AAA Proteases: Guardians of Mitochondrial Function and Homeostasis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Molecular Architecture and Mode of Action of AAA Proteases
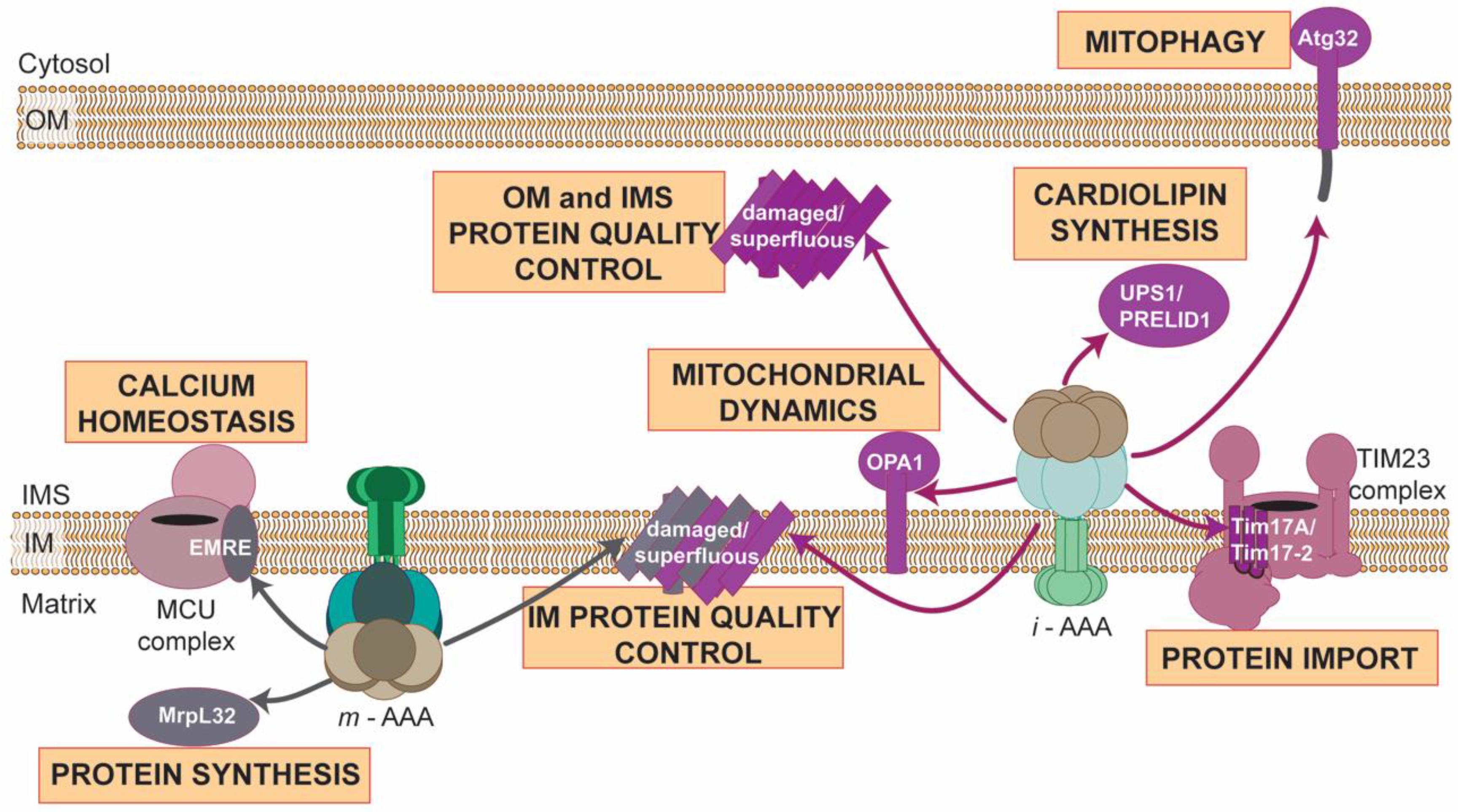
3. AAA Proteases Maintain Functional and Healthy Mitochondria
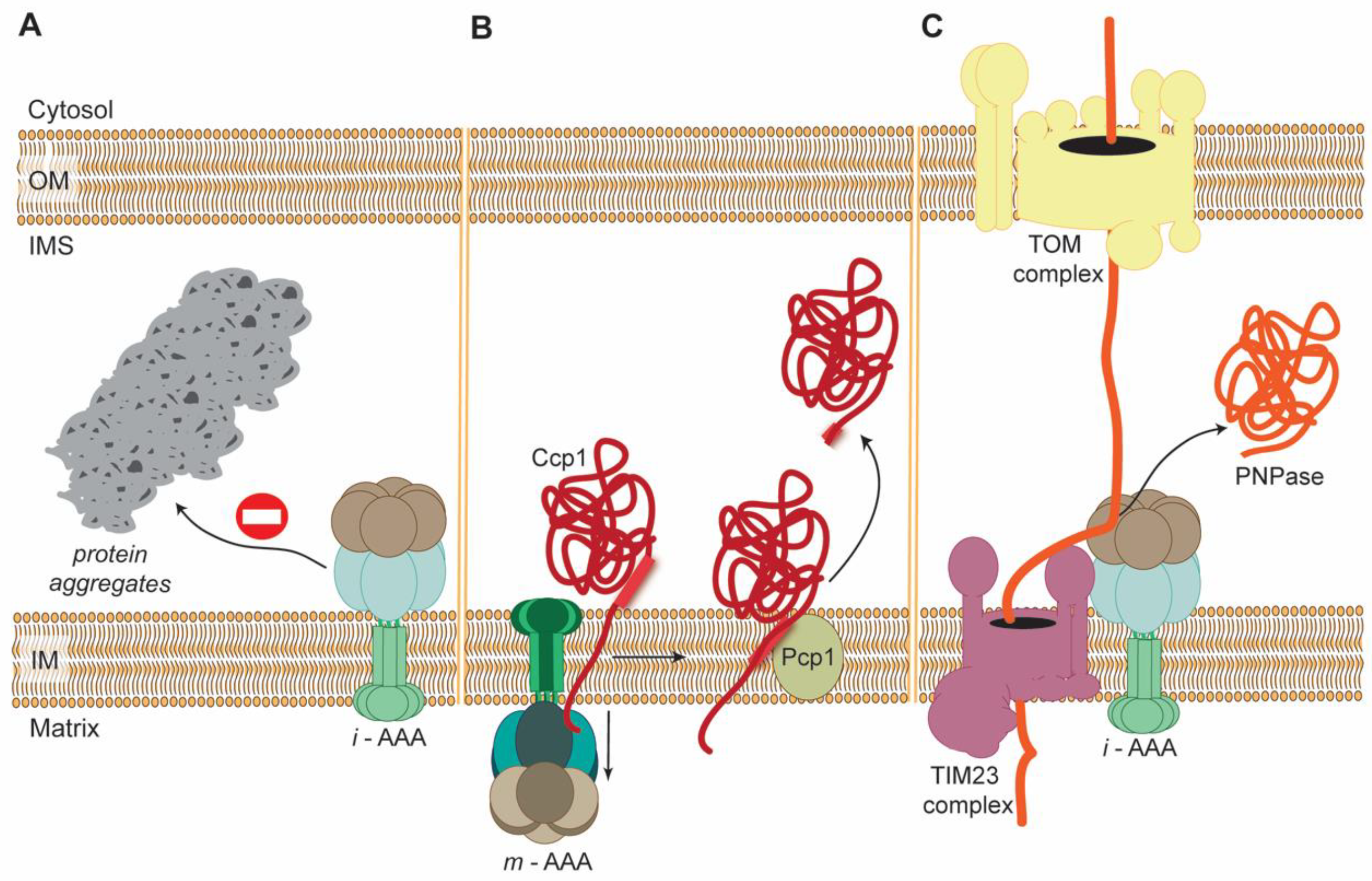
3.1. Role of AAA Proteases in Protein Mitochondrial Quality Control (PMQC)
3.2. Regulated Proteolysis: Fine-Tuning of Mitochondrial Pathways
3.3. AAA Proteases: Much More beyond the Machinesof Protein Destruction
4. AAA Proteases in the Pathogenesis of Human Diseases
5. Final Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 145–159. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 25, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Raimundo, N. Mitochondrial pathology: Stress signals from the energy factory. Trends Mol. Med. 2014, 20, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Stehling, O.; Lill, R. The Role of Mitochondria in Cellular Iron–Sulfur Protein Biogenesis: Mechanisms, Connected Processes, and Diseases. Cold Spring Harb. Perspect. Biol. 2013, 5, a011312. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Xu, H.; Chen, Z.J. Prion-Like Polymerization in Immunity and Inflammation. Cold Spring Harb. Perspect. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, J.; Venditti, P.; Adhihetty, P.J.; Ramsey, J.J.; Ascensão, A. Mitochondria in health and disease. Oxid. Med. Cell. Longev. 2014, 2014, 814042. [Google Scholar] [CrossRef] [PubMed]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 39, 2560–2574. [Google Scholar] [CrossRef]
- Gonczarowska-Jorge, H.; Zahedi, R.P.; Sickmann, A. The proteome of baker’s yeast mitochondria. Mitochondrion 2017, 33, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Langer, T.; López-Otín, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Richter, U.; Lahtinen, T.; Marttinen, P.; Suomi, F.; Battersby, B.J. Quality control of mitochondrial protein synthesis is required for membrane integrity and cell fitness. J. Cell Biol. 2015, 211, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Bohovych, I.; Chan, S.S.; Khalimonchuk, O. Mitochondrial protein quality control: The mechanisms guarding mitochondrial health. Antioxid. Redox Signal. 2015, 22, 977–994. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Gebert, M.; Pfanner, N.; van der Laan, M. Biogenesis of mitochondrial membrane proteins. Curr. Opin. Cell Biol. 2009, 21, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Mick, D.U.; Fox, T.D.; Rehling, P. Inventory control: Cytochrome c oxidase assembly regulates mitochondrial translation. Nat. Rev. Mol. Cell Biol. 2011, 12, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Dennerlein, S.; Wang, C.; Rehling, P. Plasticity of Mitochondrial Translation. Trends Cell Biol. 2017, 27, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Bode, M.; Longen, S.; Morgan, B.; Peleh, V.; Dick, T.P.; Bihlmaier, K.; Herrmann, J.M. Inaccurately assembled cytochrome c oxidase can lead to oxidative stress-induced growth arrest. Antioxid. Redox Signal. 2013, 18, 1597–1612. [Google Scholar] [CrossRef] [PubMed]
- Khalimonchuk, O.; Bird, A.; Winge, DR. Evidence for a pro-oxidant intermediate in the assembly of cytochrome oxidase. J. Biol. Chem. 2007, 282, 17442–17449. [Google Scholar] [CrossRef] [PubMed]
