
Lysine Demethylase 1 Has Demethylase-Dependent and Non-Canonical Functions in Myofibroblast Activation in Systemic Sclerosis

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cell Lines
2.2. Lentiviral Transduction
2.3. Small-Molecule Inhibitors
2.4. Treatment of Fibroblasts with Pro-Fibrotic Factors
2.5. siRNA Transfections
2.6. Western Blotting
2.7. Quantitative Real-Time PCR
2.8. Immunofluorescence
2.9. Statistical Analysis
3. Results
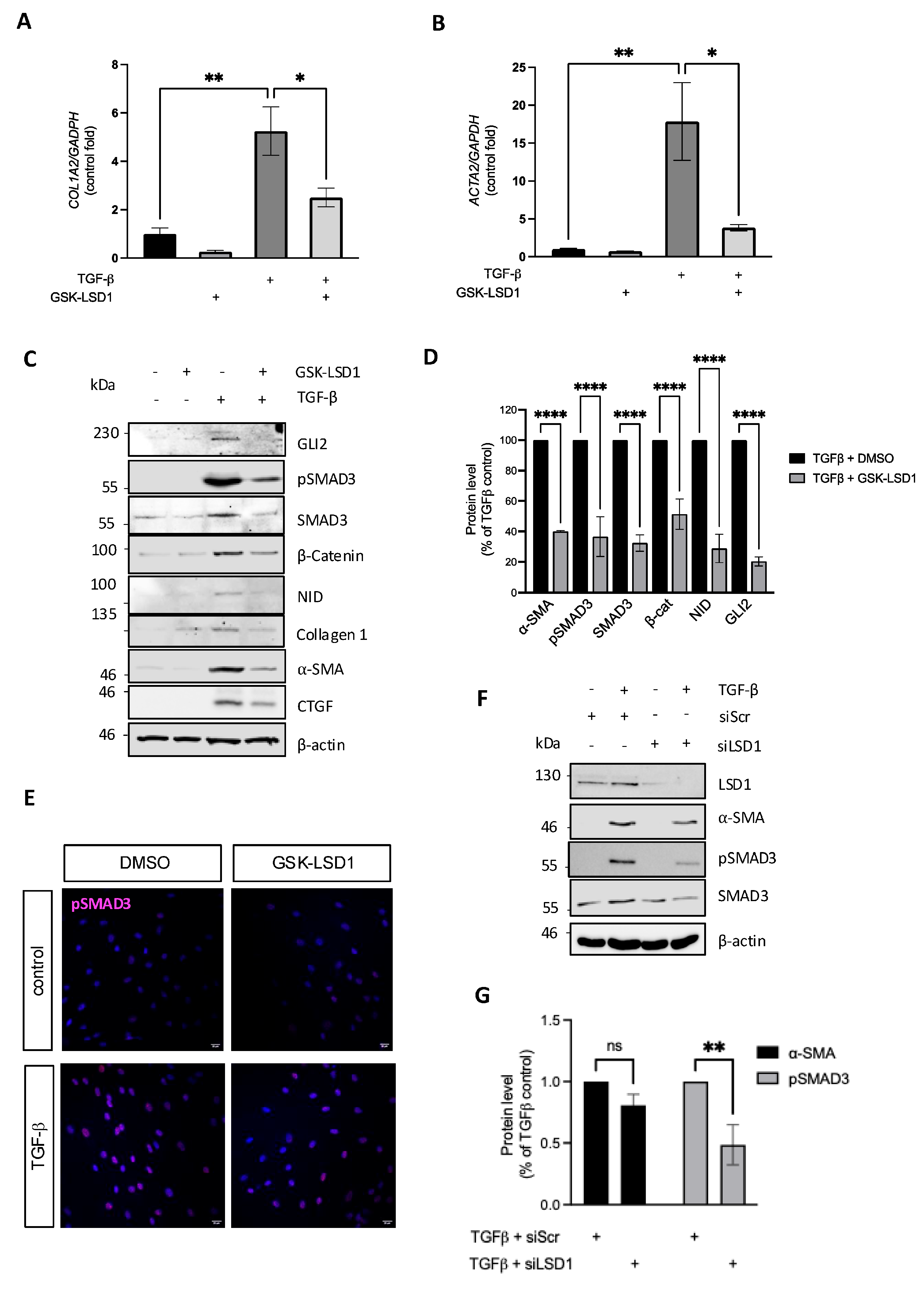
3.1. LSD1 Activity Is Required for Maximal TGF-β-Induced Dermal Fibroblast Activation
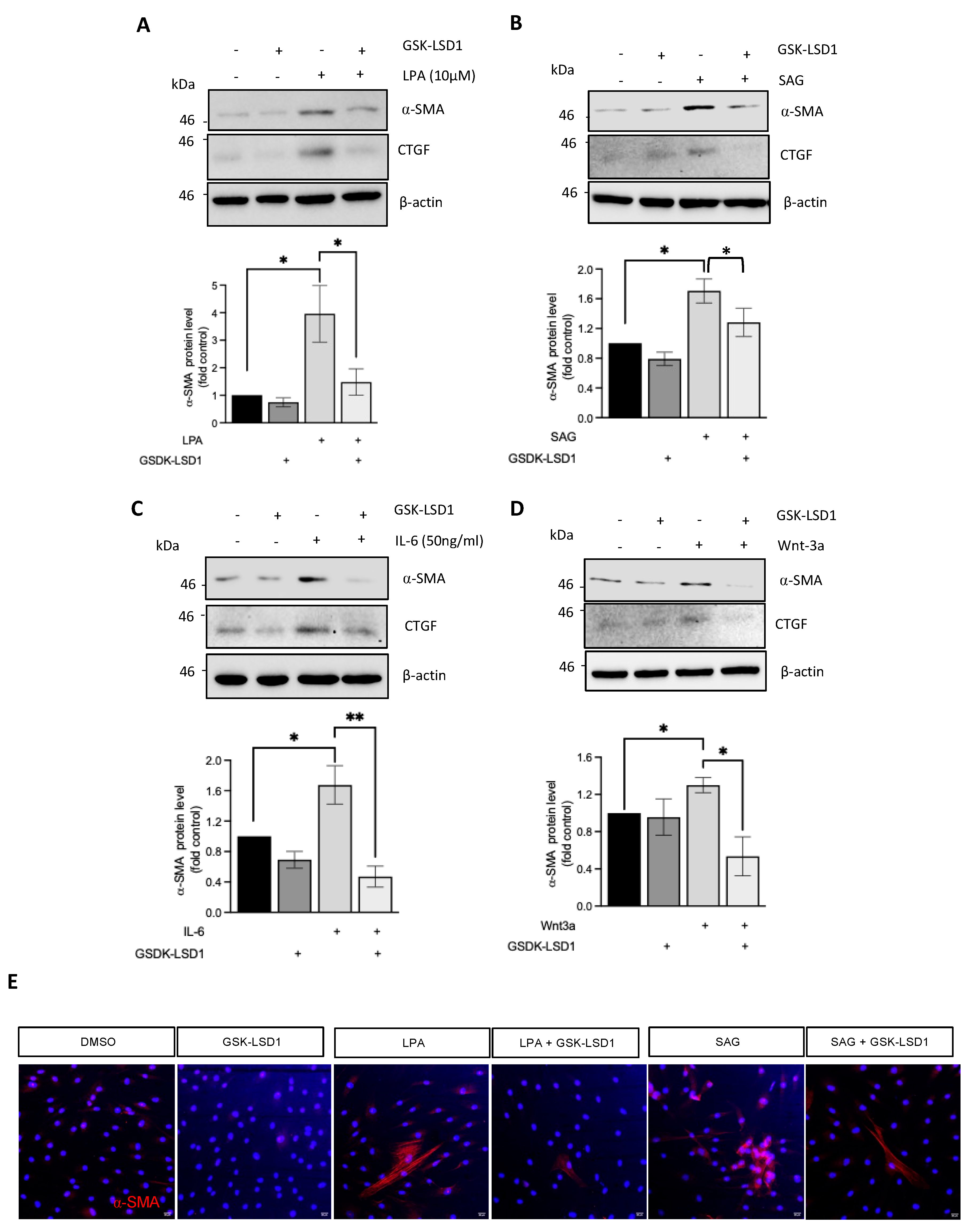
3.2. Inhibition of LSD1 Demethylase Activity Attenuates Fibroblast Activation in Response to Multiple Fibrotic Stimuli
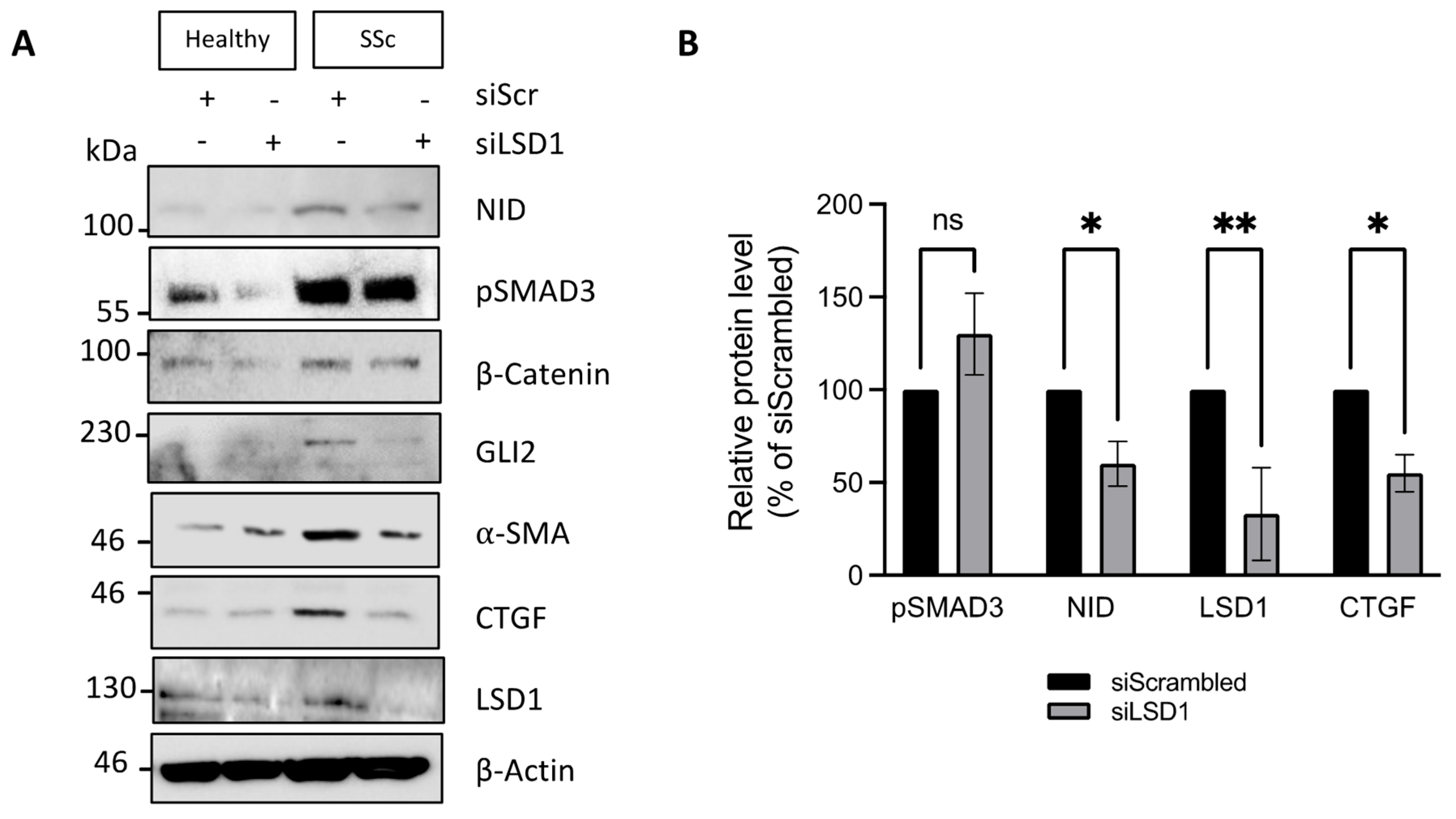
3.3. SSc Dermal Fibroblasts Require LSD1 Activity for Maximal Response to TGF-β but Not for Maintenance of the Myofibroblastic Phenotype
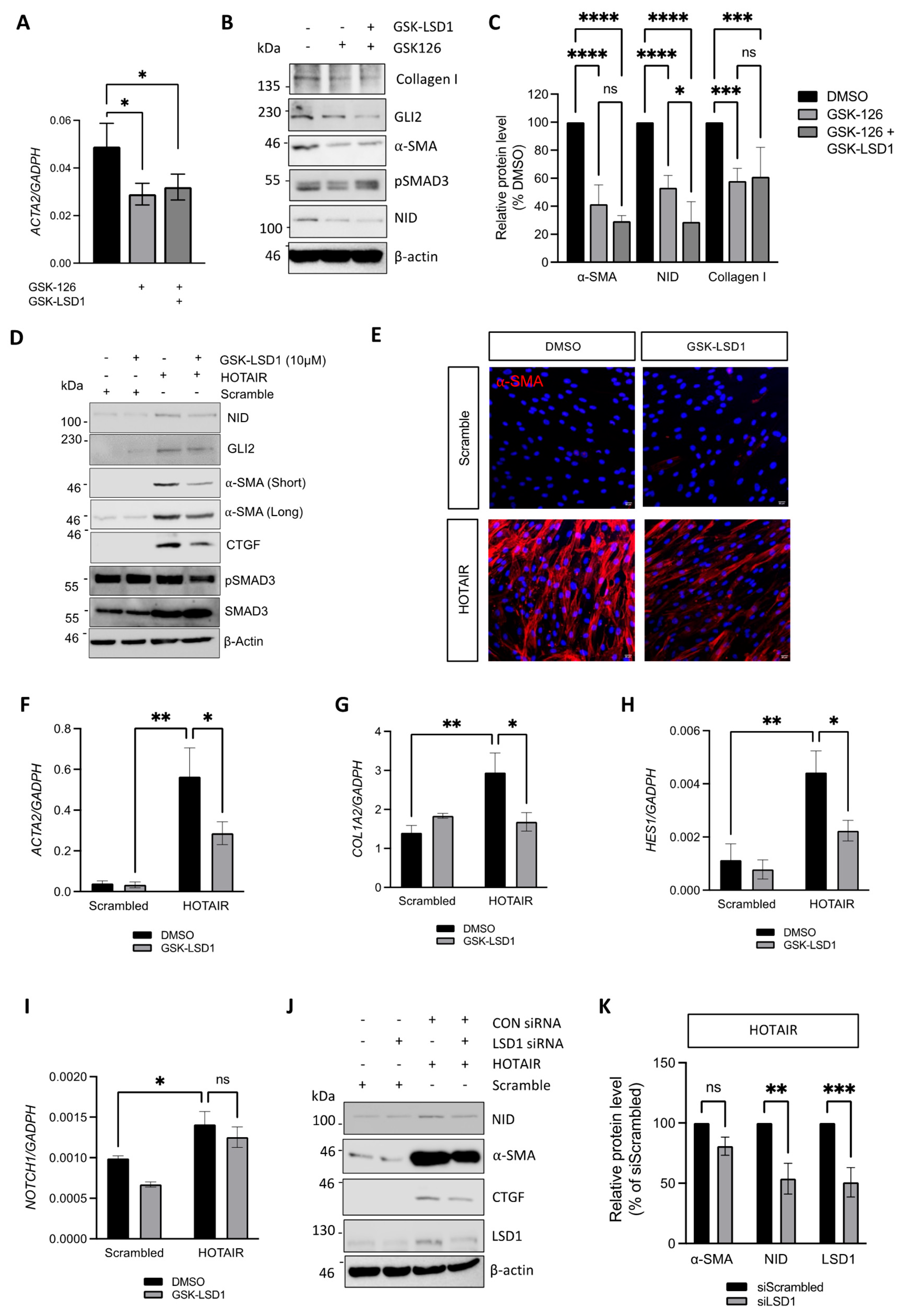
3.4. LSD1 Supports the SSc Myofibroblast Phenotype Through Non-Canonical Functions
3.5. Inhibition of LSD1 Activity Attenuates Pro-Fibrotic Gene Expression in HOTAIR Expressing Fibroblasts
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Varga, J.; Abraham, D. Systemic sclerosis: A prototypic multisystem fibrotic disorder. J. Clin. Investig. 2007, 117, 557–567. [Google Scholar] [CrossRef]
