Influence of Molecular Design on the Targeting Properties of ABD-Fused Mono- and Bi-Valent Anti-HER3 Affibody Therapeutic Constructs


 ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
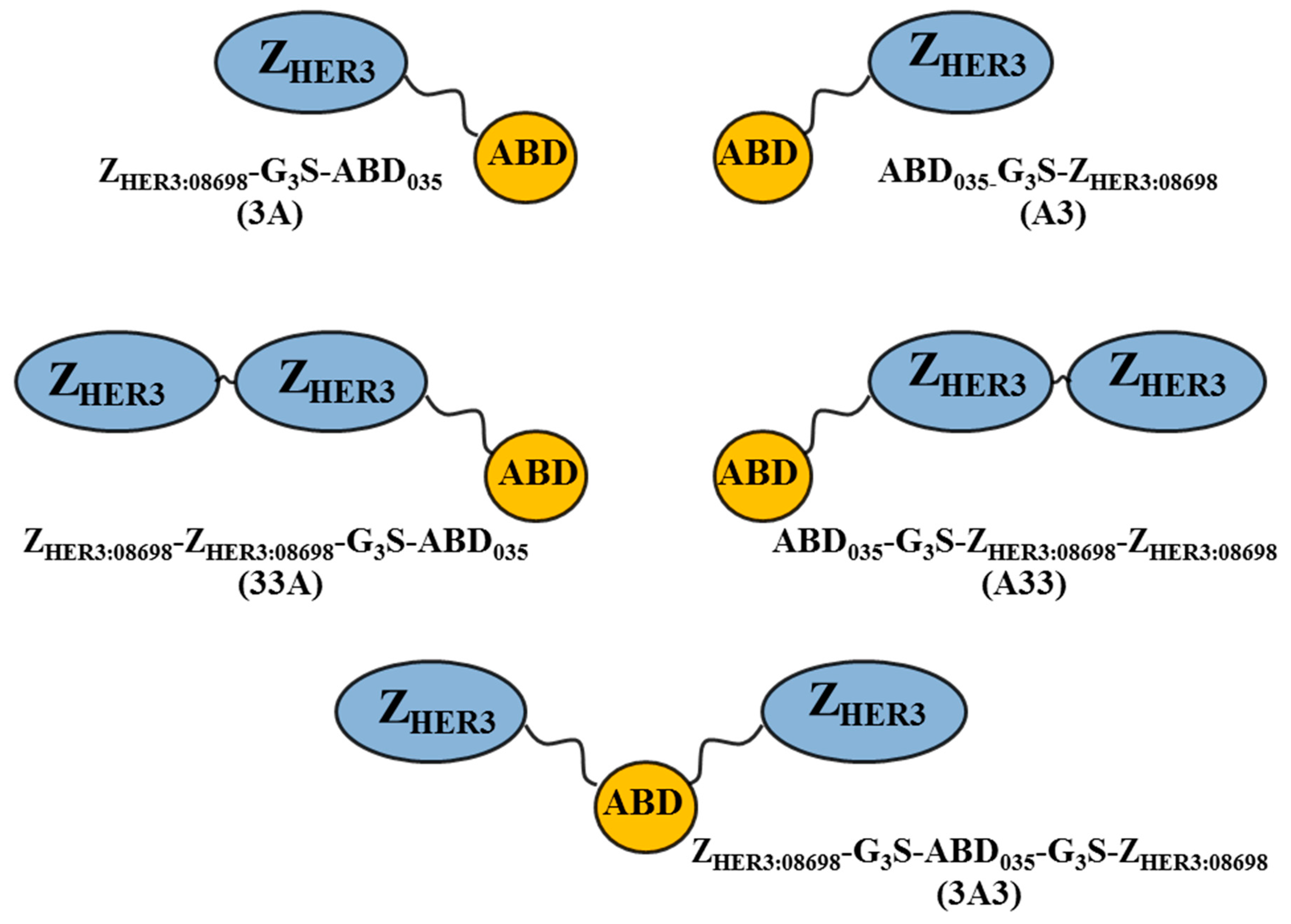
2.1. Production of Constructs, Chelator Conjugation, and Product Purification
2.2. Characterization of the Conjugated Proteins
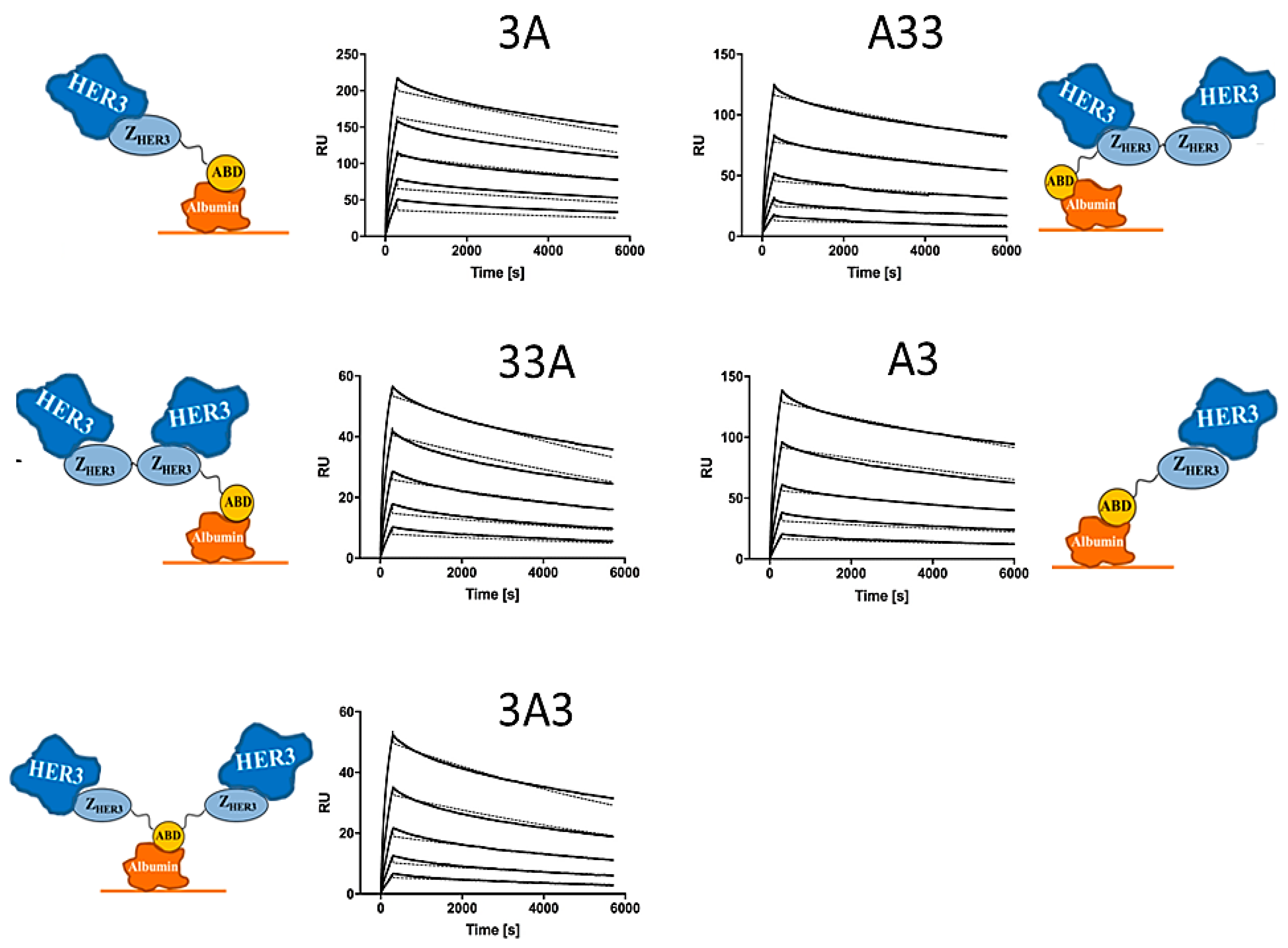
2.3. Affinity Determination
2.4. Radiolabelling of Constructs with Indium-111 and Stability Test of Radiolabelled Constructs
2.5. In Vitro Studies
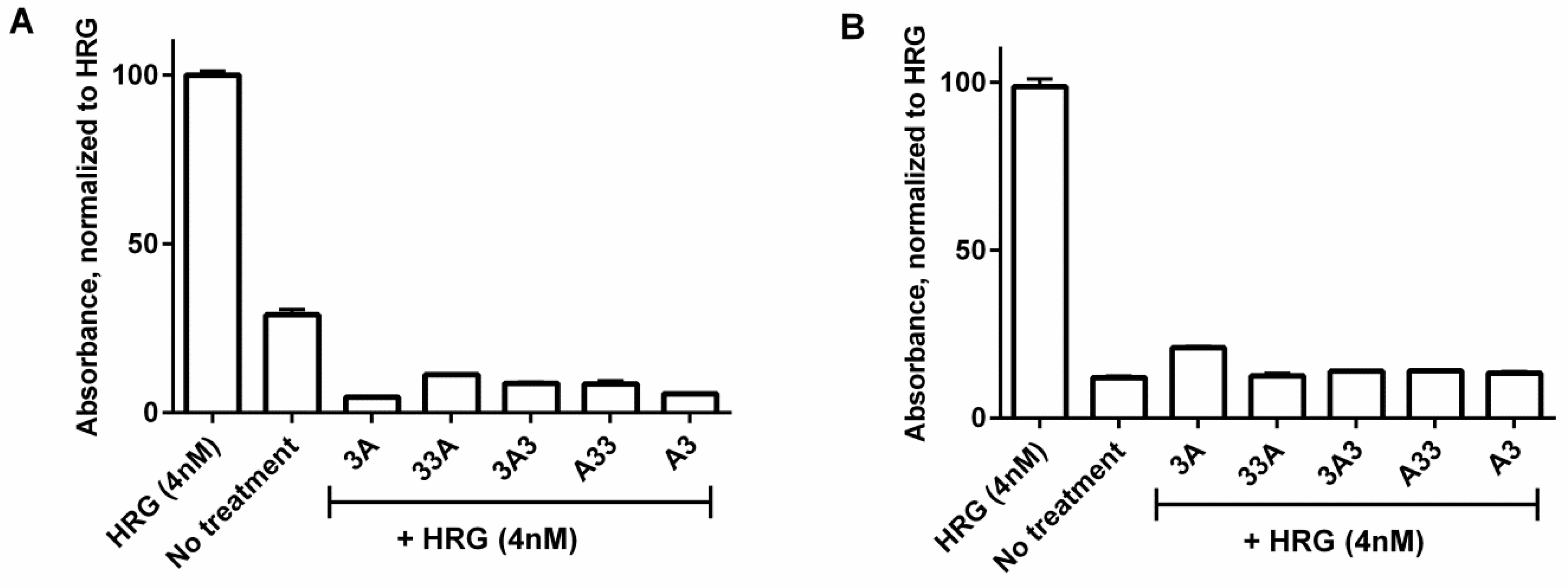
2.6. Inhibition of Phosphorylation
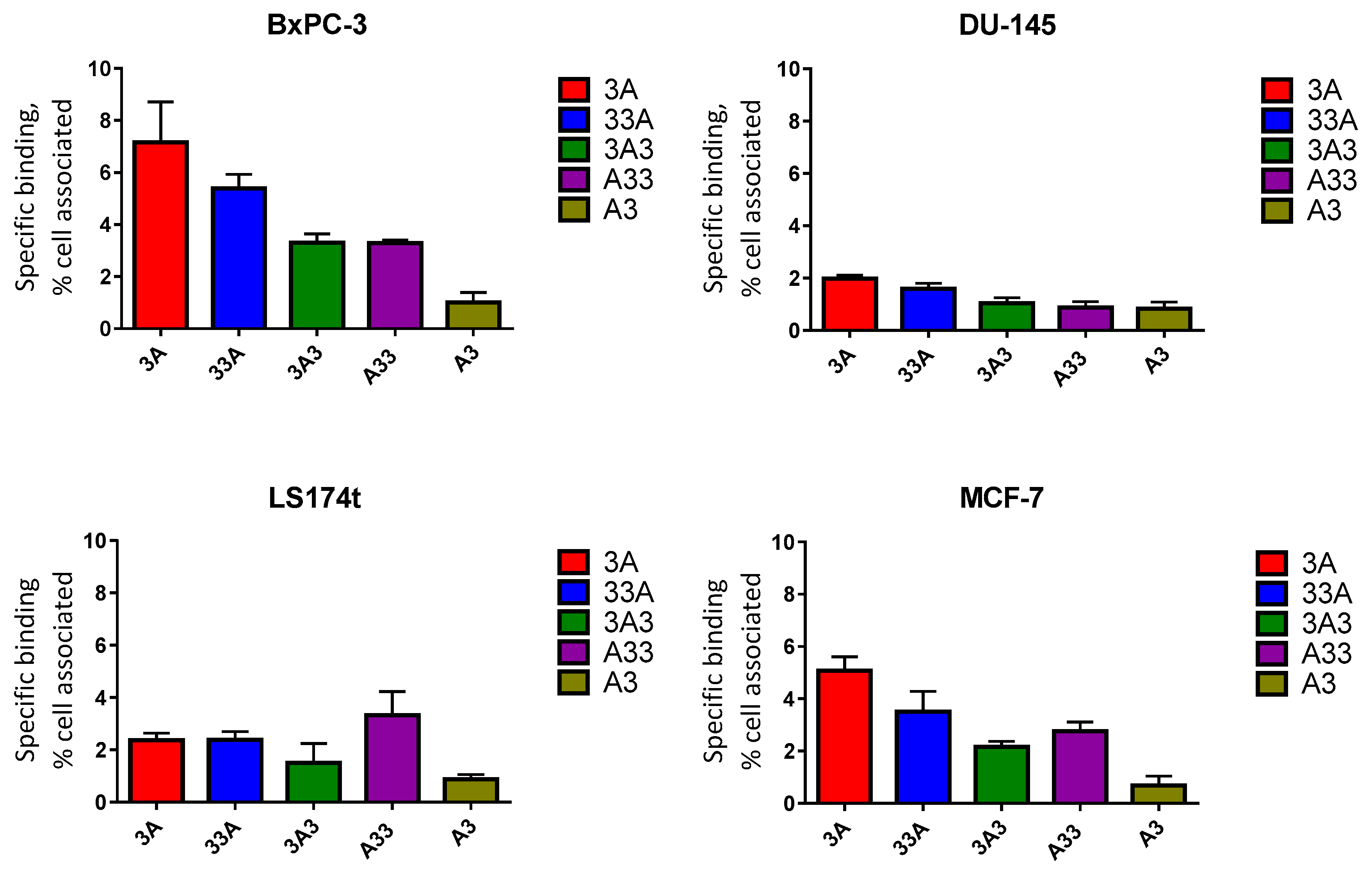
2.7. In Vitro Specificity
2.8. Cellular Processing and Internalization
2.9. In Vivo Studies
3. Results
3.1. Production of Constructs, Chelator Conjugation and Product Purification
3.2. Characterization of the Conjugated Proteins
3.3. Affinity Determination
3.4. Radiolabelling of Constructs with Indium-111 and Stability Test
3.5. Inhibition of Phosphorylation
3.6. In Vitro Specificity
3.7. Cellular Processing and Internalization
3.8. In Vivo Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Berkenblit, A. Antibody-Drug Conjugates for Cancer Treatment. Annu. Rev. Med. 2018, 69, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Lazzari, C.; Verlicchi, A.; Sosa, A.E.; Rosell, R. HER3 as a Therapeutic Target in Cancer. BioDrugs 2017, 31, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Lyu, H.; Huang, J.; Liu, B. Targeting of erbB3 receptor to overcome resistance in cancer treatment. Mol. Cancer 2014, 13, 105. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, V.J.; Levisson, M.; Eppink, M.H.; Smidt, H.; van der Oost, J. Alternative affinity tools: More attractive than antibodies? Biochem. J. 2011, 436, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nygren, P.A.; Skerra, A. Binding proteins from alternative scaffolds. J. Immunol. Methods 2004, 290, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Feldwisch, J.; Tolmachev, V. Engineering of affibody molecules for therapy and diagnostics. Methods Mol. Biol. 2012, 899, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.T.; Pehrson, R.; Tolmachev, V.; Daba, M.B.; Abrahmsén, L.; Ekblad, C. Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J. Biol. Chem. 2011, 286, 5234–5241. [Google Scholar] [CrossRef] [PubMed]