- Na, U.; Yu, W.; Cox, J.; Bricker, D.K.; Brockmann, K.; Rutter, J.; Thummel, C.S.; Winge, D.R. The LYR factors SDHAF1 and SDHAF3 mediate maturation of the iron-sulfur subunit of succinate dehydrogenase. Cell Metab. 2014, 20, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Figueira, T.R.; Barros, M.H.; Camargo, A.A.; Castilho, R.F.; Ferreira, J.C.; Kowaltowski, A.J.; Sluse, F.E.; Souza-Pinto, N.C.; Vercesi, AE. Mitochondria as a source of reactive oxygen and nitrogen species: From molecular mechanisms to human health. Antioxid. Redox Signal. 2013, 18, 2029–2074. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Haynes, C.M. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem. Sci. 2011, 36, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, F.; Tatsuta, T.; Langer, T. Mitochondrial AAA proteases—Towards a molecular understanding of membrane-bound proteolytic machines. Biochim. Biophys. Acta 2012, 1823, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Levytskyy, R.M.; Bohovych, I.; Khalimonchuk, O. Metalloproteases of the Inner Mitochondrial Membrane. Biochemistry 2017, 56, 4737–4746. [Google Scholar] [CrossRef] [PubMed]
- Glynn, S.E. Multifunctional Mitochondrial AAA Proteases. Front. Mol. Biosci. 2017, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Ammelburg, M.; Frickey, T.; Lupas, A.N. Classification of AAA+ proteins. J. Struct. Biol. 2006, 156, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Augustin, S.; Tatsuta, T.; Gerdes, F.; Langer, T.; Tsai, FT. Electron cryomicroscopy structure of a membrane-anchored mitochondrial AAA protease. J. Biol. Chem. 2011, 286, 4404–4411. [Google Scholar] [CrossRef] [PubMed]
- Puchades, C.; Rampello, A.J.; Shin, M.; Giuliano, C.J.; Wiseman, R.L.; Glynn, S.E.; Lander, G.C. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 2017, 358. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.R.; Hanekamp, T.; Thorsness, P.E. Biochemical and functional analysis of the YME1 gene product, an ATP and zinc-dependent mitochondrial protease from S. cerevisiae. Mol. Biol. Cell 1996, 7, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Urantowka, A.; Knorpp, C.; Olczak, T.; Kolodziejczak, M.; Janska, H. Plant mitochondria contain at least two i-AAA-like complexes. Plant Mol. Biol. 2005, 59, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Arlt, H.; Tauer, R.; Feldmann, H.; Neupert, W.; Langer, T. The YTA10-12 complex, an AAA protease with chaperone-like activity in the inner membrane of mitochondria. Cell 1996, 85, 875–885. [Google Scholar] [CrossRef]
- Koppen, M.; Metodiev, M.D.; Casari, G.; Rugarli, E.I.; Langer, T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol. Cell. Biol. 2007, 27, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Kremmidiotis, G.; Gardner, A.E.; Settasatian, C.; Savoia, A.; Sutherland, G.R.; Callen, D.F. Molecular and functional analyses of the human and mouse genes encoding AFG3L1, a mitochondrial metalloprotease homologous to the human spastic paraplegia protein. Genomics 2001, 76, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Piechota, J.; Kolodziejczak, M.; Juszczak, I.; Sakamoto, W.; Janska, H. Identification and characterization of high molecular weight complexes formed by matrix AAA proteases and prohibitins in mitochondria of Arabidopsis thaliana. J. Biol. Chem. 2010, 285, 12512–12521. [Google Scholar] [CrossRef] [PubMed]
