Abstract
Mitochondria are bilayer sub-cellular organelles that are an integral part of normal cellular physiology. They are responsible for producing the majority of a cell’s ATP, thus supplying energy for a variety of key cellular processes, especially in the brain. Although energy production is a key aspect of mitochondrial metabolism, its role extends far beyond energy production to cell signaling and epigenetic regulation–functions that contribute to cellular proliferation, differentiation, apoptosis, migration, and autophagy. Recent research on neurological disorders suggest a major metabolic component in disease pathophysiology, and mitochondria have been shown to be in the center of metabolic dysregulation and possibly disease manifestation. This review will discuss the basic functions of mitochondria and how alterations in mitochondrial activity lead to neurological disease progression.
1. General Mitochondrial Function
1.1. Mitochondria: Metabolic Hub of a Cell
The mitochondrion is the result of evolution from a perfect marriage of an α-proteobacterium and a precursor of a modern eukaryotic cell, evidenced by it being the only non-nuclear sub-cellular organelle that has its own DNA [1]. In mammals, mitochondria are passed down maternally [2] and they are present in every cell in the body, except red blood cells [3]. Mitochondria are bilayer organelles with an outer and inner membrane that enclose the intermembrane space and a matrix compartment, respectively [4]. Mitochondrial DNA (mtDNA) are circular and reside within the matrix. Furthermore, mtDNA are intron-free which make them more susceptible to mutagenesis than nuclear DNA [5]. The mitochondrial proteome consists of over 3300 proteins and more are being identified daily [6]. mtDNA encode for 13 proteins that are part of the electron transport chain, which produces ATP via oxidative phosphorylation [7]. Transport of nuclear-encoded proteins to either the matrix or the intermembrane space requires separate signals. Matrix localization signals are located on the N-terminal of a protein and transport to the matrix requires membrane potential and ATP hydrolysis [8]. The intermembrane translocation signal is a hydrophilic region internal to the cell membrane that directs protein localization independent of membrane potential or ATP hydrolysis [9,10]. The intermembrane space is home to proteins important for mitochondrial structural integrity and multiple proteins in the BCL-2 family that control programmed cell death or apoptosis [11,12]. The intermembrane space and mitochondrial matrix contain proteins for the tricarboxylic acid (TCA) cycle—the major metabolic hub for cellular homeostasis and the electron transport chain that generates ATP from the redox gradient [13,14,15]. During ATP production, mitochondria generate a large number of reactive oxygen species (ROS) that are contained within the matrix [16]. Controlled release of ROS supports signaling events [17,18,19]; however, ectopic release of ROS from the mitochondria can result in DNA, RNA, and protein damage that ultimately leads to cell death [20,21]. Recent advances in techniques such as real-time oxygen consumption monitoring [22] and stable isotope enriched metabolomics have revealed an enormous amount of information on mitochondrial metabolism and its connection to cellular physiology [23,24]. These studies confirm that mitochondrion is an extremely complex and dynamic organelle and is at the crossroads of cellular metabolism and signaling.
1.2. Mitochondria and Energy Production
The major biochemical pathway in the matrix of a mitochondrion is the TCA cycle, shown in Figure 1. Dr. Hans Adolf Krebs won the Nobel Prize in Physiology or Medicine in 1953 for its discovery; hence, it is also referred to as the Krebs cycle [25]. Dr. Krebs discovered that all of the enzymes in the TCA cycle are bi-directional in cell-free assays [25] which led to the illustration of the circular pathway seen in biochemical textbooks. The primary role of the TCA cycle is to extract electrons from carbon sources in the form of NADH and FADH2 and supply them to the electron transport chain (ETC) for the production of ATP [26,27]. Cytoplasmic metabolites supply the TCA cycle to facilitate its continuous operation and are divided into two categories: (1) the canonical oxidative pathway and (2) anaplerotic pathways. The canonical oxidation pathway is a continuation from glycolysis, where pyruvate enters the TCA cycle as acetyl-CoA, carried out by the enzyme pyruvate dehydrogenase (PDH), which is then converted to citrate [27,28]. This is viewed as the major pathway to supply oxidative phosphorylation or ATP production. The anaplerotic pathways involve metabolic enzymes such as pyruvate carboxylase (PCB) [29], phosphoenolpyruvate carboxykinase (PEPCK) [30], malic enzyme (ME) [31], glutaminase (GLS) [32] and glutamate dehydrogenase (GDH) [33]. While PCB, PEPCK and ME connect the TCA cycle with glycolysis; GLS/GDH supply carbon from glutamine. Anaplerotic pathways are bi-directional and most commonly believed to primarily maintain compartmentalized metabolite pools between the cytosol and mitochondria but not contribute to ATP production directly [34,35]. However, alternative studies have suggested that glutamine supports NADH production from the GLS/GDH anaplerotic pathway equally, if not more, when compared to the canonical pathway [36,37].
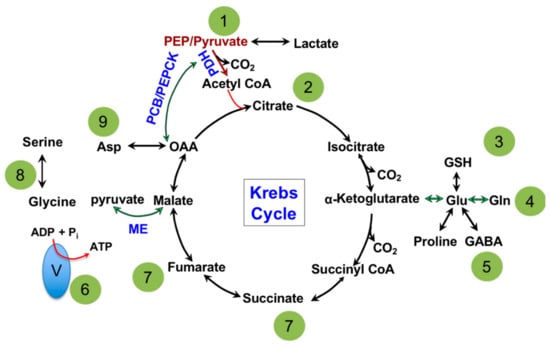
Figure 1.
Major pathways and metabolite exchange that take place in the mitochondria. 1: Glycolysis and gluconeogenesis connects to the mitochondria by phosphoenolpyruvate (PEP), pyruvate and lactate. 2: Fatty acid biosynthesis and oxidation utilize citrate and acetyl-CoA as metabolic intermediates. 3: Glutathione (GSH) is produced in the mitochondria from glutamate, which maintains cellular redox balance. 4: Protein biosynthesis uses de novo synthesized amino acids from the mitochondria. 5 Neurotransmitter biosynthesis (proline and GABA) partially takes place in the mitochondria then continues in the cytosol. 6: Electron Transport Chain subunits I-V make up the Oxidative Phosphorylation pathway and are located on the inner mitochondrial membrane and use reducing equivalents, NADH and FADH2, as electron sources. Electrons move through the subunits from I or II to IV, which creates a proton gradient in the process. The proton gradient is used by subunit V to generate ATP. 7: Fumarate and succinate are exported to the cytosol as enzyme co-factors. 8: 1-carbon metabolism supplies carbon for purine nucleotide biosynthesis and methylation of proteins and DNA. 9: Aspartate (Asp) supports pyrimidine ring biosynthesis. →: Canonical oxidative pathway. →: Anaplerotic pathway. Pyruvate dehydrogenase (PDH); pyruvate carboxylase (PCB); phosphoenolpyruvate carboxykinase (PEPCK); malic enzyme (ME); gamma-aminobutyric acid (GABA).
1.3. Mitochondria and Nucleotide Biosynthesis
To maintain homeostasis during proliferation, a cell needs to produce energy, nucleotides, and fatty acids. All three biosynthetic pathways are connected to mitochondria through metabolite exchange. Energy production mainly stems from dehydrogenase activities within the TCA cycle, as mentioned earlier. Nucleotide biosynthesis also relies on key metabolites inside mitochondria. The two major classes of nucleotides are pyrimidines and purines. Pyrimidine nucleotides include cytosine, thymine, and uracil. Its structure consists of a ribose base connected to a 4-carbon diazine ring, see Figure 2. The ribose base is produced from the pentose phosphate pathway (PPP) that takes place in the cytosol. The biosynthesis of the diazine ring requires oxaloacetate, a TCA cycle intermediate, as a precursor. The first enzyme of this biochemical pathway is aspartate aminotransferase (GOT) [38] and its dominant isoform, GOT2, is primarily a mitochondrial enzyme [39]. Purine nucleotides include the key molecules adenine and guanine crucial for DNA and RNA biosynthesis as well as other biomolecules such as AMP, GMP, and NADH, as shown in Figure 3. Similar in structure to pyrimidines, purine nucleotides also contain a ribose base connected to a nitrogenous base produced from the PPP. Purine rings consist of a pyrimidine diazine fused to an imidazole ring. The 1-carbon or folate pathway donates carbon as 5-methyltetrahydrofolate (CHO-THF) to the biosynthesis of purine rings [40]. Key enzymes involved in this metabolic pathway include serine hydroxymethyltransferase (SMHT) [41], glycine decarboxylase (GDC) [42], and methylenetetrahydrofolate dehydrogenase (MTHFD) [43]. All three of these enzymes have mitochondrial isoforms expressed in a wide range of tissue types, suggesting the production of purine rings, at least in part, takes place in the mitochondria [44,45].
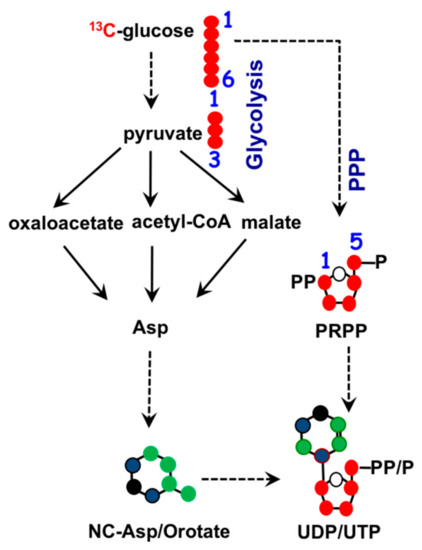
Figure 2.
Biochemistry of pyrimidine nucleotide synthesis. Ribose base biosynthesis comes from the pentose phosphate pathway (PPP) in the cytosol. Mitochondrial TCA cycle metabolites oxaloacetate and aspartate (asp) supply three out of the four carbons in the diazine ring. •: Possible cytosolic-derived carbons, •: Possible mitochondrial-derived carbons. •: Nitrogen.
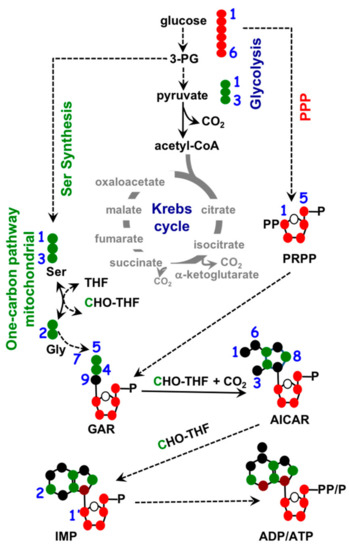
Figure 3.
Biochemistry of purine nucleotide synthesis. Ribose base biosynthesis comes from pentose phosphate pathway (PPP) in the cytosol, mitochondrial 1-carbon metabolism contributes to purine ring biosynthesis through 5-methyltetrahydrofolate (CHO-THF). •: Possible cytosolic-derived carbons, •: Possible mitochondrial-derived carbons.