- Younesi, F.S.; Miller, A.E.; Barker, T.H.; Rossi, F.M.V.; Hinz, B. Fibroblast and myofibroblast activation in normal tissue repair and fibrosis. Nat. Rev. Mol. Cell Biol. 2024, 25, 617–638. [Google Scholar] [CrossRef]
- Beyer, C.; Dees, C.; Distler, J.H. Morphogen pathways as molecular targets for the treatment of fibrosis in systemic sclerosis. Arch. Dermatol. Res. 2013, 305, 1–8. [Google Scholar] [CrossRef]
- Hu, B.; Phan, S.H. Myofibroblasts. Curr. Opin. Rheumatol. 2013, 25, 71–77. [Google Scholar] [CrossRef]
- Wasson, C.W.; De Lorenzis, E.; Clavane, E.M.; Ross, R.L.; Walker, K.A.; Caballero-Ruiz, B.; Antinozzi, C.; Wells, R.; Migneco, G.; Brown, J.M.Y.; et al. The β-secretase BACE1 drives fibroblast activation in systemic sclerosis through the APP/β-catenin/Notch signaling axis. J. Investig. Dermatol. 2024, 144, 2197–2210. [Google Scholar] [CrossRef]
- Wasson, C.W.; Caballero-Ruiz, B.; Gillespie, J.; Derrett-Smith, E.; Mankouri, J.; Denton, C.P.; Canettieri, G.; Riobo-Del Galdo, N.A.; Del Galdo, F. Induction of pro-fibrotic CLIC4 in dermal fibroblasts by TGF-β/Wnt3a is mediated by GLI2 upregulation. Cells 2022, 11, 53. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Wasson, C.W.; Abignano, G.; Hermes, H.; Malaab, M.; Ross, R.L.; Jimenez, S.A.; Chang, H.Y.; Feghali-Bostwick, C.A.; Del Galdo, F. Long non-coding RNA HOTAIR drives EZH2-dependent myofibroblast activation in systemic sclerosis through miRNA 34a-dependent activation of NOTCH. Ann. Rheum. Dis. 2020, 79, 507–517. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-caoding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Wasson, C.W.; Ross, R.L.; Wells, R.; Corinaldesi, C.; Georgiou ICh Riobo-Del Galdo, N.; Del Galdo, F. Long non-coding RNA HOTAIR induces GLI2 expression through Notch signalling in systemic sclerosis dermal fibroblasts. Arthritis Res. Ther. 2020, 22, 286. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.S.; Campbell, P.; Amin, M.A.; Coit, P.; Miller, S.; Fox, D.A.; Khanna, D.; Sawalha, A.H. Inhibition of EZH2 prevents fibrosis and restores normal angiogenesis in scleroderma. Proc. Natl. Acad. Sci. USA 2019, 116, 3695–3702. [Google Scholar] [CrossRef]
- Somarowthu, S.; Legiewicz, M.; Chillon, I.; Marcia, M.; Liu, F.; Pyle, A.M. HOTAIR forms an intricate and modular secondary structure. Mol. Cell 2015, 58, 353–361. [Google Scholar] [CrossRef]
- Wang, H.; Helin, K. Roles of H3K4 methylation in biology and disease. Trends Cell Biol. 2025, 35, 115–128. [Google Scholar] [CrossRef]
- Song, Y.; Dagil, L.; Fairall, L.; Robertson, N.; Wu, M.; Ragan, T.J.; Savva, C.G.; Saleh, A.; Morone, N.; Kunze, M.B.A.; et al. Mechanism of Crosstalk between the LSD1 Demethylase and HDAC1 Deacetylase in the CoREST Complex. Cell Rep. 2020, 30, 2699–2711.e8. [Google Scholar] [CrossRef]