- Leonhard, K.; Stiegler, A.; Neupert, W.; Langer, T. Chaperone-like activity of the AAA domain of the yeast Yme1 AAA protease. Nature 1999, 398, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Rampello, A.J.; Glynn, S.E. Engineered AAA+ proteases reveal principles of proteolysis at the mitochondrial inner membrane. Nat. Commun. 2016, 7, 13301. [Google Scholar] [CrossRef] [PubMed]
- Augustin, S.; Gerdes, F.; Lee, S.; Tsai, F.T.; Langer, T.; Tatsuta, T. An intersubunit signaling network coordinates ATP hydrolysis by m-AAA proteases. Mol. Cell. 2009, 35, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Leonhard, K.; Guiard, B.; Pellecchia, G.; Tzagoloff, A.; Neupert, W.; Langer, T. Membrane protein degradation by AAA proteases in mitochondria: Extraction of substrates from either membrane surface. Mol. Cell. 2000, 5, 629–638. [Google Scholar] [CrossRef]
- Rampello, A.J.; Glynn, S.E. Identification of a Degradation Signal Sequence within Substrates of the Mitochondrial i-AAA Protease. J. Mol. Biol. 2017, 429, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Martin, D.W.; Rampello, A.J.; Glynn, S.E. Dissecting Substrate Specificities of the Mitochondrial AFG3L2 Protease. Biochemistry 2018, 57, 4225–4235. [Google Scholar] [CrossRef] [PubMed]
- Steglich, G.; Neupert, W.; Langer, T. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol. Cell. Biol. 1999, 19, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Osman, C.; Merkwirth, C.; Langer, T. Prohibitins and the functional compartmentalization of mitochondrial membranes. J. Cell Sci. 2009, 122, 3823–3830. [Google Scholar] [CrossRef] [PubMed]
- König, T.; Tröder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Mühlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.D.; Lee, M.S.; Spencer, F.A.; Jensen, R.E. A genomewide screen for petite-negative yeast strains yields a new subunit of the i-AAA protease complex. Mol. Biol. Cell 2006, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.D.; Tamura, Y.; Sesaki, H.; Jensen, R.E. Mgr3p and Mgr1p are adaptors for the mitochondrial i-AAA protease complex. Mol. Biol. Cell 2008, 12, 5387–5397. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Saita, S.; Nolte, H.; Müller, S.; König, T.; Richter-Dennerlein, R.; Sprenger, H.G.; Madrenas, J.; Mühlmeister, M.; Bandt, U.; et al. The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L. EMBO Rep. 2016, 17, 1844–1856. [Google Scholar] [CrossRef] [PubMed]
- Opalińska, M.; Parys, K.; Jańska, H. Identification of Physiological Substrates and Binding Partners of the Plant Mitochondrial Protease FTSH4 by the Trapping Approach. Int. J. Mol. Sci. 2017, 2455. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, L.; Jiang, H. Mitochondrial inner-membrane protease Yme1 degrades outer-membrane proteins Tom22 and Om45. J. Cell Biol. 2018, 217, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Bonn, F.; Tatsuta, T.; Petrungaro, C.; Riemer, J.; Langer, T. Presequence-dependent folding ensures MrpL32 processing by the m-AAA protease in mitochondria. EMBO J. 2011, 30, 2545–2556. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Palmer, C.S.; Stojanovski, D. Mitochondrial protein quality control in health and disease. Br. J. Pharmacol. 2014, 171, 1870–1889. [Google Scholar] [CrossRef] [PubMed]