1.4. Mitochondria and Fatty Acid Metabolism.
Fatty acids store energy, are precursors to lipid bilayers, and function as signaling molecules. De novo fatty acid synthesis and β-oxidation are opposing processes that either begin or end in the mitochondria. Acetyl-CoA and citrate are two key metabolites required for fatty acid synthesis. Citrate derived from glucose is transported out of the mitochondria and converted by ATP citrate lyase back to acetyl-CoA, the building block of fatty acids [46,47]. While fasting, stored fats undergo lipolysis to produce fatty acids for ATP production. During β-oxidation, free fatty acids are added to L-carnitine and transported into the mitochondria to be broken down sequentially to acetyl-CoA, which is then fed into the TCA cycle for ATP production [48,49]. Recent reports suggest that mitochondrial lipids such as lipoic acid, a co-factor for mitochondrial dehydrogenase, and 3-hydroxymyristyl-ACP, a structural component of complex I of the electron transport chain, are made de novo inside mitochondria in yeast [50]. To our knowledge, this process has yet to be studied in mammalian systems.
1.5. Mitochondria Metabolism and Epigenetics
Epigenetic programming is the way chromatin is organized. It has been shown to dictate cell fate [51], control the cell cycle [52], and contribute to disease pathology [53]. Post-translational modifications that alter gene expression patterns include histone acetylation, glycosylation, phosphorylation, and histone or DNA methylation. Histone acetylation occurs when the mitochondrial metabolite acetyl-CoA is added to the amino group of lysine residues on a histone. Acetyl-CoA is produced from pyruvate dehydrogenase, fatty acid oxidation, and exogenous acetate, processes that all take place inside mitochondria [54,55]. However, the exact relation between compartmentalized regulation of acetyl-CoA concentration to support both mitochondrial metabolism and histone acetylation remains unclear. Acetyl-CoA is also required for the production of uridine diphosphate N-acetyl-glucosamine (UDP-GlcNAc), a precursor for histone glycosylation [56]. The functional consequence of histone glycosylation is relatively unknown compared to other histone modifications but its level seems to correlate with extracellular glucose flux [57].
Levels of mitochondrial metabolic intermediates such as ATP and NAD+ can affect histone phosphorylation and acetylation, respectively. ATP is the substrate for histone phosphorylation by histone kinases [58] and NAD+ is a co-factor for the sirtuin family of deacetylases [59]. Mitochondria regulate NAD+ levels in two ways: first, through oxidative phosphorylation, where NAD+/NADH cycling takes place. Second, NAD+ is a purine nucleotide whose biosynthesis requires the serine-glycine 1-carbon metabolic pathway mentioned above. Furthermore, hydroxylase and demethylase activities are especially sensitive to changes in α-ketoglutarate (αKG), fumarate, and succinate concentrations. All three molecules are substrates crucial for the functioning of dioxygenases including prolyl hydroxylases and histone lysine demethylases, which are important for epigenetic regulation [60,61,62]. Germline or somatic mutations in mitochondrial enzymes that produce abnormal amounts of succinate [63], fumarate [64], and 2-hydroxyglutarate (2HG) [65], have been shown to have an inhibitory effect on these enzymes, leading to DNA hypermethylation-induced epigenetic alterations.
Mitochondrial metabolism controls the epigenetic landscape by providing substrates for acetylation, phosphorylation, and methylation of histones. The balanced supply of substrates from mitochondria to the nucleus is an extremely complex system that is tightly regulated by interconnected signaling pathways. Somatic mutations or altered mitochondrial metabolism by different diseased states greatly affect histone modification and the epigenetic landscape. Detailed mechanisms of how a cell can maintain the flow of metabolites between mitochondria and the nucleus to support proliferation, differentiation, and autophagy are relatively unknown and form an area of intense research.
2. Mitochondria Dysfunction in Alzheimer’s Disease
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive and severe memory loss. AD is the most common form of dementia, affecting over 5.3 million people in the United States alone [66]. The strongest risk factor for AD is age, and as the elderly population expands, the number of individuals afflicted with this devastating disease is growing rapidly; particularly considering that effective therapies for the disease remain elusive [66]. The AD brain is characterized by two neuropathological hallmarks: extracellular senile plaques and intracellular neurofibrillary tangles [67]. The plaques are formed mostly from the deposition of amyloid β (Aβ) peptide, while neurofibrillary tangles are formed from neurofilaments and hyperphosphorylated tau protein. A small percentage of AD cases (<5%) are caused by mutations in genes encoding for either the amyloid β precursor protein (APP) or from mutations in genes encoding presenilin 1 or presenilin 2 (involved in the generation of amyloid beta (Aβ) from APP) [68]. These rare, autosomal dominantly inherited cases are typically referred to as familial or early-onset AD. However, the vast majority of AD cases occur sporadically in individuals >65 years of age and are commonly referred to as late-onset AD (LOAD) [67]. Despite the sporadic nature of LOAD, the inheritance of several well-established susceptibility genes significantly increases the risk of disease [69].
In addition to advanced age, various metabolic impairments predispose individuals to cognitive dysfunction and dementia, including LOAD. For example, metabolic dysfunction as in the case of insulin resistance and type 2 diabetes, increases the risk of dementia and shares several pathological characteristics with AD, such as neuroinflammation, increased oxidative stress, and cerebrovascular dysfunction [70,71]. Metabolic disorders also increase in incidence with age [72], meaning the number of high-risk elderly individuals suffering from metabolic disorders is expanding precipitously.
The central role of metabolism in AD is likely due to the fact that normal synaptic function requires a multitude of energy-intensive processes [73]. The first clues pointing toward a potential metabolic component of the disease were observed nearly four decades ago, as a series of neuroimaging studies utilizing flurodeoxyglucose positron emission tomography (FDG-PET) that showed that the brains of individuals with AD took up less glucose compared to those of cognitively normal controls [74,75,76,77]. Since these initial findings in the early 1980s, a multitude of additional studies have confirmed the phenomena, and a reduction in the cerebral metabolic rate of glucose (CMRglc), as measured by FDG-PET, is now considered one of the hallmarks of AD [78]. In fact, FDG-PET is able to differentiate AD from other types of dementia with a high degree of specificity due to specific regional signaling patterns [79]. Clinical AD symptoms essentially never occur without glucose hypometabolism, the extent of which strongly correlates with the severity of clinical symptoms [80,81,82]. Importantly, evidence suggests that these alterations in glucose metabolism occur very early in the neurodegenerative process [83,84,85,86], raising the possibility that metabolic dysfunction precedes, and possibly contributes causally, to the pathophysiology of AD.
Despite these important findings, the molecular mechanisms underlying AD neurodegeneration remain elusive. The field has been dominated by the “amyloid hypothesis”, which argues that sequential cleavages of APP produce Aβ, initiating a cascade of neurodegeneration [87]. In this context, mitochondrial impairments observed in AD patients are thought to be secondary effects of Aβ toxicity, as Aβ may block mitochondrial translocation of ETC components [88,89,90], and impair respiratory chain function and oxidative phosphorylation in mitochondria [91,92]. Alternatively, some evidence points to a direct effect of mitochondria on AD neuropathology, which are either independent of Aβ or, themselves, potentially drive the changes in APP and Aβ homeostasis.
In support of the latter, shortly after the initial reports demonstrating the reduced glucose uptake via FDG-PET in AD patients, a series of additional PET-based studies described reductions in oxygen consumption in AD brains [93,94]. Importantly, around this same time, several groups began to show evidence of mitochondrial dysfunction in AD brain tissue. This included reductions in cytochrome oxidase (COX) activity in AD patients [88], and decreases in mitochondrial enzymes such as PDH, and other enzymes in the early part of the TCA cycle, such as isocitrate dehydrogenase and α-ketoglutarate dehydrogenase (αKGDH) [95,96]. Additionally, the activity of late TCA cycle enzymes such as succinate dehydrogenase and malate dehydrogenase were increased in AD brains, and the alterations of TCA cycle enzyme activities correlated strongly with clinical symptoms [97]. Together, these studies may point toward TCA enzyme activity reductions, resulting in reduced cerebral glucose metabolism, which translates to clinically relevant cognitive impairments.
While age is the primary risk factor for AD, genetic factors also strongly contribute to disease risk (Bertram et al., 2007; Bertram et al., 2009). The strongest genetic risk factor for the more common late-onset form of AD is APOE [98,99]. In humans, there are three major isoforms of apoE: E2, E3, and E4 [100]. Apolipoprotein E (apoE), which is associated with circulating lipoproteins, plays several important metabolic roles [100]. In the periphery, apoE is primarily produced by the liver, while in the brain, apoE is primarily produced by astrocytes, and is responsible for neuronal maintenance and repair in addition to a number of other physiological and pathological roles [101,102,103]. E3 is the major isoform expressed in humans (~60% of the population), and the effects of E2 and E4 are typically compared to those of E3 to determine the relative risk [98,104]. Importantly, the E4 isoform confers between a 2- (heterozygous) to 15-fold (homozygous) increase in the risk of developing AD [98], and studies suggest the E4 allele may account for up to 50% of AD in the US [99].
Interestingly, a consistent pattern of brain glucose hypometabolism, similar to that seen in AD, has been noted in individuals with E4 [84,105]. Even non-demented, cognitively normal E4 carriers demonstrate this pattern of glucose hypometabolism [106]. Importantly, these metabolic deficits are present decades in advance of AD onset in E4+ and other at-risk individuals thereby lending support to this being an inherent biological feature of E4, rather than simply a byproduct of dementia [107,108]. The metabolic impairments associated with E4 extend beyond reductions in CMRglc to also include mitochondrial deficits described in several in vivo and in vitro models. For example, the expression of the subunits of several mitochondrial respiratory complexes is decreased in neurons expressing E4 compared to those expressing E3 in a mouse model of neuron-specific apoE expression [109]. Mitochondrial protein expression was also shown to be modified by APOE genotype in both the absence and presence of an ischemic injury [110]. Consistent with a role of upregulated mitochondria-associated endoplasmic reticulum membrane (MAM) function in AD, Tambini et al. recently showed that E4-containing astrocyte conditioned media increased MAM activity in vitro [111]. Additionally, a series of studies in mice show that apoE4 is cleaved by a protease in neurons to generate a toxic apoE4(1-272) fragment with a mitochondrial targeting sequence [112]. This fragment of apoE4 has been shown to bind to mitochondrial complexes and affect their activities [113]. Finally, an exciting new study by Zhao et al. shows that E4 inhibits insulin-induced mitochondrial respiration in primary neurons [114].
Two studies have reported an interaction between APOE genotype and mitochondrial DNA haplogroups in determining AD risk [115,116], and E4-associated mitochondrial deficits have also been confirmed in human autopsy tissue. Importantly, these abnormalities appear to begin early in life—at a point that precedes amyloid deposition. For example, post-mortem brains from young adult APOE ε4 carriers show reduced COX [117,118]. Interestingly, these findings extend to the periphery, as platelet mitochondria COX activity in AD subjects with E4 alleles are lower than in non-carrier AD subjects [119].