- Frejd, F.Y.; Kim, K.T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306. [Google Scholar] [CrossRef] [PubMed]
- Simeon, R.; Chen, Z. In vitro-engineered non-antibody protein therapeutics. Protein Cell 2018, 9, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Nord, K.; Gunneriusson, E.; Ringdahl, J.; Ståhl, S.; Uhlén, M.; Nygren, P.A. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat. Biotechnol. 1997, 15, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, S.; Gräslund, T.; Eriksson Karlström, A.; Frejd, F.Y.; Nygren, P.Å.; Löfblom, J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef] [PubMed]
- Sörensen, J.; Sandberg, D.; Sandström, M.; Wennborg, A.; Feldwisch, J.; Tolmachev, V.; Åström, G.; Lubberink, M.; Garske-Román, U.; Carlsson, J.; et al. First-in-human molecular imaging of HER2 expression in breast cancer metastases using the 111In-ABY-025 affibody molecule. J. Nucl. Med. 2014, 55, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Sörensen, J.; Velikyan, I.; Sandberg, D.; Wennborg, A.; Feldwisch, J.; Tolmachev, V.; Orlova, A.; Sandström, M.; Lubberink, M.; Olofsson, H.; et al. Measuring HER2-Receptor Expression In Metastatic Breast Cancer Using [68Ga]ABY-025 Affibody PET/CT. Theranostics 2016, 6, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Malm, M.; Kronqvist, N.; Lindberg, H.; Gudmundsdotter, L.; Bass, T.; Frejd, F.Y.; Höidén-Guthenberg, I.; Varasteh, Z.; Orlova, A.; Tolmachev, V.; et al. Inhibiting HER3-mediated tumor cell growth with affibody molecules engineered to low picomolar affinity by position-directed error-prone PCR-like diversification. PLoS ONE 2013, 8, e62791. [Google Scholar] [CrossRef]
- Göstring, L.; Malm, M.; Höidén-Guthenberg, I.; Frejd, F.Y.; Ståhl, S.; Löfblom, J.; Gedda, L. Cellular effects of HER3-specific affibody molecules. PLoS ONE 2012, 7, e40023. [Google Scholar] [CrossRef] [PubMed]