- Zhang, H.; Xing, J.; Zhao, L. Lysine-specific demethylase 1 induced epithelial-mesenchymal transition and promoted renal fibrosis through Jagged-1/Notch signaling pathway. Hum. Exp. Toxicol. 2021, 40 (Suppl. 12), S203–S214. [Google Scholar] [CrossRef]
- Jarroux, J.; Foretek, D.; Bertrand, C.; Gabriel, M.; Szachnowski, U.; Saci, Z.; Guo, S.; Londoño-Vallejo, A.; Pinskaya, M.; Morillon, A. HOTAIR lncRNA promotes epithelial-mesenchymal transition by redistributing LSD1 at regulatory chromatin regions. EMBO Rep. 2021, 22, e50193. [Google Scholar] [CrossRef]
- LeRoy, E.C.; Medsger, T.A.J.r. Criteria for the classification of early systemic sclerosis. J. Rheumatol. 2001, 28, 1573–1576. [Google Scholar]
- Gillespie, J.; Ross, R.L.; Corinaldesi, C.; Esteves, F.; Derrett-Smith, E.; McDermott, M.F.; Doody, G.M.; Denton, C.P.; Emery, P.; Del Galdo, F. Transforming growth factor β activation primes canonical Wnt signalling through down-regulation of axin-2. Arthritis Rheumatol. 2018, 70, 932–942. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.X.; Yu, C.; Nath, K.A.; Zhuang, S.; Li, X. Targeting lysine-specific demethylase 1A inhibits renal epithelial-mesenchymal transition and attenuates renal fibrosis. FASEB J. 2022, 36, e22122. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Yu, L.; Zhong, J. Histone lysine-specific demethylase 1 induced renal fibrosis via decreasing sirtuin 3 expression and activating TGF-B1/Smad3 pathway in diabetic nepropathy. Diabetol. Metab. Syndr. 2022, 14, 2. [Google Scholar] [CrossRef]