In addition to Alzheimer’s disease, mitochondrial dysfunction has been reported in other neurodegenerative diseases such as Huntington’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Mitochondrial metabolism in these diseases has been extensively reviewed previously [120,121,122,123,124]; therefore, we will be focusing on other neurological disorders such as traumatic brain injury and epilepsy for the remainder of the review.
3. Mitochondrial Disruption Following TBI
Traumatic brain injury (TBI) is a major cause of death and disability estimated to affect approximately 1.7 million people annually in the US [125]. The cause of injury consists of a primary insult due to acceleration and deceleration forces on neuronal structures. Following the primary injury phase is the secondary injury cascade. It is believed that it is the secondary injury, involving a complex cascade of biochemical, metabolic, and molecular changes, that leads to profound malfunctioning in cerebral cells and permanent long-term sequelae of TBI. Importantly, metabolic changes are heavily involved in this process [126,127,128]. It has been proposed that it’s the neuronal hyperexcitability, intracellular calcium concentration elevations, and mitochondrial enzymatic alterations in the secondary cascade that ultimately lead to oxidative stress and cellular death [129,130,131].
The secondary injury cascade after TBI is initiated by disruptions in the membrane potential that lead to spontaneous firing of the neuronal cells [132]. The aberrant action potentially causes the release of numerous neurotransmitters, primarily the excitatory amino acid glutamate [133,134]. Increases in extracellular glutamate concentrations following TBI are accompanied by a rise in lactate and a decline in extracellular glucose [135,136,137], suggestive of a shift from oxidative metabolism to non-oxidative methods such as glycolysis. Glutamate is an amino acid that is closely intertwined with mitochondrial energy production [138]. Its biosynthesis takes place in the mitochondria, from either αKGDH by GDH or from glutamine by GLS. Furthermore, the cytoplasmic enzyme GOT promotes the reversible conversion of glutamate to αKGDH, a key step during nucleotide biosynthesis. The aberrant release of glutamate could potentially alter all of the linked pathways described above. In addition, glutamate is one of the precursors for the production of gamma-aminobutyric acid (GABA), the major inhibitory neurotransmitter [139,140]; hence, an ectopic release of glutamate could result in the lack of GABA production, and consequently, a lack of inhibitory responses in the brain. The interplay between glutamate, the TCA cycle, and mitochondrial metabolism is only beginning to emerge. Due to the complexity of the metabolic networks affected by glutamate, further metabolomic approaches are needed to improve our understanding at a system level.
The increase in excitatory neurotransmission from increased glutamate release leads to cellular depolarization and calcium influx into neurons via AMPA, NMDA channel, and voltage-gated calcium channels (VGCC) [141,142,143], further causing energy depletion and excitotoxic-mediated cell death [144]. As the intracellular calcium concentration increases, the mitochondria attempt to buffer intracellular calcium by sequestering it into the matrix through mitochondrial permeability transition pores [143,145]. Increased mitochondrial calcium levels result in mitochondrial dysfunction in a dose-dependent manner [146,147,148]. Furthermore, studies have shown that increased calcium levels have an inhibitory effect over key mitochondrial dehydrogenase enzymes such as PDH, NAD-dependent isocitrate dehydrogenase, and αKGDH in in vitro models [149,150]. Several studies have confirmed these findings after TBI. PDH activity has been found to be significantly reduced in blood levels of TBI patients [151]. Similarly, in a rodent model, αKGDH activity was decreased after TBI, while application of a αKGDH coenzyme precursor, thiamine, rescued the activity of αKGDH and restored mitochondrial respiration [152]. Further studies are needed to confirm the effect of calcium elevations on other mitochondrial enzymes in TBI.
In addition to the aforementioned increase in excitatory signaling and intracellular calcium, several other factors may contribute to the energy crisis defined after TBI. HIF1α, a transcription factor responsive to acute hypoxia, has been found to be elevated in the brains of mice after TBI models [153,154]. Multiple studies have documented a significant role of HIF1α in elevating aquaporin (AQP)-4 and -9 further exacerbating brain edema post-TBI [153,154,155]. Pharmacological inhibition of HIF1α using 2-methoxyestradiol (2ME2) reduced brain edema, AQP-4 and -9 expression [153]. HIF1α activation also reduces glucose and glutamine flux through mitochondria in other experimental models [156,157,158]; however, it has not yet been investigated whether similar metabolic changes are also occurring post-TBI.
It has been long shown that acute mitochondrial disruption occurs following experimental TBI [142,159]. The secondary injury cascade after TBI causes detrimental cell damage and is marked by alterations in excitatory amino acids such as glutamate [160,161] increases in acute hypoxia [162], and the disruption of calcium homeostasis [141]; all of which alter mitochondrial metabolism. The vital roles for mitochondria in cellular function and survival have resulted in increased efforts to identify the molecular events associated with mitochondrial impairment in TBI.
4. Role of Mitochondria in Epilepsy
Epilepsy is a neurological disorder that affects approximately 1.2% of the US population [163]. It consists of abnormal, excessive, hypersynchronous discharges from a population of neurons that disrupt normal brain function. The causes of epilepsy are multifactorial, ranging from trauma to the brain, tumor growth, infection, malformations, or genetic abnormalities [164]. Despite the large variability in causes, mitochondrial dysfunction is frequently involved. Mitochondrial gene mutations frequently lead to seizures [165,166], while nuclear genes that alter mitochondrial function can also result in epilepsy [167]. Moreover, seizure activity triggers the release of ROS, calcium influxes, neurotransmitter imbalances that cause further alterations in mitochondrial metabolism, and neuronal death [168,169,170]. Therefore, mitochondrial dysfunction can be both a cause and a consequence of epileptic disease [171].
Research in the last few decades has highlighted numerous nuclear genes that alter mitochondrial function, over 100 of which have been linked to epilepsy [167]. Mutations in the nuclear gene POLG, which encodes for the catalytic subunit of mitochondrial DNA, polymerase gamma, has been the one of the most studied [172,173,174,175,176,177]. Mutations have resulted in numerous phenotypes ranging from generalized seizures to myopathy, hepatopathy, sensory-ataxias, and opthalmoparesis [175,178]. Iron–sulfur clusters are important for the assembly of the electron transfer complexes [179]. Disruption of multiple genes in this pathway, such as LIAS, BOLA3, and NFU1, are associated with mitochondrial-disease-induced epilepsy [180,181,182]. Furthermore, mutations in mitochondrial transporter genes SLC25A12 and SLC25A22 are another cause of epilepsy [183,184]. These are members of the solute carrier family 25 and are both used for intracellular glutamate transport between the mitochondria and the cytoplasm. Aberrant function of these transporters is thought to be responsible for excessive glutamate release leading to hyperexcitability [185,186,187]. Additional nuclear genes that alter mitochondrial function and have been shown to be associated with epilepsies have been listed in Table 1.

Table 1.
Nuclear genes involved in mitochondrial function associated with epilepsy.
The mitochondrial genome is composed of circular, double-stranded DNA containing 37 genes which are all essential for the normal function of the mitochondrion. While mtDNA represents only a small fraction of total human DNA, mutations in over half of the 37 mtDNA genes have been associated with epilepsy [167]. The most frequently occurring mutations are in mitochondrial tRNA genes that lead to the epileptic syndromes such as mitochondrial epilepsy, lactic acidosis, stroke-like episodes (MELAS), and myoclonic epilepsy with ragged red fibers (MERRF) [252,253]. Less commonly seen mutations involving oxidative phosphorylation complexes also can induce various epileptic syndromes [167,254]. For example, mutations in mitochondrial genes that result in encoding proteins forming complexes I, III, IV, and V have been noted to cause Leigh syndrome, a mitochondrial disorder resulting in a severe, progressive, necrotizing encephalopathy [196]. While the syndrome is characterized by motor dysfunction and intellectual regression in the first few years of life, epilepsy is observed in 21–39% of patients [255,256].
Mitochondrial dysfunction and seizures have been found to be closely linked not only in genetic causes of epilepsy, but are also becoming increasingly recognized in acquired epilepsy [167]. These non-genetic causes of epilepsy include physical head trauma, infectious diseases, brain tumors, or drug intoxications [257]. Irrespective of the trigger, seizure activity significantly decreases ATP levels in neurons, suggestive of energy depletion [258]. Studies have identified that synaptic transmission is the largest consumer of neuronal ATP [259,260]; therefore, with the excessive neuronal firing during seizures, it is not surprising that energy depletion occurs leading to metabolic dysfunction even in acquired epilepsies. Other characteristics of homeostatic changes occurring after seizures are the accumulation of intracellular calcium and ROS [261,262,263]. Excess ROS and Ca2+ are potent triggers causing the opening of the mitochondrial permeability transition pore, leading to irreversible mitochondrial swelling and cytochrome c release, subsequently triggering mitochondrial damage and cellular death [264,265]. Due to a neuron’s lack of glycolytic capacities, the metabolic strain post-seizure activity increases the demand for oxidative phosphorylation and is a possible trigger for the excess ROS and Ca2+ fluctuations. Furthermore, oxidative damage can have profound effects on neurotransmitter uptake and release, altering neuronal excitability [266] and further increasing the likelihood of seizure activity and exacerbating neuronal energy depletion [267].
Regardless of which came first, epilepsy and mitochondrial dysfunction interact in a vicious cycle to deplete a neurons’ energy stores, leading to extensive brain damage. Although a detailed connection between mitochondrial dysfunction, energy metabolism, and epilepsy remains to be elucidated, mitochondria are clearly an integral part of disease manifestation.
5. Concluding Remarks
AD, TBI, and epilepsy are just a few of the neurological diseases with mitochondrial involvement. Metabolic dysfunction has also been documented in numerous other neurological diseases such as stroke [268,269], glioblastomas [270], multiple sclerosis [271,272], various neurodegenerative diseases [120,121,122,123,124], and even psychiatric disorders [273,274,275]. In this article, we have highlighted the importance of mitochondria metabolism in cellular pathophysiology in a few of the major neurological disorders and hope to have shed light on areas of exploration to further unravel the complexities of mitochondrial involvement in disease states.
Mitochondrial metabolism supplies the energy demand of a cell, coordinates the balance between catabolism and anabolism, and maintains redox homeostasis. However, knowledge gaps still remain to elucidate detailed mechanisms that lead to the manifestation of disease. While high-resolution techniques, such as chromatograph-based mass spectrometry metabolomics methods, have provided an efficient way to visualize mitochondrial metabolism in different types of cancers, these methods have not been adopted in the field of neurological research. Future research should incorporate traditional metabolomics and stable tracer technology to map mitochondrial metabolic flux with high-resolution, to yield vital information for the understanding of disease pathophysiology, and to assist in the design of future therapeutic agents to treat neurological disorders.
Author Contributions
Z.Z., G.L.A., L.A.J, R.S. drafted and edited the manuscript. L.E.A.Y. assisted with the literature search and constructed the Table. R.S. conceptualized the manuscript, and made the figures. All authors approved the final manuscript.
Funding
This study was supported by the University of Kentucky Center for Cancer and Metabolism, National Institute of general medical sciences COBRE program (grant ID: P20 GM121327).
