [PubMed]
- Tegelberg, S.; Tomašić, N.; Kallijärvi, J.; Purhonen, J.; Elmér, E.; Lindberg, E.; Nord, D.G.; Soller, M.; Lesko, N.; Wedell, A. Respiratory chain complex III deficiency due to mutated BCS1L: A novel phenotype with encephalomyopathy, partially phenocopied in a Bcs1l mutant mouse model. Orphanet J. Rare Dis. 2017, 12, 73. [Google Scholar] [CrossRef] [PubMed]
- Antonicka, H.; Leary, S.C.; Guercin, G.-H.; Agar, J.N.; Horvath, R.; Kennaway, N.G.; Harding, C.O.; Jaksch, M.; Shoubridge, E.A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Antonicka, H.; Shoubridge, E.A. Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep. 2015, 10, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, M.; Ogilvie, I.; Yao, J.; Kortenhaus, G.; Bresser, H.-G.; Gerbitz, K.-D.; Shoubridge, E.A. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Antonicka, H.; Sasarman, F.; Weraarpachai, W.; Cobine, P.A.; Pan, M.; Brown, G.K.; Brown, R.; Majewski, J.; Ha, K.C. Novel Mutations in SCO 1 as a Cause of Fatal Infantile Encephalopathy and Lactic Acidosis. Hum. Mutation 2013, 34, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Smith, K.R.; Stroud, D.A.; Compton, A.G.; Tucker, E.J.; Dasvarma, A.; Gandolfo, L.C.; Marum, J.E.; McKenzie, M.; Peters, H.L. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am. J. Hum. Genet. 2014, 94, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Melchionda, L.; Haack, T.B.; Hardy, S.; Abbink, T.E.; Fernandez-Vizarra, E.; Lamantea, E.; Marchet, S.; Morandi, L.; Moggio, M.; Carrozzo, R. Mutations in APOPT1, encoding a mitochondrial protein, cause cavitating leukoencephalopathy with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 2014, 95, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Oquendo, C.; Antonicka, H.; Shoubridge, E.; Reardon, W.; Brown, G. Functional and genetic studies demonstrate that mutation in the COX15 gene can cause Leigh syndrome. J. Med. Genet. 2004, 41, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, E.; Augoustides-Savvopoulou, P.; Gregersen, N.; Haas, D.; Gkampeta, A.; Athanassiadou-Piperopoulou, F. An infant with ethylmalonic encephalopathy masquerading as a hematologic disorder. J. Child Neurol. 2013, 28, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.; Rahman, S.; Wedatilake, Y.; Polke, J.M.; Cirak, S.; Foley, A.R.; Sailer, A.; Hurles, M.E.; Stalker, J.; Hargreaves, I. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013, 3, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Braczynski, A.K.; Vlaho, S.; Müller, K.; Wittig, I.; Blank, A.-E.; Tews, D.S.; Drott, U.; Kleinle, S.; Abicht, A.; Horvath, R. ATP synthase deficiency due to TMEM70 mutation leads to ultrastructural mitochondrial degeneration and is amenable to treatment. BioMed. Res. Inter. 2015, 2015, 462592. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, A.I.; Renkema, G.H.; Bras, M.; van den Heuvel, L.P.; Hoischen, A.; Gilissen, C.; Nabuurs, S.B.; Huynen, M.A.; de Vries, M.C.; Smeitink, J.A. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain 2013, 136, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Brea-Calvo, G.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Bürgi, S. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef] [PubMed]