- Voos, W.; Jaworek, W.; Wilkening, A.; Bruderek, M. Protein quality control at the mitochondrion. Essays Biochem. 2016, 60, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Youle, R.J. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol. 2011, 23, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.B.; Rutter, J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem. Soc. Trans. 2011, 39, 1509–1513. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Zhou, C.; Jin, E.; Kucharavy, A.; Zhang, Y.; Wen, Z.; Florens, L.; Li, R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 2017, 543, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Leonhard, K.; Herrmann, J.M.; Stuart, R.A.; Mannhaupt, G.; Neupert, W.; Langer, T. AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP-dependent degradation of inner membrane proteins in mitochondria. EMBO J. 1996, 15, 4218–4229. [Google Scholar] [CrossRef] [PubMed]
- Guelin, E.; Rep, M.; Grivell, L.A. Afg3p, a mitochondrial ATP-dependent metalloprotease, is involved in the degradation of mitochondrially-encoded CoxI, Cox3, Cob, Su6, Su8 and Su9 subunits of the inner membrane complexes I, IV and V. FEBS Lett. 1996, 381, 42–46. [Google Scholar] [CrossRef]
- Pearce, D.E.; Sherman, F. Degradation of Cytochrome Oxidase Subunits in Mutants of Yeast Lacking Cytochrome c and Suppression of the Degradation by Mutation of yme1. J. Biol. Chem. 1995, 270, 20879–20882. [Google Scholar] [CrossRef] [PubMed]
- Nakai, T.; Yasuhara, T.; Fujiki, Y.; Ohashi, A. Multiple genes, including a member of the AAA family, are essential for degradation of unassembled subunit 2 of cytochrome c oxidase in yeast mitochondria. Mol. Cell. Biol. 1995, 15, 4441–4452. [Google Scholar] [CrossRef] [PubMed]
- Stiburek, L.; Cesnekova, J.; Kostkova, O.; Fornuskova, D.; Vinsova, K.; Wenchich, L.; Houstek, J.; Zeman, J. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol. Biol. Cell 2012, 23, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Kaser, M.; Kambacheld, M.; Kisters-Woike, B.; Langer, T. Oma1, a novel membrane-bound metallopeptidase in mitochondria with activities overlapping with the m-AAA protease. J. Biol. Chem. 2003, 278, 46414–46423. [Google Scholar] [CrossRef] [PubMed]
- Maltecca, F.; Magnoni, R.; Cerri, F.; Cox, G.A.; Quattrini, A.; Casari, G. Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J. Neurosci. 2009, 29, 9244–9254. [Google Scholar] [CrossRef] [PubMed]
- Kicia, M.; Gola, E.M.; Janska, H. Mitochondrial protease AtFtsH4 protects ageing Arabidopsis rosettes against oxidative damage under short-day photoperiod. Plant Signal. Behav. 2010, 5, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, Y.; Zhang, K.; Jian, F.; Hu, C.; Miao, L.; Gong, L.; Sun, L.; Zhang, X.; Chen, S.; et al. Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ. 2016, 23, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Liu, H.; Daniels, M.P.; Zhang, G.; Xu1, H. Loss of Drosophila i-AAA protease, dYME1L, causes abnormal mitochondria and apoptotic degeneration. Cell Death Differ. 