Acknowledgments
The authors would like to thank Dr. Gentry and and his lab members for vigorous discussions regarding the scope of this review. We would also like to thank O.S. for his unconditional love and support over the years. The joy you’ve spread and your excitement towards all aspects of life will be forever cherished.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gray, M.W.; Burger, G.; Lang, B.F. The origin and early evolution of mitochondria. Genome Biol. 2001, 2, reviews1018.1011. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E. The Red Cell. In Hemolytic Anemia in Disorders of Red Cell Metabolism; Beutler, E., Ed.; Springer: Boston, MA, USA, 1978. [Google Scholar]
- Palade, G.E. An electron microscope study of the mitochondrial structure. J. Histochem. Cytochem. 1953, 1, 188–211. [Google Scholar] [CrossRef] [PubMed]
- Moritz, C. Applications of mitochondrial DNA analysis in conservation: A critical review. Mol. Ecol. 1994, 3, 401–411. [Google Scholar] [CrossRef]
- Morgenstern, M.; Stiller, S.B.; Lübbert, P.; Peikert, C.D.; Dannenmaier, S.; Drepper, F.; Weill, U.; Höß, P.; Feuerstein, R.; Gebert, M. Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep. 2017, 19, 2836–2852. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.D.; Thomas, W.K.; Meyer, A.; Edwards, S.V.; Pääbo, S.; Villablanca, F.X.; Wilson, A.C. Dynamics of mitochondrial DNA evolution in animals: Amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. USA 1989, 86, 6196. [Google Scholar] [CrossRef] [PubMed]
- Heijne, G.; Steppuhn, J.; Herrmann, R.G. Domain structure of mitochondrial and chloroplast targeting peptides. Eur. J. Biochem. 1989, 180, 535–545. [Google Scholar] [CrossRef]
- Mesecke, N.; Terziyska, N.; Kozany, C.; Baumann, F.; Neupert, W.; Hell, K.; Herrmann, J.M. A Disulfide Relay System in the Intermembrane Space of Mitochondria that Mediates Protein Import. Cell 2005, 121, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Chacinska, A.; Pfannschmidt, S.; Wiedemann, N.; Kozjak, V.; Sanjuán Szklarz, L.K.; Schulze-Specking, A.; Truscott, K.N.; Guiard, B.; Meisinger, C.; Pfanner, N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO. J. 2004, 23, 3735. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, T.; Schlipf, S.; Sanz, J.; Neubert, K.; Stein, R.; Borner, C. Characterization of the signal that directs Bcl-x7, but not Bcl-2, to the mitochondrial outer membrane. J. Cell Biol. 2003, 160, 53. [Google Scholar] [CrossRef] [PubMed]
- van Gurp, M.; Festjens, N.; van Loo, G.; Saelens, X.; Vandenabeele, P. Mitochondrial intermembrane proteins in cell death. Biochem. Biophys. Res. Commun. 2003, 304, 487–497. [Google Scholar] [CrossRef]
- Nazaret, C.; Heiske, M.; Thurley, K.; Mazat, J.-P. Mitochondrial energetic metabolism: A simplified model of TCA cycle with ATP production. J. Theor. Biol. 2009, 258, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Srere, P.A. The infrastructure of the mitochondrial matrix. Trends Biochem. Sci. 1980, 5, 120–121. [Google Scholar] [CrossRef]
- Hatefi, Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu. Rev. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar] [CrossRef]
- Tattoli, I.; Carneiro, L.A.; Jéhanno, M.; Magalhaes, J.G.; Shu, Y.; Philpott, D.J.; Arnoult, D.; Girardin, S.E. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-κB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, M.; Clément, M.V.; Pervaiz, S.; Brenner, C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 2005, 20, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2004, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, A.S.; Rogers, G.W.; Murphy, A.N. Measuring Mitochondrial Function in Permeabilized Cells Using the Seahorse XF Analyzer or a Clark-Type Oxygen Electrode. Curr. Protocols Toxicol. 2014, 60, 21–25. [Google Scholar] [CrossRef]
- Sun, R.C.; Fan, T.W.M.; Deng, P.; Higashi, R.M.; Lane, A.N.; Le, A.-T.; Scott, T.L.; Sun, Q.; Warmoes, M.O.; Yang, Y. Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat. Commun. 2017, 8, 1646. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Mielke, M.M. Recent advances in the application of metabolomics to Alzheimer’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A.; Johnson, W.A. The role of citric acid in intermediate metabolism in animal tissues. Enzymologia 1937, 4, 148–156. [Google Scholar]
- Chance, B.; Williams, G.R. The respiratory chain and oxidative phosphorylation. Adv. Enzymol. Relat. Sub. Biochem. 1956, 17, 65–134. [Google Scholar]
- Patel, M.S.; Roche, T.E. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990, 4, 3224–3233. [Google Scholar] [CrossRef] [PubMed]
- Linn, T.C.; Pettit, F.H.; Reed, L.J. α-keto acid dehydrogenase complexes, X. Regulation of the activity of the pyruvate dehydrogenase complex from beef keidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. USA 1969, 62, 234. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.C.H.; Drejer, J.; Hertz, L.; Schousboe, A. Pyruvate Carboxylase Activity in Primary Cultures of Astrocytes and Neurons. J. Neurochem. 1983, 41, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Nordlie, R.C.; Lardy, H.A. Mammalian Liver Phosphoenolpyruvate Carboxykinase Activities. J. Biol. Chem. 1963, 238, 2259–2263. [Google Scholar] [PubMed]
- Hsu, R.Y.; Lardy, H.A. Malic enzyme. In Methods in Enzymology; Cademic Press: Cambridge, MA, USA, 1969; Volume 13, pp. 230–235. [Google Scholar]
- Bradford, H.F.; Ward, H.K. On glutaminase activity in mammalian synaptosomes. Brain Res. 1976, 110, 115–125. [Google Scholar] [CrossRef]
- Frieden, C. Glutamate Dehydrogenase: V. The relation of enzyme structure to the catalytic function. J. Biol. Chem. 1963, 238, 3286–3299. [Google Scholar] [PubMed]
- Yamaya, T.; Oaks, A. Synthesis of glutamate by mitochondria—An anaplerotic function for glutamate dehydrogenase. Physiol. Plant. 1987, 70, 749–756. [Google Scholar] [CrossRef]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The Key Role of Anaplerosis and Cataplerosis for Citric Acid Cycle Function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B. Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Reitzer, L.J.; Wice, B.M.; Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [Google Scholar] [PubMed]
- Lowenstein, J. Ammonia production in muscle and other tissues: The purine nucleotide cycle. Physiol. Rev. 1972, 52, 382–414. [Google Scholar] [CrossRef] [PubMed]
- Jeremiah, S.; Povey, S.; Burley, M.; Kielty, C.; Lee, M.; Spowart, G.; Corney, G.; Cook, P. Mapping studies on human mitochondrial glutamate oxaloacetate transaminase. Ann. Hum. Genet. 1982, 46, 145–152. [Google Scholar] [CrossRef]
- Beardsley, G.; Moroson, B.; Taylor, E.; Moran, R. A new folate antimetabolite, 5, 10-dideaza-5, 6, 7, 8-tetrahydrofolate is a potent inhibitor of de novo purine synthesis. J. Biol. Chem. 1989, 264, 328–333. [Google Scholar] [PubMed]
- Schirch, L. Serine hydroxymethyltransferase. Adv. Enzymol. Relat. Areas Mol. Biol. 1982, 53, 83–112. [Google Scholar] [PubMed]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Drury, E.J.; MacKenzie, R.E. Methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase-formyltetrahydrofolate synthetase. A multifunctional protein from porcine liver. J. Biol. Chem. 1977, 252, 1117–1122. [Google Scholar] [PubMed]
- Appling, D.R. Compartmentation of folate-mediated one-carbon metabolism in eukaryotes. FASEB J. 1991, 5, 2645–2651. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Wakil, S.J. Mechanism of fatty acid synthesis. J. Lipid Res. 1961, 2, 1–24. [Google Scholar]
- Watson, J.A.; Lowenstein, J.M. Citrate and the conversion of carbohydrate into fat fatty acid synthesis by a combination of cytoplasm and mitochondria. J. Biol. Chem. 1970, 245, 5993–6002. [Google Scholar] [PubMed]
- McGarry, J.D.; Leatherman, G.F.; Foster, D.W. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J. Biol. Chem. 1978, 253, 4128–4136. [Google Scholar] [PubMed]
- McGarry, J.D.; Mannaerts, G.; Foster, D.W. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Invest. 1977, 60, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, J.K.; Schonauer, M.S.; Autio, K.J.; Mittelmeier, T.M.; Kastaniotis, A.J.; Dieckmann, C.L. Mitochondrial fatty acid synthesis type II: More than just fatty acids. J. Biol. Chem. 2009, 284, 9011–9015. [Google Scholar] [CrossRef] [PubMed]
- Moris, N.; Pina, C.; Arias, A.M. Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 2016, 17, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009, 10, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-Mediated Changes in Acetyl-CoA and Histone Acetylation Control the Early Differentiation of Embryonic Stem Cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Mews, P.; Donahue, G.; Drake, A.M.; Luczak, V.; Abel, T.; Berger, S.L. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 2017, 546, 381. [Google Scholar] [CrossRef] [PubMed]
- Moseley, H.N.; Lane, A.N.; Belshoff, A.C.; Higashi, R.M.; Fan, T.W. A novel deconvolution method for modeling UDP-N-acetyl-D-glucosamine biosynthetic pathways based on 13 C mass isotopologue profiles under non-steady-state conditions. BMC Biol. 2011, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, Y.; Bian, C.; Fujiki, R.; Yu, X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 2012, 493, 561. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.-S.; Duggan, L.; Tolga, N.; Belotserkovskya, R.; Lane, W.S.; Shiekhattar, R.; Berger, S.L. Snf1–a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 2001, 293, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins—Emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Gonçalves, E.; Isaac Johnson, T.; Roberto Zecchini, V.; da Costa, A.S.H.; Gaude, E.; Vercauteren Drubbel, A.; Julian Theobald, S.; Abbo, S.; Tran, M.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Soga, T.; Pollard, P.J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Invest. 2013, 123, 3652–3658. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Toro, J.R.; Nickerson, M.L.; Wei, M.-H.; Warren, M.B.; Glenn, G.M.; Turner, M.L.; Stewart, L.; Duray, P.; Tourre, O.; Sharma, N.; et al. Mutations in the Fumarate Hydratase Gene Cause Hereditary Leiomyomatosis and Renal Cell Cancer in Families in North America. Am. J. Hum. Genet. 2003, 73, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s, A. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015, 11, 332. [Google Scholar]
- Ramirez-Bermudez, J. Alzheimer’s disease: Critical notes on the history of a medical concept. Arch. Med. Res. 2012, 43, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xie, N.; Tang, B.; Li, R.; Shen, Y. Alzheimer’s Disease: From Genetic Variants to the Distinct Pathological Mechanisms. Front. Mol. Neurosci. 2017, 10, 319. [Google Scholar] [CrossRef] [PubMed]