- Kronqvist, N.; Malm, M.; Göstring, L.; Gunneriusson, E.; Nilsson, M.; Höidén Guthenberg, I.; Gedda, L.; Frejd, F.Y.; Ståhl, S.; Löfblom, J. Combining phage and staphylococcal surface display for generation of ErbB3-specific Affibody molecules. Protein Eng. Des. Sel. 2011, 24, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Malm, M.; Bass, T.; Gudmundsdotter, L.; Lord, M.; Frejd, F.Y.; Ståhl, S.; Löfblom, J. Engineering of a bispecific affibody molecule towards HER2 and HER3 by addition of an albumin-binding domain allows for affinity purification and in vivo half-life extension. Biotechnol. J. 2014, 9, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Rosestedt, M.; Andersson, K.G.; Mitran, B.; Tolmachev, V.; Löfblom, J.; Orlova, A.; Ståhl, S. Affibody-mediated PET imaging of HER3 expression in malignant tumours. Sci. Rep. 2015, 5, 15226. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.G.; Rosestedt, M.; Varasteh, Z.; Malm, M.; Sandström, M.; Tolmachev, V.; Löfblom, J.; Ståhl, S.; Orlova, A. Comparative evaluation of 111In-labeled NOTA-conjugated affibody molecules for visualization of HER3 expression in malignant tumors. Oncol. Rep. 2015, 34, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Frejd, F. Half-Life Extension by Binding to Albumin through an Albumin Binding Domain. In Therapeutic Proteins: Strategies to Modulate Their Plasma Half-Lives; Kontermann, R., Ed.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2012; pp. 269–283. ISBN 978-3-527-32849-9. [Google Scholar]
- Jonsson, A.; Dogan, J.; Herne, N.; Abrahmsén, L.; Nygren, P.A. Engineering of a femtomolar affinity binding protein to human serum albumin. Protein Eng. Des. Sel. 2008, 21, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Frejd, F.; Klint, S.; Gudmundsdotter, L.; Höidén-Guthenberg, I.; Feldwisch, J.; Wennborg, A.; Bejker, D.; Reich, K. Blocking IL-17A with femtomolar affinity using the novel engineered Affibody ligand trap ABY-035: Interim results from a phase I, first in human study. In Proceedings of the 26th EADV Congress, Geneva, Switzerland, 13–17 September 2017. [Google Scholar]