2016, 23, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Sprenger, H.G.; Langer, T. m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell Res. 2018, 28, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Maltecca, F.; Baseggio, E.; Consolato, F.; Mazza, D.; Podini, P.; Young, S.M., Jr.; Drago, I.; Bahr, B.A.; Puliti, A.; Codazzi, F.; et al. Purkinje neuron Ca2+ influx reduction rescues ataxia in SCA28 model. J. Clin. Investig. 2015, 125, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jacquemyn, J.; Murru, S.; Martinelli, P.; Barth, E.; Langer, T.; Niessen, C.M.; Rugarli, E.I. The Mitochondrial m-AAA Protease Prevents Demyelination and Hair Greying. PLoS Genet. 2016, 12, e1006463. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, P.; Mattina, V.; Bernacchia, A.; Magnoni, R.; Cerri, F.; Cox, G.; Quattrini, A.; Casari, G.; Rugarli, E.I. Genetic interaction between the m-AAA protease isoenzymes reveals novel roles in cerebellar degeneration. Hum. Mol. Genet. 2009, 18, 2001–2013. [Google Scholar] [CrossRef] [PubMed]
- Ferreirinha, F.; Quattrini, A.; Pirozzi, M.; Valsecchi, V.; Dina, G.; Broccoli, V.; Auricchio, A.; Piemonte, F.; Tozzi, G.; Gaeta, L.; et al. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J. Clin. Investig. 2004, 113, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Maltecca, F.; Aghaie, A.; Schroeder, D.G.; Cassina, L.; Taylor, B.A.; Phillips, S.J.; Malaguti, M.; Previtali, S.; Guénet, J.L.; Quattrini, A.; et al. The mitochondrial protease AFG3L2 is essential for axonal development. J. Neurosci. 2008, 28, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.W.; Wu, Y.; Willliams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Vögtle, F.N.; Burkhart, J.M.; Rao, S.; Gerbeth, C.; Hinrichs, J.; Martinou, J.C.; Chacinska, A.; Sickmann, A.; Zahedi, R.P.; Meisinger, C. Intermembrane space proteome of yeast mitochondria. Mol. Cell. Proteom. 2012, 11, 1840–1852. [Google Scholar] [CrossRef] [PubMed]
- Hung, V.; Zou, P.; Rhee, H.W.; Udeshi, N.D.; Cracan, V.; Svinkina, T.; Carr, S.A.; Mootha, V.K.; Ting, A.Y. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 2014, 55, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Backes, S.; Herrmann, J.M. Protein Translocation into the Intermembrane Space and Matrix of Mitochondria: Mechanisms and Driving Forces. Front. Mol. Biosci. 2017, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, B.; Westerburg, H.; Forné, I.; Imhof, A.; Neupert, W.; Mokranjac, D. Role of the AAA protease Yme1 in folding of proteins in the intermembrane space of mitochondria. Mol. Biol. Cell 2012, 23, 4335–4346. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Mooga, V.P.; Guiard, B.; Langer, T.; Ryan, M.T.; Stojanovski, D. Impaired folding of the mitochondrial small TIM chaperones induces clearance by the i-AAA protease. J. Mol. Biol. 2012, 424, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Spiller, M.P.; Guo, L.; Wang, Q.; Tran, P.; Lu, H. Mitochondrial Tim9 protects Tim10 from degradation by the protease Yme1. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Bittner, L.M.; Arends, J.; Narberhaus, F. When, how and why? Regulated proteolysis by the essential FtsH protease in Escherichia coli. Biol. Chem. 2017, 398, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Mårtensson, C.U.; Doan, K.N.; Becker, T. Effects of lipids on mitochondrial functions. Biochim. Biophys. Acta 2017, 1862, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Greenberg, M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta 2017, 1862, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Cardiolipin and mitochondrial function in health and disease. Antioxid. Redox Signal. 2014, 20, 1925–1953. [Google Scholar] [CrossRef] [PubMed]
- Osman, C.; Voelker, D.R.; Langer, T. Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 2011, 192, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Potting, C.; Wilmes, C.; Engmann, T.; Osman, C.; Langer, T. Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 2010, 29, 2888–2898. [Google Scholar] [CrossRef] [PubMed]
- Potting, C.; Tatsuta, T.; König, T.; Haag, M.; Wai, T.; Aaltonen, M.J.; Langer, T. TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 2013, 18, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Connerth, M.; Tatsuta, T.; Haag, M.; Klecker, T.; Westermann, B.; Langer, T. Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 2012, 338, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Sesaki, H.; Endo, T. Phospholipid transport via mitochondria. Traffic 2014, 15, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Smakowska, E.; Skibior-Blaszczyk, R.; Czarna, M.; Kolodziejczak, M.; Kwasniak-Owczarek, M.; Parys, K.; Funk, C.; Janska, H. Lack of FTSH4 Protease Affects Protein Carbonylation, Mitochondrial Morphology, and Phospholipid Content in Mitochondria of Arabidopsis: New Insights into a Complex Interplay. Plant Physiol. 2016, 171, 2516–2535. [Google Scholar] [CrossRef] [PubMed]
- Harbauer, A.B.; Zahedi, R.P.; Sickmann, A.; Pfanner, N.; Meisinger, C. The protein import machinery of mitochondria—A regulatory hub in metabolism, stress, and disease. Cell Metab. 2014, 19, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Atanassova, N.; Genereux, J.C.; Wiseman, R.L. Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 2013, 18, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Goemans, C.G.; Boya, P.; Skirrow, C.J.; Tolkovsky, A.M. Intra-mitochondrial degradation of Tim23 curtails the survival of cells rescued from apoptosis by caspase inhibitors. Cell Death Differ. 2008, 15, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.N.; Hood, D.A. Effect of Tim23 Knockdown in vivo on Mitochondrial Protein Import and Retrograde Signaling to the UPRmt in Muscle. Am. J. Physiol. Cell Physiol. 2018, 315, C516–C526. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Schendzielorz, A.; Rehling, P. Unlocking the presequence import pathway. Trends Cell Biol. 2015, 25, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Opalińska, M.; Parys, K.; Murcha, M.W.; Jańska, H. The plant i-AAA protease controls the turnover of an essential mitochondrial protein import component. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Ghifari, A.S.; Gill-Hille, M.; Murcha, M.W. Plant mitochondrial protein import: The ins and outs. Biochem. J. 2018, 475, 2191–2208. [Google Scholar] [CrossRef] [PubMed]