- Craft, S. The role of metabolic disorders in Alzheimer disease and vascular dementia: Two roads converged. Arch. Neurol. 2009, 66, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Harrison, F.E. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients 2015, 7, 7332–7357. [Google Scholar] [CrossRef] [PubMed]
- Narayan, K.M.; Boyle, J.P.; Geiss, L.S.; Saaddine, J.B.; Thompson, T.J. Impact of recent increase in incidence on future diabetes burden: U.S., 2005–2050. Diabetes Care 2006, 29, 2114–2116. [Google Scholar] [CrossRef] [PubMed]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.L.; Chase, T.N.; Fedio, P.; Patronas, N.J.; Brooks, R.A.; Di Chiro, G. Alzheimer’s disease: Focal cortical changes shown by positron emission tomography. Neurology 1983, 33, 961–965. [Google Scholar] [CrossRef] [PubMed]
- de Leon, M.J.; Ferris, S.H.; George, A.E.; Christman, D.R.; Fowler, J.S.; Gentes, C.; Reisberg, B.; Gee, B.; Emmerich, M.; Yonekura, Y.; et al. Positron emission tomographic studies of aging and Alzheimer disease. AJNR Am. J. Neuroradiol. 1983, 4, 568–571. [Google Scholar] [PubMed]
- Ferris, S.H.; de Leon, M.J.; Wolf, A.P.; Farkas, T.; Christman, D.R.; Reisberg, B.; Fowler, J.S.; Macgregor, R.; Goldman, A.; George, A.E.; et al. Positron emission tomography in the study of aging and senile dementia. Neurobiol. Aging 1980, 1, 127–131. [Google Scholar] [CrossRef]
- Friedland, R.P.; Budinger, T.F.; Ganz, E.; Yano, Y.; Mathis, C.A.; Koss, B.; Ober, B.A.; Huesman, R.H.; Derenzo, S.E. Regional cerebral metabolic alterations in dementia of the Alzheimer type: Positron emission tomography with [18F]fluorodeoxyglucose. J. Comput. Assist. Tomogr. 1983, 7, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Small, G.W.; Ercoli, L.M.; Silverman, D.H.; Huang, S.C.; Komo, S.; Bookheimer, S.Y.; Lavretsky, H.; Miller, K.; Siddarth, P.; Rasgon, N.L.; et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 6037–6042. [Google Scholar] [CrossRef] [PubMed]
- Laforce, R., Jr.; Rabinovici, G.D. Amyloid imaging in the differential diagnosis of dementia: Review and potential clinical applications. Alzheimers Res. Ther. 2011, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Grady, C.L.; Haxby, J.V.; Schlageter, N.L.; Berg, G.; Rapoport, S.I. Stability of metabolic and neuropsychological asymmetries in dementia of the Alzheimer type. Neurology 1986, 36, 1390–1392. [Google Scholar] [CrossRef] [PubMed]
- Haxby, J.V.; Grady, C.L.; Koss, E.; Horwitz, B.; Heston, L.; Schapiro, M.; Friedland, R.P.; Rapoport, S.I. Longitudinal study of cerebral metabolic asymmetries and associated neuropsychological patterns in early dementia of the Alzheimer type. Arch. Neurol. 1990, 47, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Blass, J.P. Alzheimer’s disease and Alzheimer’s dementia: Distinct but overlapping entities. Neurobiol. Aging 2002, 23, 1077–1084. [Google Scholar] [CrossRef]
- Small, G.W.; Mazziotta, J.C.; Collins, M.T.; Baxter, L.R.; Phelps, M.E.; Mandelkern, M.A.; Kaplan, A.; La Rue, A.; Adamson, C.F.; Chang, L.; et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 1995, 273, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Caselli, R.J.; Yun, L.S.; Chen, K.; Bandy, D.; Minoshima, S.; Thibodeau, S.N.; Osborne, D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl. J. Med. 1996, 334, 752–758. [Google Scholar] [CrossRef] [PubMed]
- De Leon, M.J.; Convit, A.; Wolf, O.T.; Tarshish, C.Y.; DeSanti, S.; Rusinek, H.; Tsui, W.; Kandil, E.; Scherer, A.J.; Roche, A.; et al. Prediction of cognitive decline in normal elderly subjects with 2-[18F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc. Natl. Acad. Sci. USA 2001, 98, 10966–10971. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; De Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; de Leon, M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 1990, 40, 1302–1302. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.K. Cytochrome C Oxidase in Alzheimer’s Disease Brain Purification and Characterization. Neurology 1995, 45, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Aβ toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Hauptmann, S.; Scherping, I.; Meinhardt, J.; Rhein, V.; Dröse, S.; Brandt, U.; Fändrich, M.; Müller, W.E.; Götz, J. Oligomeric and fibrillar species of β-amyloid (Aβ42) both impair mitochondrial function in P301L tau transgenic mice. J. Mol. Med. 2008, 86, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Frackowiak, R.S.; Pozzilli, C.; Legg, N.J.; Du Boulay, G.H.; Marshall, J.; Lenzi, G.L.; Jones, T. Regional cerebral oxygen supply and utilization in dementia. A clinical and physiological study with oxygen-15 and positron tomography. Brain 1981, 104, 753–778. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, H.; Ogawa, M.; Yamauchi, H.; Yamaguchi, S.; Kimura, J.; Yonekura, Y.; Konishi, J. Altered cerebral energy metabolism in Alzheimer’s disease: A PET study. J. Nucl. Med. 1994, 35, 1–6. [Google Scholar] [PubMed]
- Sorbi, S.; Bird, E.D.; Blass, J.P. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann. Neurol. 1983, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Starkov, A.; Blass, J.P.; Ratan, R.R.; Beal, M.F. Cause and consequence: Mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A. meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Raber, J.; Huang, Y.; Ashford, J.W. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 2004, 25, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet 2000, 1, 507–537. [Google Scholar] [CrossRef] [PubMed]
- Eichner, J.E.; Dunn, S.T.; Perveen, G.; Thompson, D.M.; Stewart, K.E.; Stroehla, B.C. Apolipoprotein E polymorphism and cardiovascular disease: A HuGE review. Am. J. Epidemiol. 2002, 155, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Bernardo, A.; Walker, D.; Kanegawa, T.; Mahley, R.W.; Huang, Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 2006, 26, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C. Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 2014, 66, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Altmann, A.; Ng, B.; Landau, S.M.; Jagust, W.J.; Greicius, M.D. Alzheimer’s Disease Neuroimaging, I. Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain 2015, 138, 3734–3746. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Caselli, R.J.; Chen, K.; Alexander, G.E.; Bandy, D.; Frost, J. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 3334–3339. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Benzinger, T.L.; Blazey, T.; Jack, C.R., Jr.; Koeppe, R.A.; Su, Y.; Xiong, C.; Raichle, M.E.; Snyder, A.Z.; Ances, B.M.; Bateman, R.J.; et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, E4502–4509. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.K.; Ji, Z.S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, S.B.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem. 2011, 286, 5215–5221. [Google Scholar] [CrossRef] [PubMed]
- James, R.; Searcy, J.L.; Le Bihan, T.; Martin, S.F.; Gliddon, C.M.; Povey, J.; Deighton, R.F.; Kerr, L.E.; McCulloch, J.; Horsburgh, K. Proteomic analysis of mitochondria in APOE transgenic mice and in response to an ischemic challenge. J. Cereb. Blood Flow. Metab. 2012, 32, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Tambini, M.D.; Pera, M.; Kanter, E.; Yang, H.; Guardia-Laguarta, C.; Holtzman, D.; Sulzer, D.; Area-Gomez, E.; Schon, E.A. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016, 17, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Ran Ma, T.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Watanabe, A.; Fujino, T.; Hosono, T.; Michikawa, M. Apolipoprotein E4 (1-272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol. Neurodegener. 2009, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, G.; Bonafe, M.; De Luca, M.; Rose, G.; Varcasia, O.; Bruni, A.; Maletta, R.; Nacmias, B.; Sorbi, S.; Corsonello, F.; et al. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s disease. Hum. Genet. 2001, 108, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Maruszak, A.; Safranow, K.; Branicki, W.; Gaweda-Walerych, K.; Pospiech, E.; Gabryelewicz, T.; Canter, J.A.; Barcikowska, M.; Zekanowski, C. The impact of mitochondrial and nuclear DNA variants on late-onset Alzheimer’s disease risk. J. Alzheimers Dis. 2011, 27, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.; Wolf, A.B.; Chavira, B.; Shonebarger, D.; Meckel, J.P.; Leung, L.; Ballina, L.; Ly, S.; Saini, A.; Jones, T.B.; et al. Altered Energy Metabolism Pathways in the Posterior Cingulate in Young Adult Apolipoprotein E varepsilon4 Carriers. J. Alzheimers Dis. 2016, 53, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Valla, J.; Yaari, R.; Wolf, A.B.; Kusne, Y.; Beach, T.G.; Roher, A.E.; Corneveaux, J.J.; Huentelman, M.J.; Caselli, R.J.; Reiman, E.M. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer’s susceptibility gene. J. Alzheimers Dis. 2010, 22, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Koppel, S.J.; Bothwell, R.; Mahnken, J.; Burns, J.M.; Swerdlow, R.H. Platelet cytochrome oxidase and citrate synthase activities in APOE epsilon4 carrier and non-carrier Alzheimer’s disease patients. Redox. Biol. 2017, 12, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Albers, D.S.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. In Advances in Dementia Research; Springer: Berlin, Germany, 2000; pp. 133–154. [Google Scholar]
- Trushina, E.; McMurray, C. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta (BBA)-Bioenerg. 1998, 1366, 211–223. [Google Scholar] [CrossRef]
- Emerit, J.; Edeas, M.; Bricaire, F. Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 2004, 58, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Faul, M.; Coronado, V. Epidemiology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.; Tavazzi, B.; Signoretti, S.; Amorini, A.M.; Belli, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Finocchiaro, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Mitochondrial-related impairment—Part I. Neurosurgery 2007, 61, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Kong, R.H.; Zhang, L.M.; Zhang, J.N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef] [PubMed]
- Bulstrode, H.; Nicoll, J.A.; Hudson, G.; Chinnery, P.F.; Di Pietro, V.; Belli, A. Mitochondrial DNA and traumatic brain injury. Ann. Neurol. 2014, 75, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Mautes, A.E.; Thome, D.; Steudel, W.I.; Nacimiento, A.C.; Yang, Y.; Shohami, E. Changes in regional energy metabolism after closed head injury in the rat. J. Mol. Neurosci. 2001, 16, 33–39. [Google Scholar] [CrossRef]