- Schardt, J.S.; Oubaid, J.M.; Williams, S.C.; Howard, J.L.; Aloimonos, C.M.; Bookstaver, M.L.; Lamichhane, T.N.; Sokic, S.; Liyasova, M.S.; O’Neill, M.; et al. Engineered Multivalency Enhances Affibody-Based HER3 Inhibition and Downregulation in Cancer Cells. Mol. Pharm. 2017, 14, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Bass, T.Z.; Rosestedt, M.; Mitran, B.; Frejd, F.Y.; Löfblom, J.; Tolmachev, V.; Ståhl, S.; Orlova, A. In vivo evaluation of a novel format of a bivalent HER3-targeting and albumin-binding therapeutic affibody construct. Sci. Rep. 2017, 7, 43118. [Google Scholar] [CrossRef] [PubMed]
- Orlova, A.; Bass, T.Z.; Rinne, S.S.; Leitao, C.D.; Rosestedt, M.; Atterby, C.; Gudmundsdotter, L.; Frejd, F.Y.; Löfblom, J.; Tolmachev, V.; et al. Evaluation of the Therapeutic Potential of a HER3-Binding Affibody Construct TAM-HER3 in Comparison with a Monoclonal Antibody, Seribantumab. Mol. Pharm. 2018, 15, 3394–3403. [Google Scholar] [CrossRef] [PubMed]
- Tolmachev, V.; Mume, E.; Sjöberg, S.; Frejd, F.Y.; Orlova, A. Influence of valency and labelling chemistry on in vivo targeting using radioiodinated HER2-binding Affibody molecules. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Tolmachev, V.; Feldwisch, J.; Lindborg, M.; Baastrup, B.; Sandström, M.; Orlova, A. Influence of an aliphatic linker between DOTA and synthetic Z(HER2:342) Affibody molecule on targeting properties of the (111)In-labeled conjugate. Nucl. Med. Biol. 2011, 38, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Wållberg, H.; Orlova, A.; Altai, M.; Hosseinimehr, S.J.; Widström, C.; Malmberg, J.; Ståhl, S.; Tolmachev, V. Molecular design and optimization of 99mTc-labeled recombinant affibody molecules improves their biodistribution and imaging properties. J. Nucl. Med. 2011, 52, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Altai, M.; Liu, H.; Orlova, A.; Tolmachev, V.; Gräslund, T. Influence of molecular design on biodistribution and targeting properties of an Affibody-fused HER2-recognising anticancer toxin. Int. J. Oncol. 2016, 49, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Wållberg, H.; Orlova, A. Slow internalization of anti-HER2 synthetic affibody monomer 111In-DOTA-ZHER2:342-pep2: Implications for development of labeled tracers. Cancer Biother. Radiopharm. 