- Pan, X.; Li, J.; Tu, X.; Wu, C.; Liu, H.; Luo, Y.; Dong, X.; Li, X.; Pan, L.-L.; Sun, J. Lysine-specific demetylase-1 regulates fibroblast activation in pulmonary fibrosis via TGF-B/Smad3 pathway. Pharmacol. Res. 2020, 152, 104592. [Google Scholar] [CrossRef]
- Huo, J.L.; Jiao, L.; An, Q.; Chen, X.; Qi, Y.; Wei, B.; Zheng, Y.; Shi, X.; Gao, E.; Liu, H.M.; et al. Myofibroblast Deficiency of LSD1 Alleviates TAC-Induced Heart Failure. Circ. Res. 2021, 129, 400–413. [Google Scholar] [CrossRef]
- Zeng, C.; Chen, J.; Cooke, E.W.; Subuddhi, A.; Roodman, E.T.; Chen, F.X.; Cao, K. Demethylase-independent roles of LSD1 in regulating enhancers and cell fate transition. Nat. Commun. 2023, 14, 4944. [Google Scholar] [CrossRef]
- Malla, S.; Kumari, K.; García-Prieto, C.A.; Caroli, J.; Nordin, A.; Phan, T.T.T.; Bhattarai, D.P.; Martinez-Gamero, C.; Dorafshan, E.; Stransky, S.; et al. The scaffolding function of LSD1 controls DNA methylation in mouse ESCs. Nat. Commun. 2024, 15, 7758. [Google Scholar] [CrossRef]
- Casey, M.J.; Call, A.M.; Thorpe, A.V.; Jette, C.A.; Engel, M.E.; Stewart, R.A. The scaffolding function of LSD1/KDM1A reinforces a negative feedback loop to repress stem cell gene expression during primitive hematopoiesis. iScience 2022, 26, 105737. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef]