- García-Corzo, L.; Luna-Sánchez, M.; Doerrier, C.; García, J.A.; Guarás, A.; Acín-Pérez, R.; Bullejos-Peregrín, J.; López, A.; Escames, G.; Enríquez, J.A. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet. 2012, 22, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Hikmat, O.; Tzoulis, C.; Knappskog, P.M.; Johansson, S.; Boman, H.; Sztromwasser, P.; Lien, E.; Brodtkorb, E.; Ghezzi, D.; Bindoff, L.A. ADCK 3 mutations with epilepsy, stroke-like episodes and ataxia: A POLG mimic? Eur. J. Neurol. 2016, 23, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; DiMauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Scalais, E.; Chafai, R.; Van Coster, R.; Bindl, L.; Nuttin, C.; Panagiotaraki, C.; Seneca, S.; Lissens, W.; Ribes, A.; Geers, C. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur. J. Paediatric Neurol. 2013, 17, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Farhan, S.M.; Wang, J.; Robinson, J.F.; Lahiry, P.; Siu, V.M.; Prasad, C.; Kronick, J.B.; Ramsay, D.A.; Rupar, C.A.; Hegele, R.A. Exome sequencing identifies NFS 1 deficiency in a novel Fe-S. cluster disease, infantile mitochondrial complex II/III deficiency. Mol. Genet.Genomic Med. 2014, 2, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.L.; Vanderver, A.; Yang, S.; Chang, T.; Cramp, L.; Vezina, G.; Lichter-Konecki, U.; Cusmano-Ozog, K.P.; Smpokou, P.; Chapman, K.A. Thiamine pyrophosphokinase deficiency causes a Leigh disease like phenotype in a sibling pair: Identification through whole exome sequencing and management strategies. Mol. Genet. Metabol. Rep. 2014, 1, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, F.; Ardissone, A.; Lamantea, E.; Garavaglia, B.; Zeviani, M.; Farina, L.; Ghezzi, D.; Moroni, I. Cavitating leukoencephalopathy with multiple mitochondrial dysfunction syndrome and NFU1 mutations. Front. Genet. 2014, 5, 412. [Google Scholar] [CrossRef] [PubMed]
- Wimplinger, I.; Morleo, M.; Rosenberger, G.; Iaconis, D.; Orth, U.; Meinecke, P.; Lerer, I.; Ballabio, A.; Gal, A.; Franco, B. Mutations of the Mitochondrial Holocytochrome c–Type Synthase in X-Linked Dominant Microphthalmia with Linear Skin Defects Syndrome. Am. J. Hum. Genet. 2006, 79, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Almalki, A.; Alston, C.L.; Parker, A.; Simonic, I.; Mehta, S.G.; He, L.; Reza, M.; Oliveira, J.M.; Lightowlers, R.N.; McFarland, R. Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. Biochimica. Et Biophysica. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Alsemari, A.; Al-Younes, B.; Goljan, E.; Jaroudi, D.; BinHumaid, F.; Meyer, B.F.; Arold, S.T.; Monies, D. Recessive VARS2 mutation underlies a novel syndrome with epilepsy, mental retardation, short stature, growth hormone deficiency, and hypogonadism. Hum. Genet. 2017, 11, 28. [Google Scholar]
- Coughlin, C.R.; Scharer, G.H.; Friederich, M.W.; Yu, H.-C.; Geiger, E.A.; Creadon-Swindell, G.; Collins, A.E.; Vanlander, A.V.; Van Coster, R.; Powell, C.A. Mutations in the mitochondrial cysteinyl-tRNA synthase gene, CARS2, lead to a severe epileptic encephalopathy and complex movement disorder. J. Med. Genet. 2015, 52, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Elo, J.M.; Yadavalli, S.S.; Euro, L.; Isohanni, P.; Götz, A.; Carroll, C.J.; Valanne, L.; Alkuraya, F.S.; Uusimaa, J.; Paetau, A. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum. Mol. Genet. 2012, 21, 4521–4529. [Google Scholar] [CrossRef] [PubMed]
- Scheper, G.C.; Van Der Klok, T.; Van Andel, R.J.; Van Berkel, C.G.; Sissler, M.; Smet, J.; Muravina, T.I.; Serkov, S.V.; Uziel, G.; Bugiani, M. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nature Genet. 2007, 39, 534. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Richard, E.M.; Wang, X.; Shahzad, M.; Huang, V.H.; Qaiser, T.A.; Potluri, P.; Mahl, S.E.; Davila, A.; Nazli, S. Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet. 2015, 11, e1005097. [Google Scholar] [CrossRef] [PubMed]