2008, 23, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Schoeberl, B.; Kudla, A.; Masson, K.; Kalra, A.; Curley, M.; Finn, G.; Pace, E.; Harms, B.; Kim, J.; Kearns, J.; et al. Systems biology driving drug development: From design to the clinical testing of the anti-ErbB3 antibody seribantumab (MM-121). NPJ Syst. Biol. Appl. 2017, 3, 16034. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Milenic, D.E.; Whitlow, M.; Schlom, J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992, 52, 3402–3408. [Google Scholar] [PubMed]
- Da Pieve, C.; Allott, L.; Martins, C.D.; Vardon, A.; Ciobota, D.M.; Kramer-Marek, G.; Smith, G. Efficient [(18)F]AlF Radiolabeling of ZHER3:8698 Affibody Molecule for Imaging of HER3 Positive Tumors. Bioconjug. Chem. 2016, 27, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Lindzen, M.; Lavi, S.; Leitner, O.; Yarden, Y. Tailored cancer immunotherapy using combinations of chemotherapy and a mixture of antibodies against EGF-receptor ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 12559–12563. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.B.; Johnson, B.W.; Baum, J.; Adams, S.; Iadevaia, S.; Tang, J.; Rimkunas, V.; Xu, L.; Kohli, N.; Rennard, R.; et al. MM-141, an IGF-IR- and ErbB3-directed bispecific antibody, overcomes network adaptations that limit activity of IGF-IR inhibitors. Mol. Cancer Ther. 2014, 13, 410–425. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Ray-Coquard, I.; Selle, F.; Poveda, A.M.; Cibula, D.; Hirte, H.; Hilpert, F.; Raspagliesi, F.; Gladieff, L.; Harter, P.; et al. Randomized Phase II Trial of Seribantumab in Combination with Paclitaxel in Patients with Advanced Platinum-Resistant or -Refractory Ovarian Cancer. J. Clin. Oncol. 2016, 34, 4345–4353. [Google Scholar] [CrossRef] [PubMed]