- Lifshitz, J.; Friberg, H.; Neumar, R.W.; Raghupathi, R.; Welsh, F.A.; Janmey, P.; Saatman, K.E.; Wieloch, T.; Grady, M.S.; McIntosh, T.K. Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: Evidence for differentially sensitive populations in the cortex and hippocampus. J. Cereb. Blood Flow. Metab. 2003, 23, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Keller, J.N.; Mattson, M.P.; Scheff, S.W. Traumatic brain injury alters synaptic homeostasis: Implications for impaired mitochondrial and transport function. J. Neurotrauma. 1998, 15, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.T.; Soltesz, I. Selective depolarization of interneurons in the early posttraumatic dentate gyrus: Involvement of the Na(+)/K.(+)-ATPase. J. Neurophysiol. 2000, 83, 2916–2930. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Becker, D.P.; Tamura, T.; Hovda, D.A. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 1990, 73, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Katayama, Y.; Hovda, D.A.; Yoshino, A.; Becker, D.P. Lactate accumulation following concussive brain injury: The role of ionic fluxes induced by excitatory amino acids. Brain Res. 1995, 674, 196–204. [Google Scholar] [CrossRef]
- Bentzer, P.; Davidsson, H.; Grande, P.O. Microdialysis-based long-term measurements of energy-related metabolites in the rat brain following a fluid percussion trauma. J. Neurotrauma. 2000, 17, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Qian, Y.Z.; Di, X.; Rice, A.; Zhu, J.P.; Bullock, R. Lactate/glucose dynamics after rat fluid percussion brain injury. J. Neurotrauma. 2000, 17, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Hertz, L.; Huang, R.; Sonnewald, U.; Petersen, S.B.; Westergaard, N.; Larsson, O.; Schousboe, A. Utilization of glutamine and of TCA cycle constituents as precursors for transmitter glutamate and GABA. Dev. Neurosci. 1993, 15, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Tapia, R.; Gonzalez, R.M. Glutamine and glutamate as precursors of the releasable pool of GABA in brain cortex slices. Neurosci. Lett. 1978, 10, 165–169. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Peterson, P.L.; Muizelaar, J.P.; Lee, C.P. Amelioration of mitochondrial function by a novel antioxidant U-101033E following traumatic brain injury in rats. J. Neurotrauma. 1997, 14, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Lifshitz, J.; Sullivan, P.G.; Hovda, D.A.; Wieloch, T.; McIntosh, T.K. Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion 2004, 4, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Rabchevsky, A.G.; Waldmeier, P.C.; Springer, J.E. Mitochondrial permeability transition in CNS trauma: Cause or effect of neuronal cell death? J. Neurosci. Res. 2005, 79, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Slater, E.C.; Cleland, K.W. The effect of calcium on the respiratory and phosphorylative activities of heart-muscle sarcosomes. Biochem. J. 1953, 55, 566–590. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.; Nicholls, D.G. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 2003, 278, 19062–19070. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Qian, L.; Zhou, T.; Iadecola, C. Mitochondria are involved in the neurogenic neuroprotection conferred by stimulation of cerebellar fastigial nucleus. J. Neurochem. 2005, 95, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Brustovetsky, T.; Purl, K.J.; Capano, M.; Crompton, M.; Dubinsky, J.M. Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J. Neurosci. 2003, 23, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.D.; Nukala, V.N.; Sullivan, P.G. Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Front. Neuroenergetics 2013, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; LaNoue, K.F.; Cheung, J.Y.; Scaduto, R.C., Jr. Regulation of citric acid cycle by calcium. J. Biol. Chem. 1989, 264, 13430–13439. [Google Scholar] [PubMed]
- Denton, R.M.; Rutter, G.A.; Midgley, P.J.; McCormack, J.G. Effects of Ca2+ on the activities of the calcium-sensitive dehydrogenases within the mitochondria of mammalian tissues. J. Cardiovasc. Pharmacol. 1988, 12 Suppl 5, S69–S72. [Google Scholar] [CrossRef]
- Sharma, P.; Benford, B.; Li, Z.Z.; Ling, G.S. Role of pyruvate dehydrogenase complex in traumatic brain injury and Measurement of pyruvate dehydrogenase enzyme by dipstick test. J. Emerg. Trauma. Shock 2009, 2, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchyan, G.V.; Ucal, M.; Mullebner, A.; Dumitrescu, S.; Kames, M.; Moldzio, R.; Molcanyi, M.; Schaefer, S.; Weidinger, A.; Schaefer, U.; et al. Thiamine preserves mitochondrial function in a rat model of traumatic brain injury, preventing inactivation of the 2-oxoglutarate dehydrogenase complex. Biochim. Biophys. Acta 2018, 1859, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.Y.; Kreipke, C.W.; Speirs, S.L.; Schafer, P.; Schafer, S.; Rafols, J.A. Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci. Lett. 2009, 453, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Bell, J.D.; Siddiq, I.P.; Baker, A.J. An analysis of regional microvascular loss and recovery following two grades of fluid percussion trauma: A role for hypoxia-inducible factors in traumatic brain injury. J. Cereb. Blood Flow. Metab. 2009, 29, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Higashida, T.; Kreipke, C.W.; Rafols, J.A.; Peng, C.; Schafer, S.; Schafer, P.; Ding, J.Y.; Dornbos, D., 3rd; Li, X.; Guthikonda, M.; et al. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J. Neurosurg. 2011, 114, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Golias, T.; Papandreou, I.; Sun, R.; Kumar, B.; Brown, N.V.; Swanson, B.J.; Pai, R.; Jaitin, D.; Le, Q.T.; Teknos, T.N.; et al. Hypoxic repression of pyruvate dehydrogenase activity is necessary for metabolic reprogramming and growth of model tumours. Sci. Rep. 2016, 6, 31146. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Gu, Q.; Peterson, P.L.; Muizelaar, J.P.; Lee, C.P. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J. Neurotrauma. 1997, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Rossi, S.; Stiefel, M.; Doppenberg, E.; Zauner, A.; Bullock, R.; Marmarou, A. CSF and ECF glutamate concentrations in head injured patients. Acta Neurochir. Suppl. 1999, 75, 17–19. [Google Scholar] [PubMed]
- Rose, M.E.; Huerbin, M.B.; Melick, J.; Marion, D.W.; Palmer, A.M.; Schiding, J.K.; Kochanek, P.M.; Graham, S.H. Regulation of interstitial excitatory amino acid concentrations after cortical contusion injury. Brain Res. 2002, 943, 15–22. [Google Scholar] [CrossRef]
- Tavazzi, B.; Signoretti, S.; Lazzarino, G.; Amorini, A.M.; Delfini, R.; Cimatti, M.; Marmarou, A.; Vagnozzi, R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 2005, 56, 582–589. [Google Scholar] [CrossRef] [PubMed]
- CDC. Epilepsy Data and Statistics. Available online: https://www.cdc.gov/epilepsy/data/index.html (accessed on 3 November 2018).
- Gavvala, J.R.; Schuele, S.U. JAMA Patient Page: Epilepsy. JAMA 2016, 316, 2686. [Google Scholar] [CrossRef] [PubMed]
- Debray, F.G.; Lambert, M.; Chevalier, I.; Robitaille, Y.; Decarie, J.C.; Shoubridge, E.A.; Robinson, B.H.; Mitchell, G.A. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics 2007, 119, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Khurana, D.; Salganicoff, L.; Melvin, J.; Hobdell, E.; Valencia, I.; Hardison, H.; Marks, H.; Grover, W.; Legido, A. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathies. Epilepsia 2008, 49, 1972. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Davis, R.E.; Williams, M. Mitochondrial function and dysfunction: An update. J. Pharmacol. Exp. Ther. 2012, 342, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef] [PubMed]
- March, P.A. Seizures: Classification, etiologies, and pathophysiology. Clin. Tech. Small Anim. Prac. 1998, 13, 119–131. [Google Scholar] [CrossRef]
- Rahman, S. Mitochondrial disease and epilepsy. Dev. Med. Child Neurol. 2012, 54, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Dermaut, B.; Löfgren, A.; Martin, J.-J.; Van Broeckhoven, C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001, 28, 211. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Luoma, P.; Rantamäki, M.; Al Memar, A.; Kaakkola, S.; Hackman, P.; Krahe, R.; Löfgren, A.; Martin, J.-J.; De Jonghe, P. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology 2004, 63, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Martin, J.; Dermaut, B.; Löfgren, A.; Wibail, A.; Ververken, D.; Tack, P.; Dehaene, I.; Van Zandijcke, M.; Moonen, M. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscular Disorders 2003, 13, 133–142. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Nguyen, K.V. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann. Neurol. 2004, 55, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Hudson, G.; Ferrari, G.; Fütterer, N.; Ahola, S.; Lamantea, E.; Prokisch, H.; Lochmüller, H.; McFarland, R.; Ramesh, V. Phenotypic spectrum associated with mutations of the mitochondrial polymerase γ gene. Brain 2006, 129, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Zeviani, M. 155th ENMC workshop: Polymerase gamma and disorders of mitochondrial DNA synthesis, 21–23 September 2007, Naarden, The Netherlands. Neuromuscular Disorders 2008, 18, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Neeve, V.C.; Samuels, D.C.; Bindoff, L.A.; van den Bosch, B.; Van Goethem, G.; Smeets, H.; Lombès, A.; Jardel, C.; Hirano, M.; DiMauro, S. What is influencing the phenotype of the common homozygous polymerase-γ mutation p. Ala467Thr? Brain 2012, 135, 3614–3626. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Walter, P.B.; Ames, B.N. The role of heme and iron-sulfur clusters in mitochondrial biogenesis, maintenance, and decay with age. Arch. Biochem. Biophys. 2002, 397, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.M.; Janer, A.; Levandovskiy, V.; MacKay, N.; Rouault, T.A.; Tong, W.-H.; Ogilvie, I.; Shoubridge, E.A.; Robinson, B.H. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am. J. Hum. Genet. 2011, 89, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.R.; Friederich, M.W.; Swanson, M.A.; Shaikh, T.; Bhattacharya, K.; Scharer, G.H.; Aicher, J.; Creadon-Swindell, G.; Geiger, E.; MacLean, K.N. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 2013, 137, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Zimmermann, F.A.; Fauth, C.; Bergheim, C.; Meierhofer, D.; Radmayr, D.; Zschocke, J.; Koch, J.; Sperl, W. Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Am. J. Hum. Genet. 2011, 89, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Raas-Rothschild, A.; Rio, M.; Fiermonte, G.; Encha-Razavi, F.; Palmieri, L.; Palmieri, F.; Ben-Neriah, Z.; Kadhom, N.; Vekemans, M. Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am. J. Hum. Genet. 2005, 76, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Li, D.; Gai, X.; McCormick, E.; Place, E.; Lasorsa, F.M.; Otieno, F.G.; Hou, C.; Kim, C.E.; Abdel-Magid, N. AGC1 deficiency causes infantile epilepsy, abnormal myelination, and reduced N.-acetylaspartate. In JIMD Reports, Volume 14; Springer: Berlin, Germany, 2014; pp. 77–85. [Google Scholar]