- Nolden, M.; Ehses, S.; Koppen, M.; Bernacchia, A.; Rugarli, E.I.; Langer, T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 2005, 123, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Woellhaf, M.W.; Hansen, K.G.; Garth, C.; Herrmann, J.M. Import of ribosomal proteins into yeast mitochondria. Biochem. Cell Biol. 2014, 92, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Almajan, E.R.; Richter, R.; Paeger, L.; Martinelli, P.; Barth, E.; Decker, T.; Larsson, N.G.; Kloppenburg, P.; Langer, T.; Rugarli, E.I. AFG3L2 supports mitochondrial protein synthesis and Purkinje cell survival. J. Clin. Investig. 2012, 122, 4048–4058. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczak, M.; Skibior-Blaszczyk, R.; Janska, H. m-AAA Complexes Are Not Crucial for the Survival of Arabidopsis under Optimal Growth Conditions despite Their Importance for Mitochondrial Translation. Plant Cell Physiol. 2018, 59, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Koppen, M.; Bonn, F.; Ehses, S.; Langer, T. Autocatalytic processing of m-AAA protease subunits in mitochondria. Mol. Biol. Cell 2009, 20, 4216–4224. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jin, M.; Liu, X.; Klionsky, D.J. Proteolytic processing of Atg32 by the mitochondrial i-AAA protease Yme1 regulates mitophagy. Autophagy 2013, 9, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.B.; Pekkurnaz, G. Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J. Mol. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Augustin, S.; Nolden, M.; Friedrichs, B.; Langer, T. m-AAA protease-driven membrane dislocation allows intramembrane cleavage by rhomboid in mitochondria. EMBO J. 2007, 26, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Rainey, R.N.; Glavin, J.D.; Chen, H.W.; French, S.W.; Teitell, M.A.; Koehler, C.M. A new function in translocation for the mitochondrial i-AAA protease Yme1: Import of polynucleotide phosphorylase into the intermembrane space. Mol. Cell. Biol. 2006, 26, 8488–8497. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, B.; Wai, T.; Hu, H.; MacVicar, T.; Musante, L.; Fischer-Zirnsak, B.; Stenzel, W.; Gräf, R.; van den Heuvel, L.; Ropers, H.H.; et al. Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. eLife 2016, 5, e16078. [Google Scholar] [CrossRef] [PubMed]
- Cagnoli, C.; Stevanin, G.; Brussino, A.; Barberis, M.; Mancini, C.; Margolis, R.L.; Holmes, S.E.; Nobili, M.; Forlani, S.; Padovan, S. Missense mutations in the AFG3L2 proteolytic domain account for ~1.5% of European autosomal dominant cerebellar ataxias. Hum. Mutat. 2010, 31, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, D.; Lazzaro, F.; Brusco, A.; Plumari, M.; Battaglia, G.; Pastore, A.; Finardi, A.; Cagnoli, C.; Tempia, F.; Frontali, M.; et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 2010, 42, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.M.; Adams, D.; Bonn, F.; Martinelli, P.; Cherukuri, P.F.; Teer, J.K.; Hansen, N.F.; Cruz, P.; Mulliki, J.C.; Blakesley, R.W.; et al. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011, 10, e1002325. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, C.; Brusco, A.; Di Bella, D.; Cagnoli, C.; Seri, M.; Gellera, C.; Di Donato, S.; Taroni, F. Spinocerebellar ataxia type 28: A novel autosomal dominant cerebellar ataxia characterized by slow progression and ophthalmoparesis. Cerebellum 2008, 7, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Löbbe, A.M.; Kang, J.S.; Hilker, R.; Hackstein, H.; Müller, U.; Nolte, D. A novel missense mutation in AFG3L2 associated with late onset and slow progression of spinocerebellar ataxia type 28. J. Mol. Neurosci. 2014, 52, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Wu, C.K.; Zuzuárregui, J.R.; Hohler, A.D. A novel AFG3L2 mutation in a Somalian patient with spinocerebellar ataxia type 28. J. Neurol. Sci. 2015, 358, 530–531. [Google Scholar] [CrossRef] [PubMed]
- Zühlke, C.; Mikat, B.; Timmann, D.; Wieczorek, D.; Gillessen-Kaesbach, G.; Bürk, K. Spinocerebellar ataxia 28: A novel AFG3L2 mutation in a German family with young onset, slow progression and saccadic slowing. Cerebellum Ataxias 2015, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Svenstrup, K.; Nielsen, T.T.; Aidt, F.; Rostgaard, N.; Duno, M.; Wibrand, F.; Vinther-Jensen, T.; Law, I.; Vissing, J.; Roos, P.; et al. SCA28: Novel Mutation in the AFG3L2 Proteolytic Domain Causes a Mild Cerebellar Syndrome with Selective Type-1 Muscle Fiber Atrophy. Cerebellum 2017, 16, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Casari, G.; De Fusco, M.; Ciarmatori, S.; Zeviani, M.; Mora, M.; Fernandez, P.; De Michele, G.; Filla, A.; Cocozza, S.; Marconi, R.; et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998, 93, 973–983. [Google Scholar] [CrossRef]
- Cagnoli, C.; Mariotti, C.; Taroni, F.; Seri, M.; Brussino, A.; Michielotto, C.; Grisoli, M.; Di Bella, D.; Migone, N.; Gellera, C.; et al. SCA28, a novel form of autosomal dominant cerebellar ataxia on chromosome 18p11.22-q11.2. Brain 2006, 129, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Gorman, G.S.; Griffin, H.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.L.; et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137, 1323–1336. [Google Scholar] [CrossRef] [PubMed]
- Wedding, I.M.; Koht, J.; Tran, G.T.; Misceo, D.; Selmer, K.K.; Holmgren, A.; Frengen, E.; Bindoff, L.; Tallaksen, C.M.; Tzoulis, C. Spastic paraplegia type 7 is associated with multiple mitochondrial DNA deletions. PLoS ONE 2014, 9, e86340. [Google Scholar] [CrossRef] [PubMed]
- Almontashiri, N.A.; Chen, H.H.; Mailloux, R.J.; Tatsuta, T.; Teng, A.C.; Mahmoud, A.B.; Ho, T.; Stewart, N.A.; Rippstein, P.; Harper, M.E.; et al. SPG7 variant escapes phosphorylation-regulated processing by AFG3L2, elevates mitochondrial ROS, and is associated with multiple clinical phenotypes. Cell Rep. 2014, 7, 834–847. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Opalińska, M.; Jańska, H. AAA Proteases: Guardians of Mitochondrial Function and Homeostasis. Cells 2018, 7, 163. https://doi.org/10.3390/cells7100163
Opalińska M, Jańska H. AAA Proteases: Guardians of Mitochondrial Function and Homeostasis. Cells. 2018; 7(10):163. https://doi.org/10.3390/cells7100163
Chicago/Turabian StyleOpalińska, Magdalena, and Hanna Jańska. 2018. "AAA Proteases: Guardians of Mitochondrial Function and Homeostasis" Cells 7, no. 10: 163. https://doi.org/10.3390/cells7100163
APA StyleOpalińska, M., & Jańska, H. (2018). AAA Proteases: Guardians of Mitochondrial Function and Homeostasis. Cells, 7(10), 163. https://doi.org/10.3390/cells7100163