- Barta, P.; Malmberg, J.; Melicharova, L.; Strandgård, J.; Orlova, A.; Tolmachev, V.; Laznicek, M.; Andersson, K. Protein interactions with HER-family receptors can have different characteristics depending on the hosting cell line. Int. J. Oncol. 2012, 40, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Hettmann, T.; Schneider, M.; Ogbagabriel, S.; Xie, J.; Juan, G.; Hartmann, S.; Radinsky, R.; Freeman, D.J. U3-1287 (AMG 888), a fully human anti-HER3 mAb, inhibits HER3 activation and induces HER3 internalization and degradation. Cancer Res. 2010, 70, LB–306. [Google Scholar] [CrossRef]
- Hopp, J.; Hornig, N.; Zettlitz, K.A.; Schwarz, A.; Fuss, N.; Müller, D.; Kontermann, R.E. The effects of affinity and valency of an albumin-binding domain (ABD) on the half-life of a single-chain diabody-ABD fusion protein. Protein Eng. Des. Sel. 2010, 23, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Tolmachev, V.; Orlova, A.; Pehrson, R.; Galli, J.; Baastrup, B.; Andersson, K.; Sandström, M.; Rosik, D.; Carlsson, J.; Lundqvist, H.; et al. Radionuclide therapy of HER2-positive microxenografts using a 177Lulabeled HER2-specific Affibody molecule. Cancer Res. 2007, 67, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- Orlova, A.; Jonsson, A.; Rosik, D.; Lundqvist, H.; Lindborg, M.; Abrahmsen, L.; Ekblad, C.; Frejd, F.Y.; Tolmachev, V. Site-specific radiometal labeling and improved biodistribution using ABY-027, a novel HER2-targeting affibody molecule-albumin-binding domain fusion protein. J. Nucl. Med. 2013, 54, 961–968. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
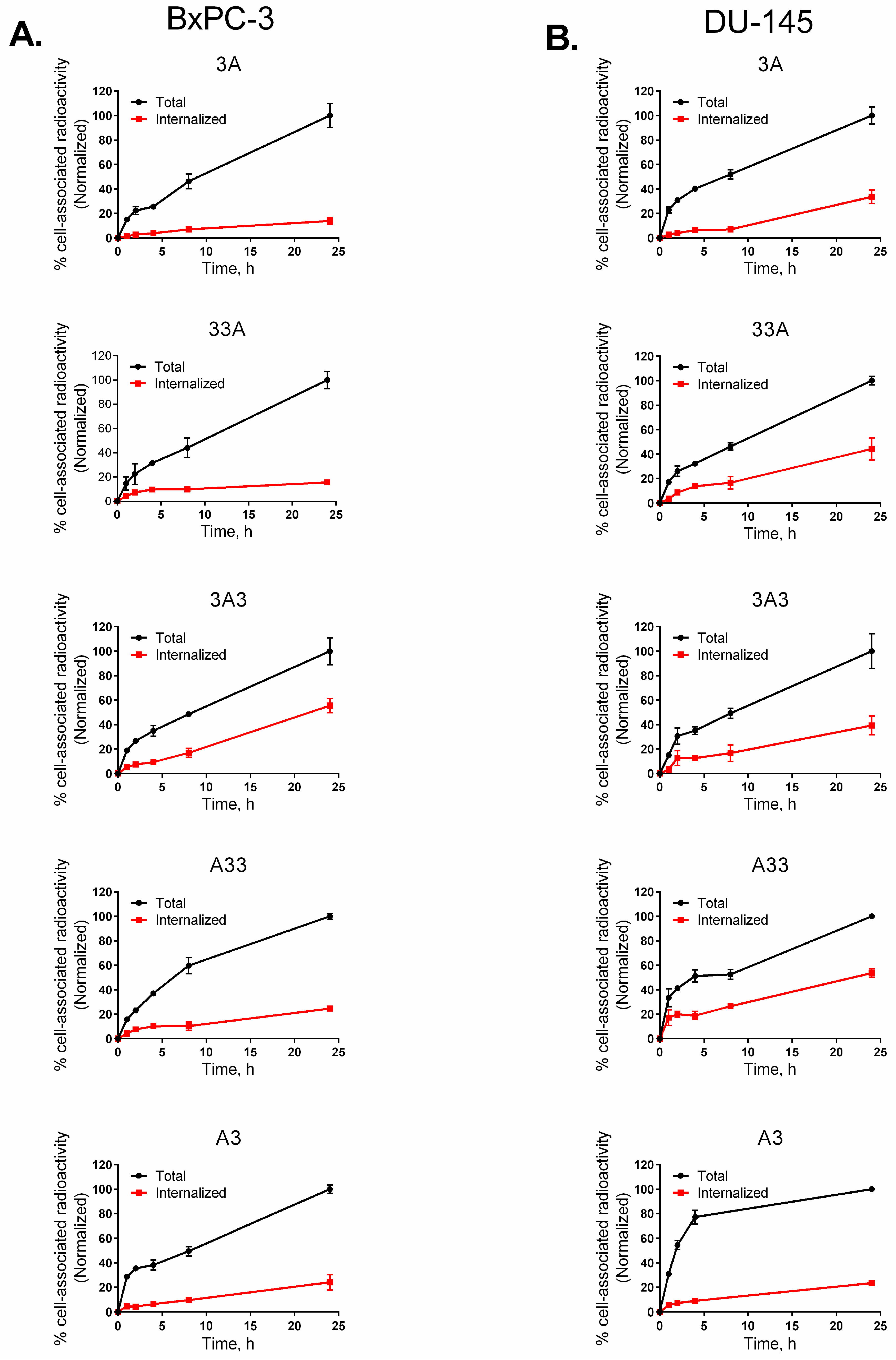{kind=link}
| Construct | MW (Da) | Tm (°C) | KD, HER3 (nM, Mean ± SD) | KD, HSA (nM, Mean ± SD) |
|---|---|---|---|---|
| 3A | 12,670.9 | 61.3 | 0.3 ± 0.03 | 0.06 ± 0.02 |
| 33A | 19,601.6 | 66.1 | 0.6 ± 0.03 | 0.24 ± 0.22 |
| 3A3 | 19,601.6 | 62.4 | 1.1 ± 0.1 | 0.68 ± 0.66 |
| A33 | 19,732.8 | 62.7 | 0.8 ± 0.15 | 0.35 ± 0.24 |
| A3 | 12,802.1 | 61.8 | 0.6 ± 0.15 | 0.15 ± 0.08 |
| Construct | Radiolabelling Yield (%) | Radiochemical Purity of Conjugates (%) | Protein Associated Activity after 1 h of Incubation with a 500-Fold Molar Excess of EDTA (%) |
|---|---|---|---|
| 3A | 89 ± 8 | 99 ± 1 | 99.6 ± 0.3 |
| 33A | 85.1 ± 0.1 | 98.6 ± 0.8 | 99.4 ± 0.1 |
| 3A3 | 51 ± 12 | 98 ± 1 | 98 ± 1 |
| A33 | 28 ± 6 | 97 ± 2 | 96 ± 1 |
| A3 | 45 ± 4 | 97 ± 2 | 96 ± 1 |