- Simons, C.; Griffin, L.B.; Helman, G.; Golas, G.; Pizzino, A.; Bloom, M.; Murphy, J.L.; Crawford, J.; Evans, S.H.; Topper, S. Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am. J. Hum. Genet. 2015, 96, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Steenweg, M.E.; Ghezzi, D.; Haack, T.; Abbink, T.E.; Martinelli, D.; van Berkel, C.G.; Bley, A.; Diogo, L.; Grillo, E.; Te Water Naudé, J. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’caused by EARS2 mutations. Brain 2012, 135, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Janer, A.; Antonicka, H.; Lalonde, E.; Nishimura, T.; Sasarman, F.; Brown, G.K.; Brown, R.M.; Majewski, J.; Shoubridge, E.A. An RMND1 Mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am. J. Hum. Genet. 2012, 91, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Kernohan, K.D.; Dyment, D.A.; Pupavac, M.; Cramer, Z.; McBride, A.; Bernard, G.; Straub, I.; Tetreault, M.; Hartley, T.; Huang, L. Matchmaking facilitates the diagnosis of an autosomal-recessive mitochondrial disease caused by biallelic mutation of the tRNA isopentenyltransferase (TRIT1) gene. Hum. Mutation 2017, 38, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.A.; Elpeleg, O.; Antonicka, H.; Diepstra, H.; Saada, A.; Smits, P.; Sasarman, F.; Vriend, G.; Jacob-Hirsch, J.; Shaag, A. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am. J. Hum. Genet. 2006, 79, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Smits, P.; Antonicka, H.; van Hasselt, P.M.; Weraarpachai, W.; Haller, W.; Schreurs, M.; Venselaar, H.; Rodenburg, R.J.; Smeitink, J.A.; van den Heuvel, L.P. Mutation in subdomain G’of mitochondrial elongation factor G1 is associated with combined OXPHOS deficiency in fibroblasts but not in muscle. Eur. J. Hum. Genet. 2011, 19, 275. [Google Scholar] [CrossRef] [PubMed]
- Vedrenne, V.; Gowher, A.; De Lonlay, P.; Nitschke, P.; Serre, V.; Boddaert, N.; Altuzarra, C.; Mager-Heckel, A.-M.; Chretien, F.; Entelis, N. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am. J. Hum. Genet. 2012, 91, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Wedatilake, Y.; Niazi, R.; Fassone, E.; Powell, C.A.; Pearce, S.; Plagnol, V.; Saldanha, J.W.; Kleta, R.; Chong, W.K.; Footitt, E. TRNT1 deficiency: Clinical, biochemical and molecular genetic features. Orphanet J. Rare Dis. 2016, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Hikmat, O.; Eichele, T.; Tzoulis, C.; Bindoff, L.A. Understanding the epilepsy in POLG related disease. Int. J. Mol. Sci. 2017, 18, 1845. [Google Scholar] [CrossRef] [PubMed]
- Kasapkara, Ç.S.; Tümer, L.; Küçükçongar, A.; Hasanoglu, A.; Seneca, S.; De Meirleir, L. DGUOK-related mitochondrial DNA depletion syndrome in a child with an early diagnosis of glycogen storage disease. J. Pediatric gastroenterol. Nutr. 2013, 57, e28–e29. [Google Scholar] [CrossRef] [PubMed]
- Lesko, N.; Naess, K.; Wibom, R.; Solaroli, N.; Nennesmo, I.; von Döbeln, U.; Karlsson, A.; Larsson, N.-G. Two novel mutations in thymidine kinase-2 cause early onset fatal encephalomyopathy and severe mtDNA depletion. Neuromuscular Disorders 2010, 20, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.; Smith, C.; Fratter, C.; Alston, C.L.; He, L.; Craig, K.; Blakely, E.L.; Evans, J.C.; Taylor, J.; Shabbir, Z. Adults with RRM2B-related mitochondrial disease have distinct clinical and molecular characteristics. Brain 2012, 135, 3392–3403. [Google Scholar] [CrossRef] [PubMed]