- Fiermonte, G.; Palmieri, L.; Todisco, S.; Agrimi, G.; Palmieri, F.; Walker, J.E. Identification of the mitochondrial glutamate transporter: Bacterial expression, reconsitution, functional characterization, tissue distribution of two human isoforms. J. Biol. Chem. 2002, 277, 19289–19294. [Google Scholar] [CrossRef] [PubMed]
- Thangaratnarajah, C.; Ruprecht, J.J.; Kunji, E.R. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nature Commun. 2014, 5, 5491. [Google Scholar] [CrossRef] [PubMed]
- Niciu, M.J.; Kelmendi, B.; Sanacora, G. Overview of glutamatergic neurotransmission in the nervous system. Pharmacol. Biochem. Behav. 2012, 100, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Bénit, P.; Chretien, D.; Kadhom, N.; de Lonlay-Debeney, P.; Cormier-Daire, V.; Cabral, A.; Peudenier, S.; Rustin, P.; Munnich, A.; Rötig, A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I. deficiency. Am. J. Hum. Genet. 2001, 68, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Benit, P.; Slama, A.; Cartault, F.; Giurgea, I.; Chretien, D.; Lebon, S.; Marsac, C.; Munnich, A.; Rötig, A.; Rustin, P. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J. Med. Genet. 2004, 41, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Scacco, S.; Piccoli, C.; Bellomo, F.; Petruzzella, V.; Trentadue, R.; Minuto, M.; Ripoli, M.; Capitanio, N.; Zeviani, M. Dysfunctions of cellular oxidative metabolism in patients with mutations in the NDUFS1 and NDUFS4 genes of complex I. J. Biol. Chem. 2006, 281, 10374–10380. [Google Scholar] [CrossRef] [PubMed]
- Kirby, D.M.; Salemi, R.; Sugiana, C.; Ohtake, A.; Parry, L.; Bell, K.M.; Kirk, E.P.; Boneh, A.; Taylor, R.W.; Dahl, H.-H.M. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I. deficiency. J. Clin. Invest. 2004, 114, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.; Hogan, V.E.; He, L.; Blakely, E.L.; Worgan, L.; Al-Dosary, M.; Saretzki, G.; Alston, C.L.; Morris, A.A.; Clarke, M. The p. M292T NDUFS2 mutation causes complex I-deficient Leigh syndrome in multiple families. Brain 2010, 133, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Berger, I.; Hershkovitz, E.; Shaag, A.; Edvardson, S.; Saada, A.; Elpeleg, O. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann. Neurol. 2008, 63, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Moreira, D.; Ugalde, C.; Smeets, R.; Rodenburg, R.J.; Lopez-Laso, E.; Ruiz-Falco, M.L.; Briones, P.; Martin, M.A.; Smeitink, J.A.; Arenas, J. X-linked NDUFA1 gene mutations associated with mitochondrial encephalomyopathy. Ann. Neurol. 2007, 61, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Loeffen, J.; Smeitink, J.; Triepels, R.; Smeets, R.; Schuelke, M.; Sengers, R.; Trijbels, F.; Hamel, B.; Mullaart, R.; van den Heuvel, L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am. J. Hum. Genet. 1998, 63, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Ruhoy, I.S.; Saneto, R.P. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2014, 7, 221. [Google Scholar] [PubMed]
- Bianciardi, L.; Imperatore, V.; Fernandez-Vizarra, E.; Lopomo, A.; Falabella, M.; Furini, S.; Galluzzi, P.; Grosso, S.; Zeviani, M.; Renieri, A. Exome sequencing coupled with mRNA analysis identifies NDUFAF6 as a Leigh gene. Mol.Genet. Metabol. 2016, 119, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Tucker, E.J.; Compton, A.G.; Kirby, D.M.; Crawford, G.; Burtt, N.P.; Rivas, M.; Guiducci, C.; Bruno, D.L.; Goldberger, O.A. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I. deficiency. Nature Genet. 2010, 42, 851. [Google Scholar] [CrossRef] [PubMed]
- Herzer, M.; Koch, J.; Prokisch, H.; Rodenburg, R.; Rauscher, C.; Radauer, W.; Forstner, R.; Pilz, P.; Rolinski, B.; Freisinger, P. Leigh disease with brainstem involvement in complex I deficiency due to assembly factor NDUFAF2 defect. Europediatrics 2010, 41, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Mimaki, M.; Wang, X.; McKenzie, M.; Thorburn, D.R.; Ryan, M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1817, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Prabhu, S.P.; Lidov, H.G.; Shi, J.; Anselm, I.; Brownstein, C.A.; Bainbridge, M.N.; Beggs, A.H.; Vargas, S.O.; Agrawal, P.B. AIFM1 mutation presenting with fatal encephalomyopathy and mitochondrial disease in an infant. Mol. Case Stud. 2017, 3, a001560. [Google Scholar] [CrossRef] [PubMed]
- Saada, A.; Vogel, R.O.; Hoefs, S.J.; van den Brand, M.A.; Wessels, H.J.; Willems, P.H.; Venselaar, H.; Shaag, A.; Barghuti, F.; Reish, O. Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I. assembly protein, cause fatal neonatal mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Sugiana, C.; Pagliarini, D.J.; McKenzie, M.; Kirby, D.M.; Salemi, R.; Abu-Amero, K.K.; Dahl, H.-H.M.; Hutchison, W.M.; Vascotto, K.A.; Smith, S.M. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am. J. Hum. Genet. 2008, 83, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Wortmann, S.B.; Smeets, R.J.; Venselaar, H.; Antoine, M.; Visser, G.; Ben-Omran, T.; Van Den Heuvel, L.P.; Timmers, H.J.; Smeitink, J.A. SDHA mutations causing a multisystem mitochondrial disease: Novel mutations and genetic overlap with hereditary tumors. Eur. J. Hum. Genet. 2015, 23, 202. [Google Scholar] [CrossRef] [PubMed]
- Tucker, E.J.; Wanschers, B.F.; Szklarczyk, R.; Mountford, H.S.; Wijeyeratne, X.W.; van den Brand, M.A.; Leenders, A.M.; Rodenburg, R.J.; Reljić, B.; Compton, A.G. Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet. 2013, 9, e1004034. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Garcia, S.; Padgett, K.R.; Moraes, C.T. A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum. Mol. Genet. 2012, 21, 5066–5077. [Google Scholar] [CrossRef] [PubMed]
- Tegelberg, S.; Tomašić, N.; Kallijärvi, J.; Purhonen, J.; Elmér, E.; Lindberg, E.; Nord, D.G.; Soller, M.; Lesko, N.; Wedell, A. Respiratory chain complex III deficiency due to mutated BCS1L: A novel phenotype with encephalomyopathy, partially phenocopied in a Bcs1l mutant mouse model. Orphanet J. Rare Dis. 2017, 12, 73. [Google Scholar] [CrossRef] [PubMed]
- Antonicka, H.; Leary, S.C.; Guercin, G.-H.; Agar, J.N.; Horvath, R.; Kennaway, N.G.; Harding, C.O.; Jaksch, M.; Shoubridge, E.A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Antonicka, H.; Shoubridge, E.A. Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep. 2015, 10, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, M.; Ogilvie, I.; Yao, J.; Kortenhaus, G.; Bresser, H.-G.; Gerbitz, K.-D.; Shoubridge, E.A. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Antonicka, H.; Sasarman, F.; Weraarpachai, W.; Cobine, P.A.; Pan, M.; Brown, G.K.; Brown, R.; Majewski, J.; Ha, K.C. Novel Mutations in SCO 1 as a Cause of Fatal Infantile Encephalopathy and Lactic Acidosis. Hum. Mutation 2013, 34, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Smith, K.R.; Stroud, D.A.; Compton, A.G.; Tucker, E.J.; Dasvarma, A.; Gandolfo, L.C.; Marum, J.E.; McKenzie, M.; Peters, H.L. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am. J. Hum. Genet. 2014, 94, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Melchionda, L.; Haack, T.B.; Hardy, S.; Abbink, T.E.; Fernandez-Vizarra, E.; Lamantea, E.; Marchet, S.; Morandi, L.; Moggio, M.; Carrozzo, R. Mutations in APOPT1, encoding a mitochondrial protein, cause cavitating leukoencephalopathy with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 2014, 95, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Oquendo, C.; Antonicka, H.; Shoubridge, E.; Reardon, W.; Brown, G. Functional and genetic studies demonstrate that mutation in the COX15 gene can cause Leigh syndrome. J. Med. Genet. 2004, 41, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, E.; Augoustides-Savvopoulou, P.; Gregersen, N.; Haas, D.; Gkampeta, A.; Athanassiadou-Piperopoulou, F. An infant with ethylmalonic encephalopathy masquerading as a hematologic disorder. J. Child Neurol. 2013, 28, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.; Rahman, S.; Wedatilake, Y.; Polke, J.M.; Cirak, S.; Foley, A.R.; Sailer, A.; Hurles, M.E.; Stalker, J.; Hargreaves, I. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013, 3, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Braczynski, A.K.; Vlaho, S.; Müller, K.; Wittig, I.; Blank, A.-E.; Tews, D.S.; Drott, U.; Kleinle, S.; Abicht, A.; Horvath, R. ATP synthase deficiency due to TMEM70 mutation leads to ultrastructural mitochondrial degeneration and is amenable to treatment. BioMed. Res. Inter. 2015, 2015, 462592. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, A.I.; Renkema, G.H.; Bras, M.; van den Heuvel, L.P.; Hoischen, A.; Gilissen, C.; Nabuurs, S.B.; Huynen, M.A.; de Vries, M.C.; Smeitink, J.A. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain 2013, 136, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Brea-Calvo, G.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Bürgi, S. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef] [PubMed]
- García-Corzo, L.; Luna-Sánchez, M.; Doerrier, C.; García, J.A.; Guarás, A.; Acín-Pérez, R.; Bullejos-Peregrín, J.; López, A.; Escames, G.; Enríquez, J.A. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet. 2012, 22, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Hikmat, O.; Tzoulis, C.; Knappskog, P.M.; Johansson, S.; Boman, H.; Sztromwasser, P.; Lien, E.; Brodtkorb, E.; Ghezzi, D.; Bindoff, L.A. ADCK 3 mutations with epilepsy, stroke-like episodes and ataxia: A POLG mimic? Eur. J. Neurol. 2016, 23, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; DiMauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Scalais, E.; Chafai, R.; Van Coster, R.; Bindl, L.; Nuttin, C.; Panagiotaraki, C.; Seneca, S.; Lissens, W.; Ribes, A.; Geers, C. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur. J. Paediatric Neurol. 2013, 17, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Farhan, S.M.; Wang, J.; Robinson, J.F.; Lahiry, P.; Siu, V.M.; Prasad, C.; Kronick, J.B.; Ramsay, D.A.; Rupar, C.A.; Hegele, R.A. Exome sequencing identifies NFS 1 deficiency in a novel Fe-S. cluster disease, infantile mitochondrial complex II/III deficiency. Mol. Genet.Genomic Med. 2014, 2, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.L.; Vanderver, A.; Yang, S.; Chang, T.; Cramp, L.; Vezina, G.; Lichter-Konecki, U.; Cusmano-Ozog, K.P.; Smpokou, P.; Chapman, K.A. Thiamine pyrophosphokinase deficiency causes a Leigh disease like phenotype in a sibling pair: Identification through whole exome sequencing and management strategies. Mol. Genet. Metabol. Rep. 2014, 1, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, F.; Ardissone, A.; Lamantea, E.; Garavaglia, B.; Zeviani, M.; Farina, L.; Ghezzi, D.; Moroni, I. Cavitating leukoencephalopathy with multiple mitochondrial dysfunction syndrome and NFU1 mutations. Front. Genet. 2014, 5, 412. [Google Scholar] [CrossRef] [PubMed]