| Organ | 3A | 33A | 3A3 | A33 | A3 |
|---|---|---|---|---|---|
| 1 h | |||||
| Blood | 33 ± 3 c,d | 25 ± 2 | 24 ± 1 | 20.2 ± 0.9 | 21 ± 1 |
| Salivary gland | 3.6 ± 1.0 | 2.8 ± 0.4 | 2.6 ± 0.6 | 2.4 ± 0.5 | 2.7 ± 0.5 |
| Lung | 12 ± 2 | 10 ± 2 f | 8 ± 1 | 8 ± 2 | 8.2 ± 0.8 |
| Liver | 7 ± 2 | 13 ± 1 g | 7 ± 1 | 9 ± 2 | 5.8 ± 0.6 |
| Spleen | 6 ± 1 | 8 ± 2 | 5.0 ± 0.6 | 5.9 ± 0.4 j | 4.0 ± 0.2 |
| Stomach | 2.0 ± 0.3 | 1.9 ± 0.5 | 1.5 ± 0.3 | 1.5 ± 0.4 | 1.6 ± 0.2 |
| Small intestine | 5 ± 1 | 5.5 ± 0.4 | 3.6 ± 0.3 | 3.4 ± 0.6 | 3.6 ± 0.8 |
| Kidney | 10 ± 2 b,c,d | 11 ± 1 e,f,g | 22 ± 2 h | 41 ± 3 | 33 ± 2 |
| Tumour | 4.1 ± 0.5 | 4.3 ± 0.4 | 3.2 ± 0.7 | 4 ± 1 | 4 ± 2 |
| Muscle | 1.0 ± 0.1 | 0.9 ± 0.2 | 0.9 ± 0.1 | 0.7 ± 0.1 | 0.7 ± 0.1 |
| Bone | 2.4 ± 0.3 | 2.4 ± 0.4 | 1.9 ± 0.7 | 1.7 ± 0.5 | 2.0 ± 0.9 |
| GI tract * | 4 ± 1 | 4.7 ± 0.7 k | 3.1 ± 0.7 | 2.7 ± 0.3 | 2.9 ± 0.5 |
| Carcass * | 32 ± 6 | 30 ± 2 f,k | 27 ± 2 | 21 ± 2 | 25.2 ± 0.7 |
| 6 h | |||||
| Blood | 19 ± 2 a,c,k | 9.5 ± 0.8 k | 14 ± 2 k | 9 ± 1 k | 16 ± 2 |
| Salivary gland | 4.3 ± 0.4 c | 3.35 ± 0.08 f | 3.2 ± 0.5 h | 2.1 ± 0.2 | 3.8 ± 0.7 |
| Lung | 9.0 ± 0.6 a,c | 6.8 ± 1.0 f | 6 ± 1 | 4.3 ± 0.6 j | 8.4 ± 0.9 |
| Liver | 6.2 ± 0.3 a | 19 ± 1.5 e,f,g,k | 6.6 ± 0.6 | 9.1 ± 0.6 | 5.6 ± 0.3 |
| Spleen | 4.4 ± 0.8 a | 14.0 ± 0.3 e,f,g,k | 5.7 ± 0.7 | 8 ± 2 | 5 ± 1 |
| Stomach | 2.5 ± 0.4 k | 2.5 ± 0.2 f | 1.6 ± 0.3 | 1.5 ± 0.2 j | 2.24 ± 0.09 |
| Small intestine | 4.3 ± 0.7 | 9 ± 3 | 3.5 ± 0.9 | 4 ± 1 | 4.2 ± 0.7 |
| Kidney | 9 ± 1 a,b,c,d | 12.0 ± 0.6 f,g,k | 21 ± 2 h | 41 ± 4 | 44 ± 8 |
| Tumour | 8 ± 2 a | 13 ± 2 e,f | 7 ± 1 | 7 ± 1 | 8 ± 1 |
| Muscle | 1.6 ± 0.2 c,k | 1.08 ± 0.09 | 1.0 ± 0.2 | 0.72 ± 0.07 k | 1.3 ± 0.1 |
| Bone | 2.0 ± 0.2 | 4 ± 1 | 1.8 ± 0.4 | 1.68 ± 0.07 | 2.4 ± 0.5 |
| GI tract * | 5.8 ± 0.7 a | 8.7 ± 0.8 f,g | 4 ± 1 | 3.97 ± 0.07 k | 4.1 ± 0.2 |
| Carcass * | 40 ± 3 c | 36 ± 3 f | 32 ± 2 h | 20.8 ± 0.1 j,k | 33.9 ± 0.3 |
| 24 h | |||||
| Blood | 10 ± 2 a,c,l,m | 1.0 ± 0.2 e,g,l,m | 6.7 ± 0.8 h,l,m | 2.0 ± 0.3 j,l,m | 6.3 ± 0.6 l,m |
| Salivary gland | 4.4 ± 0.7 | 2.5 ± 0.5 | 3.3 ± 0.2 h | 2.1 ± 0.3 j | 3.2 ± 0.2 |