- Carrozzo, R.; Dionisi-Vici, C.; Steuerwald, U.; Lucioli, S.; Deodato, F.; Di Giandomenico, S.; Bertini, E.; Franke, B.; Kluijtmans, L.A.; Meschini, M.C. SUCLA 2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain 2007, 130, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Ohlenbusch, A.; Edvardson, S.; Skorpen, J.; Bjornstad, A.; Saada, A.; Elpeleg, O.; Gärtner, J.; Brockmann, K. Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet J. Rare Dis. 2012, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Valayannopoulos, V.; Haudry, C.; Serre, V.; Barth, M.; Boddaert, N.; Arnoux, J.-B.; Cormier-Daire, V.; Rio, M.; Rabier, D.; Vassault, A. New SUCLG1 patients expanding the phenotypic spectrum of this rare cause of mild methylmalonic aciduria. Mitochondrion 2010, 10, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.D.; Isomursu, A.; Mennerich, D.; Vapola, M.H.; Weiher, H.; Kietzmann, T.; Hiltunen, J.K. The human mtDNA depletion syndrome gene MPV17 encodes a non-selective channel that modulates membrane potential. J. Biol. Chem. 2015, 290, 13840–13861. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Ghezzi, D.; Johnson, M.A.; Biagosch, C.A.; Shamseldin, H.E.; Haack, T.B.; Reyes, A.; Tsukikawa, M.; Sheldon, C.A.; Srinivasan, S. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 2013, 93, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Zeviani, M.; Muntoni, F.; Savarese, N.; Serra, G.; Tiranti, V.; Carrara, F.; Mariotti, C.; DiDonato, S. A MERRF/MELAS overlap syndrome associated with a new point mutation in the mitochondrial DNA tRNA^ Lys gene. Eur. J. Hum. Genet. 1993, 1, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Zsurka, G.; Elger, C.E.; Kunz, W.S. Mitochondrial involvement in temporal lobe epilepsy. Exper. Neurol. 2009, 218, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Koene, S.; Rodenburg, R.; Van Der Knaap, M.; Willemsen, M.; Sperl, W.; Laugel, V.; Ostergaard, E.; Tarnopolsky, M.; Martin, M.; Nesbitt, V. Natural disease course and genotype-phenotype correlations in Complex I deficiency caused by nuclear gene defects: What we learned from 130 cases. J. Inher. Metab. Dis. 2012, 35, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Sofou, K.; De Coo, I.F.; Isohanni, P.; Ostergaard, E.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; De Angst, I.B.; Lönnqvist, T. A multicenter study on Leigh syndrome: Disease course and predictors of survival. Orphanet J. Rare Dis. 2014, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Annegers, J.F.; Rocca, W.A.; Hauser, W.A. Causes of epilepsy: Contributions of the Rochester epidemiology project. Mayo Clinic Proc 1996, 71, 570–575. [Google Scholar] [CrossRef]
- Kovac, S.; Domijan, A.-M.; Walker, M.C.; Abramov, A.Y. Prolonged seizure activity impairs mitochondrial bioenergetics and induces cell death. J. Cell Sci. 2012, 125, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Ly, C.V.; Venken, K.J.; Koh, T.-W.; Zhou, Y.; Bellen, H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, T.; Evans, M.; Meldrum, B. Intracellular calcium accumulation in rat hippocampus during seizures induced by bicuculline orl-allylglycine. Neuroscience 1983, 10, 385–395. [Google Scholar] [CrossRef]