- Baron, R.; Binda, C.; Tortorici, M.; McCammon, J.A.; Mattevi, A. Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure 2011, 19, 212–220. [Google Scholar] [CrossRef]
- Pezone, A.; Taddei, M.L.; Tramontano, A.; Dolcini, J.; Boffo, F.L.; De Rosa, M.; Parri, M.; Stinziani, S.; Comito, G.; Porcellini, A.; et al. Targeted DNA oxidation by LSD1-SMAD2/3 primes TGF-beta1/ EMT genes for activation or repression. Nucleic Acids Res. 2020, 48, 8943–8958. [Google Scholar] [CrossRef]
- Carnesecchi, J.; Cerutti, C.; Vanacker, J.M.; Forcet, C. 2017 ERRα protein is stabilized by LSD1 in a demethylation-independent manner. PLoS ONE 2017, 12, e0188871. [Google Scholar] [CrossRef]
- Hong, Y.; Li, X.; Zhu, J. LSD1-mediated stabilization of SEPT6 protein activates the TGF-β1 pathway and regulates non-small-cell lung cancer metastasis. Cancer Gene Ther. 2022, 29, 189–201. [Google Scholar] [CrossRef]
- Tinazzi, I.; Mulipa, P.; Colato, C.; Abignano, G.; Ballarin, A.; Biasi, D.; Emery, P.; Ross, R.L.; Del Galdo, F. SFRP4 expression is linked to immune-driven fibrotic conditions, correlates with skin and lung fibrosis in SSc and a potential EMT biomarker. J. Clin. Med. 2021, 10, 5820. [Google Scholar] [CrossRef]
- Hou, G.; Zhao, Q.; Zhang, M.; Wang, P.; Ye, H.; Wang, Y.; Ren, Y.; Zhang, J.; Lu, Z. LSD1 regulates Notch and PI3K/Akt/mTOR pathways through binding the promoter regions of Notch target genes in esophageal squamous cell carcinoma. OncoTargets Ther. 2019, 12, 5215–5225. [Google Scholar] [CrossRef]