| Lung | 5.8 ± 0.7 c,l,m | 2.7 ± 0.8 l,m | 4.5 ± 0.8 | 1.9 ± 0.3 l,m | 3.6 ± 0.7 l,m |
| Liver | 5.9 ± 0.6 a,c | 19 ± 2 e,f,g | 8 ± 1 | 10.1 ± 0.3 j | 5.3 ± 0.6 |
| Spleen | 5.0 ± 0.8 | 15 ± 5 | 6.6 ± 0.8 | 6.7 ± 0.8 | 3.4 ± 0.7 |
| Stomach | 2.3 ± 0.3 c,m | 1.7 ± 0.7 | 1.7 ± 0.2 | 1.2 ± 0.1 | 1.7 ± 0.3 |
| Small intestine | 6 ± 1 | 7 ± 3 | 4.4 ± 0.3 | 3.3 ± 0.2 | 4.1 ± 0.6 |
| Kidney | 7.5 ± 0.7 b,c,d | 9 ± 2 f,g | 19 ± 1 h,i | 32.7 ± 0.6 l,m | 31 ± 3 |
| Tumour | 9 ± 2 m | 12 ± 2 f,m | 9 ± 1 h,i | 6.5 ± 0.9 | 5.9 ± 0.4 |
| Muscle | 1.2 ± 0.2 l | 0.6 ± 0.1 | 1.0 ± 0.1 | 0.6 ± 0.1 | 0.9 ± 0.2 |
| Bone | 2.3 ± 0.4 | 3 ± 1 | 1.7 ± 0.6 | 1.7 ± 0.3 l | 1.5 ± 0.3 |
| GI tract * | 5.8 ± 0.8 c,m | 6 ± 1 | 4.1 ± 0.4 | 3.2 ± 0.3 m | 5.2 ± 0.4 |
| Carcass * | 36 ± 3 c | 27 ± 3 l | 29 ± 3 | 16 ± 1 l | 25 ± 2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altai, M.; Leitao, C.D.; Rinne, S.S.; Vorobyeva, A.; Atterby, C.; Ståhl, S.; Tolmachev, V.; Löfblom, J.; Orlova, A. Influence of Molecular Design on the Targeting Properties of ABD-Fused Mono- and Bi-Valent Anti-HER3 Affibody Therapeutic Constructs. Cells 2018, 7, 164. https://doi.org/10.3390/cells7100164
Altai M, Leitao CD, Rinne SS, Vorobyeva A, Atterby C, Ståhl S, Tolmachev V, Löfblom J, Orlova A. Influence of Molecular Design on the Targeting Properties of ABD-Fused Mono- and Bi-Valent Anti-HER3 Affibody Therapeutic Constructs. Cells. 2018; 7(10):164. https://doi.org/10.3390/cells7100164
Chicago/Turabian StyleAltai, Mohamed, Charles Dahlsson Leitao, Sara S. Rinne, Anzhelika Vorobyeva, Christina Atterby, Stefan Ståhl, Vladimir Tolmachev, John Löfblom, and Anna Orlova. 2018. "Influence of Molecular Design on the Targeting Properties of ABD-Fused Mono- and Bi-Valent Anti-HER3 Affibody Therapeutic Constructs" Cells 7, no. 10: 164. https://doi.org/10.3390/cells7100164
APA StyleAltai, M., Leitao, C. D., Rinne, S. S., Vorobyeva, A., Atterby, C., Ståhl, S., Tolmachev, V., Löfblom, J., & Orlova, A. (2018). Influence of Molecular Design on the Targeting Properties of ABD-Fused Mono- and Bi-Valent Anti-HER3 Affibody Therapeutic Constructs. Cells, 7(10), 164. https://doi.org/10.3390/cells7100164