- Wimplinger, I.; Morleo, M.; Rosenberger, G.; Iaconis, D.; Orth, U.; Meinecke, P.; Lerer, I.; Ballabio, A.; Gal, A.; Franco, B. Mutations of the Mitochondrial Holocytochrome c–Type Synthase in X-Linked Dominant Microphthalmia with Linear Skin Defects Syndrome. Am. J. Hum. Genet. 2006, 79, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Almalki, A.; Alston, C.L.; Parker, A.; Simonic, I.; Mehta, S.G.; He, L.; Reza, M.; Oliveira, J.M.; Lightowlers, R.N.; McFarland, R. Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. Biochimica. Et Biophysica. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Alsemari, A.; Al-Younes, B.; Goljan, E.; Jaroudi, D.; BinHumaid, F.; Meyer, B.F.; Arold, S.T.; Monies, D. Recessive VARS2 mutation underlies a novel syndrome with epilepsy, mental retardation, short stature, growth hormone deficiency, and hypogonadism. Hum. Genet. 2017, 11, 28. [Google Scholar]
- Coughlin, C.R.; Scharer, G.H.; Friederich, M.W.; Yu, H.-C.; Geiger, E.A.; Creadon-Swindell, G.; Collins, A.E.; Vanlander, A.V.; Van Coster, R.; Powell, C.A. Mutations in the mitochondrial cysteinyl-tRNA synthase gene, CARS2, lead to a severe epileptic encephalopathy and complex movement disorder. J. Med. Genet. 2015, 52, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Elo, J.M.; Yadavalli, S.S.; Euro, L.; Isohanni, P.; Götz, A.; Carroll, C.J.; Valanne, L.; Alkuraya, F.S.; Uusimaa, J.; Paetau, A. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum. Mol. Genet. 2012, 21, 4521–4529. [Google Scholar] [CrossRef] [PubMed]
- Scheper, G.C.; Van Der Klok, T.; Van Andel, R.J.; Van Berkel, C.G.; Sissler, M.; Smet, J.; Muravina, T.I.; Serkov, S.V.; Uziel, G.; Bugiani, M. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nature Genet. 2007, 39, 534. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Richard, E.M.; Wang, X.; Shahzad, M.; Huang, V.H.; Qaiser, T.A.; Potluri, P.; Mahl, S.E.; Davila, A.; Nazli, S. Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet. 2015, 11, e1005097. [Google Scholar] [CrossRef] [PubMed]
- Simons, C.; Griffin, L.B.; Helman, G.; Golas, G.; Pizzino, A.; Bloom, M.; Murphy, J.L.; Crawford, J.; Evans, S.H.; Topper, S. Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am. J. Hum. Genet. 2015, 96, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Steenweg, M.E.; Ghezzi, D.; Haack, T.; Abbink, T.E.; Martinelli, D.; van Berkel, C.G.; Bley, A.; Diogo, L.; Grillo, E.; Te Water Naudé, J. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’caused by EARS2 mutations. Brain 2012, 135, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Janer, A.; Antonicka, H.; Lalonde, E.; Nishimura, T.; Sasarman, F.; Brown, G.K.; Brown, R.M.; Majewski, J.; Shoubridge, E.A. An RMND1 Mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am. J. Hum. Genet. 2012, 91, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Kernohan, K.D.; Dyment, D.A.; Pupavac, M.; Cramer, Z.; McBride, A.; Bernard, G.; Straub, I.; Tetreault, M.; Hartley, T.; Huang, L. Matchmaking facilitates the diagnosis of an autosomal-recessive mitochondrial disease caused by biallelic mutation of the tRNA isopentenyltransferase (TRIT1) gene. Hum. Mutation 2017, 38, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.A.; Elpeleg, O.; Antonicka, H.; Diepstra, H.; Saada, A.; Smits, P.; Sasarman, F.; Vriend, G.; Jacob-Hirsch, J.; Shaag, A. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am. J. Hum. Genet. 2006, 79, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Smits, P.; Antonicka, H.; van Hasselt, P.M.; Weraarpachai, W.; Haller, W.; Schreurs, M.; Venselaar, H.; Rodenburg, R.J.; Smeitink, J.A.; van den Heuvel, L.P. Mutation in subdomain G’of mitochondrial elongation factor G1 is associated with combined OXPHOS deficiency in fibroblasts but not in muscle. Eur. J. Hum. Genet. 2011, 19, 275. [Google Scholar] [CrossRef] [PubMed]
- Vedrenne, V.; Gowher, A.; De Lonlay, P.; Nitschke, P.; Serre, V.; Boddaert, N.; Altuzarra, C.; Mager-Heckel, A.-M.; Chretien, F.; Entelis, N. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am. J. Hum. Genet. 2012, 91, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Wedatilake, Y.; Niazi, R.; Fassone, E.; Powell, C.A.; Pearce, S.; Plagnol, V.; Saldanha, J.W.; Kleta, R.; Chong, W.K.; Footitt, E. TRNT1 deficiency: Clinical, biochemical and molecular genetic features. Orphanet J. Rare Dis. 2016, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Hikmat, O.; Eichele, T.; Tzoulis, C.; Bindoff, L.A. Understanding the epilepsy in POLG related disease. Int. J. Mol. Sci. 2017, 18, 1845. [Google Scholar] [CrossRef] [PubMed]
- Kasapkara, Ç.S.; Tümer, L.; Küçükçongar, A.; Hasanoglu, A.; Seneca, S.; De Meirleir, L. DGUOK-related mitochondrial DNA depletion syndrome in a child with an early diagnosis of glycogen storage disease. J. Pediatric gastroenterol. Nutr. 2013, 57, e28–e29. [Google Scholar] [CrossRef] [PubMed]
- Lesko, N.; Naess, K.; Wibom, R.; Solaroli, N.; Nennesmo, I.; von Döbeln, U.; Karlsson, A.; Larsson, N.-G. Two novel mutations in thymidine kinase-2 cause early onset fatal encephalomyopathy and severe mtDNA depletion. Neuromuscular Disorders 2010, 20, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.; Smith, C.; Fratter, C.; Alston, C.L.; He, L.; Craig, K.; Blakely, E.L.; Evans, J.C.; Taylor, J.; Shabbir, Z. Adults with RRM2B-related mitochondrial disease have distinct clinical and molecular characteristics. Brain 2012, 135, 3392–3403. [Google Scholar] [CrossRef] [PubMed]
- Carrozzo, R.; Dionisi-Vici, C.; Steuerwald, U.; Lucioli, S.; Deodato, F.; Di Giandomenico, S.; Bertini, E.; Franke, B.; Kluijtmans, L.A.; Meschini, M.C. SUCLA 2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain 2007, 130, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Ohlenbusch, A.; Edvardson, S.; Skorpen, J.; Bjornstad, A.; Saada, A.; Elpeleg, O.; Gärtner, J.; Brockmann, K. Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet J. Rare Dis. 2012, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Valayannopoulos, V.; Haudry, C.; Serre, V.; Barth, M.; Boddaert, N.; Arnoux, J.-B.; Cormier-Daire, V.; Rio, M.; Rabier, D.; Vassault, A. New SUCLG1 patients expanding the phenotypic spectrum of this rare cause of mild methylmalonic aciduria. Mitochondrion 2010, 10, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.D.; Isomursu, A.; Mennerich, D.; Vapola, M.H.; Weiher, H.; Kietzmann, T.; Hiltunen, J.K. The human mtDNA depletion syndrome gene MPV17 encodes a non-selective channel that modulates membrane potential. J. Biol. Chem. 2015, 290, 13840–13861. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Ghezzi, D.; Johnson, M.A.; Biagosch, C.A.; Shamseldin, H.E.; Haack, T.B.; Reyes, A.; Tsukikawa, M.; Sheldon, C.A.; Srinivasan, S. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 2013, 93, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Zeviani, M.; Muntoni, F.; Savarese, N.; Serra, G.; Tiranti, V.; Carrara, F.; Mariotti, C.; DiDonato, S. A MERRF/MELAS overlap syndrome associated with a new point mutation in the mitochondrial DNA tRNA^ Lys gene. Eur. J. Hum. Genet. 1993, 1, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Zsurka, G.; Elger, C.E.; Kunz, W.S. Mitochondrial involvement in temporal lobe epilepsy. Exper. Neurol. 2009, 218, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Koene, S.; Rodenburg, R.; Van Der Knaap, M.; Willemsen, M.; Sperl, W.; Laugel, V.; Ostergaard, E.; Tarnopolsky, M.; Martin, M.; Nesbitt, V. Natural disease course and genotype-phenotype correlations in Complex I deficiency caused by nuclear gene defects: What we learned from 130 cases. J. Inher. Metab. Dis. 2012, 35, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Sofou, K.; De Coo, I.F.; Isohanni, P.; Ostergaard, E.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; De Angst, I.B.; Lönnqvist, T. A multicenter study on Leigh syndrome: Disease course and predictors of survival. Orphanet J. Rare Dis. 2014, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Annegers, J.F.; Rocca, W.A.; Hauser, W.A. Causes of epilepsy: Contributions of the Rochester epidemiology project. Mayo Clinic Proc 1996, 71, 570–575. [Google Scholar] [CrossRef]
- Kovac, S.; Domijan, A.-M.; Walker, M.C.; Abramov, A.Y. Prolonged seizure activity impairs mitochondrial bioenergetics and induces cell death. J. Cell Sci. 2012, 125, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Ly, C.V.; Venken, K.J.; Koh, T.-W.; Zhou, Y.; Bellen, H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, T.; Evans, M.; Meldrum, B. Intracellular calcium accumulation in rat hippocampus during seizures induced by bicuculline orl-allylglycine. Neuroscience 1983, 10, 385–395. [Google Scholar] [CrossRef]
- Kovac, S.; Abramov, A.Y.; Walker, M.C. Energy depletion in seizures: Anaplerosis as a strategy for future therapies. Neuropharmacology 2013, 69, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Domijan, A.; Walker, M.; Abramov, A. Seizure activity results in calcium-and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 2014, 5, e1442. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 1992, 267, 8834–8839. [Google Scholar] [PubMed]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blalchy-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef]
- Gulati, A. Endothelin receptors, mitochondria and neurogenesis in cerebral ischemia. Curr. Neuropharmacol. 2016, 14, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Guntuku, L.; Naidu, V.; Yerra, V.G. Mitochondrial dysfunction in gliomas: Pharmacotherapeutic potential of natural compounds. Curr. Neuropharmacol. 2016, 14, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Kuhad, A. Mitochondrial dysfunction in depression. Curr. Neuropharmacol. 2016, 14, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kato, N. Mitochondrial dysfunction in bipolar disorder. Bipolar Disorders 2000, 2, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Rezin, G.T.; Amboni, G.; Zugno, A.I.; Quevedo, J.; Streck, E.L. Mitochondrial dysfunction and psychiatric disorders. Neurochem. Res. 2009, 34, 1021. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).