- Augert, A.; Eastwood, E.; Ibrahim, A.H.; Wu, N.; Grunblatt, E.; Basom, R.; Liggitt, D.; Eaton, K.D.; Martins, R.; Poirier, J.T.; et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci. Signal. 2019, 12, eaau2922. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wasson, C.W.; Perez Barreiro, E.; Del Galdo, F.; Riobo-Del Galdo, N.A. Lysine Demethylase 1 Has Demethylase-Dependent and Non-Canonical Functions in Myofibroblast Activation in Systemic Sclerosis. Cells 2025, 14, 433. https://doi.org/10.3390/cells14060433
Wasson CW, Perez Barreiro E, Del Galdo F, Riobo-Del Galdo NA. Lysine Demethylase 1 Has Demethylase-Dependent and Non-Canonical Functions in Myofibroblast Activation in Systemic Sclerosis. Cells. 2025; 14(6):433. https://doi.org/10.3390/cells14060433
Chicago/Turabian StyleWasson, Christopher W., Esther Perez Barreiro, Francesco Del Galdo, and Natalia A. Riobo-Del Galdo. 2025. "Lysine Demethylase 1 Has Demethylase-Dependent and Non-Canonical Functions in Myofibroblast Activation in Systemic Sclerosis" Cells 14, no. 6: 433. https://doi.org/10.3390/cells14060433
APA StyleWasson, C. W., Perez Barreiro, E., Del Galdo, F., & Riobo-Del Galdo, N. A. (2025). Lysine Demethylase 1 Has Demethylase-Dependent and Non-Canonical Functions in Myofibroblast Activation in Systemic Sclerosis. Cells, 14(6), 433. https://doi.org/10.3390/cells14060433