- Kovac, S.; Abramov, A.Y.; Walker, M.C. Energy depletion in seizures: Anaplerosis as a strategy for future therapies. Neuropharmacology 2013, 69, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Domijan, A.; Walker, M.; Abramov, A. Seizure activity results in calcium-and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 2014, 5, e1442. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 1992, 267, 8834–8839. [Google Scholar] [PubMed]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blalchy-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef]
- Gulati, A. Endothelin receptors, mitochondria and neurogenesis in cerebral ischemia. Curr. Neuropharmacol. 2016, 14, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Guntuku, L.; Naidu, V.; Yerra, V.G. Mitochondrial dysfunction in gliomas: Pharmacotherapeutic potential of natural compounds. Curr. Neuropharmacol. 2016, 14, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Kuhad, A. Mitochondrial dysfunction in depression. Curr. Neuropharmacol. 2016, 14, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kato, N. Mitochondrial dysfunction in bipolar disorder. Bipolar Disorders 2000, 2, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Rezin, G.T.; Amboni, G.; Zugno, A.I.; Quevedo, J.; Streck, E.L. Mitochondrial dysfunction and psychiatric disorders. Neurochem. Res. 2009, 34, 1021. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | Source | |
|---|---|---|
| Complex I Subunits Assembly factors | NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS6, NDUFS7, NDUFS8, NDUFV1, NDUFA1, NDUFA11 | [188,189,190,191,192,193,194,195,196] |
| Complex I Assembly Factors | NDUFAF1, NDUFAF2, NDUFAF3, NDUFAF4, NDUFAF5, NDUFAF6, AIFM1, FOXRED1, NUBPL | [197,198,199,200,201,202,203] |
| Complex II Subunits | SDHA | [204] |
| Complex III Subunits and Assembly Factors | UQCC2, UQCRC2, BCS1L | [205,206,207] |
| Complex IV Subunits and Assembly Factors | NDUFA4, ETHE1, APOPT1, SURF1, SCO1, SCO2, COX10, COX15, FASTKD2, PET100 | [208,209,210,211,212,213,214,215,216] |
| Complex V Subunits and Assembly Factors | ATP5A1, ATPAF2, TMEM70 | [210,217,218] |
| Solute Carriers | SLC25A12, SLC25A22 | [183,184] |
| Coenzyme Q10 Biosynthesis | ADCK3, COQ2, COQ4, COQ6, COQ9, PDSS2 | [219,220,221,222,223,224] |
| ETC Cofactor Biosynthesis | LIAS, BOLA3, NFU1, NFS1, TPK, HCCS | [181,182,225,226,227,228] |
| tRNA Aminoacylation | AARS2, CARS2, DARS2, EARS2, FARS2, NARS2, RARS2, VARS2 | [229,230,231,232,233,234,235,236] |
| Posttranslational Regulators | GFM1, TSFM, PNPT1, RMND1, TRIT1, TRNT1 | [237,238,239,240,241,242] |
| Mitochondrial DNA Replication | C10orf2, POLG | [197,243] |
| Nucleotide Pool | DGUOK, TYMP, TK, RRMB | [244,245,246] |
| TCA Cycle | SUCLA2, SULG1, SDHAF1 | [247,248,249] |
| Other Mitochondrial DNA Depletion Syndromes | FBXL4, MPV17 | [250,251] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Austin, G.L.; Young, L.E.A.; Johnson, L.A.; Sun, R. Mitochondrial Metabolism in Major Neurological Diseases. Cells 2018, 7, 229. https://doi.org/10.3390/cells7120229
Zhou Z, Austin GL, Young LEA, Johnson LA, Sun R. Mitochondrial Metabolism in Major Neurological Diseases. Cells. 2018; 7(12):229. https://doi.org/10.3390/cells7120229
Chicago/Turabian StyleZhou, Zhengqiu, Grant L. Austin, Lyndsay E. A. Young, Lance A. Johnson, and Ramon Sun. 2018. "Mitochondrial Metabolism in Major Neurological Diseases" Cells 7, no. 12: 229. https://doi.org/10.3390/cells7120229
APA StyleZhou, Z., Austin, G. L., Young, L. E. A., Johnson, L. A., & Sun, R. (2018). Mitochondrial Metabolism in Major Neurological Diseases. Cells, 7(12), 229. https://doi.org/10.3390/cells7120229