

Update of Aging Hallmarks in Idiopathic Pulmonary Fibrosis

 ,
,  , , , ,
, , , ,  and
and 
Abstract
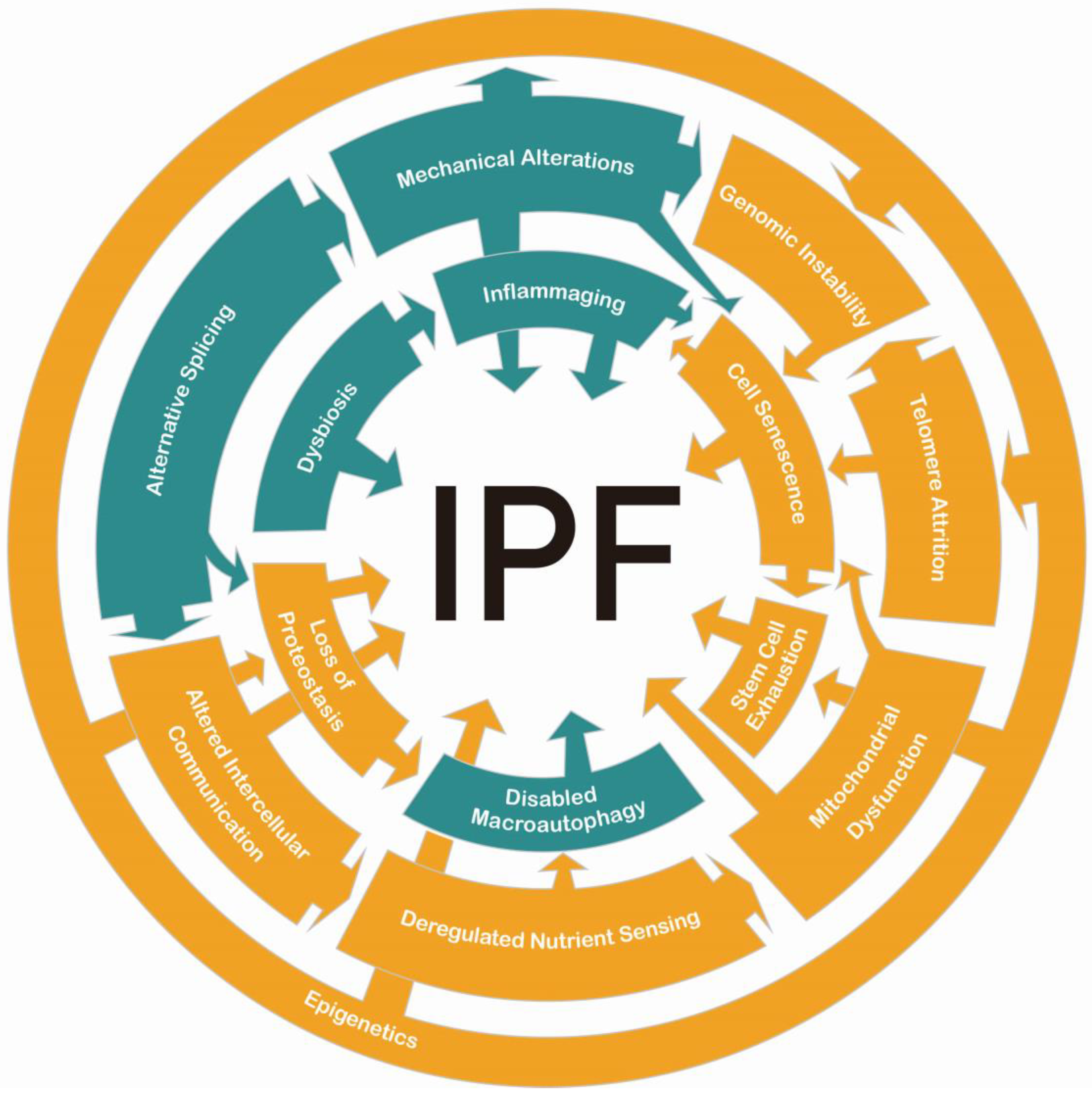
1. Introduction
2. Complementary Hallmarks
2.1. Mechanical Alterations
2.2. Alternative Splicing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Isoforms | Ensemble Reference | Function | In IPF vs. Controls | Domain Lost | Domain Gained | Additional Information | Ref. |
|---|---|---|---|---|---|---|---|---|
| AGER | Advanced Glycosylation End-Product Receptor | ENST00000375076.9 | Immuno-globulin superfamily cell-surface receptor | [40] | ||||
| Endogenous secretory (es) | No | Secreted, non-signaling receptor | Down | Trans-membrane | Considered to have an anti-inflammatory function. | [29] | ||
| POSTN | Periostin, osteoblast-specific factor | ENST00000379747.9 | ECM protein involved in tissue development and regeneration | [41] | ||||
| Shorter variant | No | Proposed reduced ECM protein binding and altered cell invasiveness | Down | C-ter portion | Exon 21 is more likely to be spliced out in IPF. | [28] | ||
| COL6A3 | Collagen Type VI Alpha 3 Chain, α | ENST00000295550.9 | Cell adhesion and integrin signaling | Exon 4 inclusion was previously associated with colon and pancreatic cancer. | [42,43] | |||
| Shorter variant | ENST00000347401.8 | Down | Exon 4 is more likely to be spliced out in IPF. | [28] | ||||
| TOM1L1 | Target Of Myb1-Like 1 Membrane Trafficking Protein | ENST00000445275 | Probable adapter protein, clathrin, and kinase binding activity | Down | Exon 6, γ | [27,44] | ||
| Shorter variant | ENST00000348161 | Upregulated | Exon 6 | Exon 6 skipping is more likely in IPF. | ||||
| CMTM4 | CKLF-Like MARVEL Transmembrane Domain Containing 4 | ENST00000330687 | Chemokine-like factor, cell growth, and cell cycle regulation | Upregulated | C-ter region | Alternative 3′ splice sites generate two protein isoforms with 26aac difference. | [27,45] | |
| Shorter variant | ENST00000394106 | Believed to contribute to stress-induced senescence | Down | C-ter region | [27] | |||
| PEX11B | Peroxisomal Biogenesis Factor 11 Beta | ENST00000369306 | Peroxisome metabolic pathway and oxidative stress protection | Down | 5′ end, δ | [27,46] | ||
| Shorter variant | ENST00000428634 | May contribute to peroxisomal dysfunction, generating oxidative stress | Upregulated | 5′ end | Truncated 5′ end by alternative promoter. | [27] | ||
| SCL38A10 | Solute carrier family 38 member 10 | ENST00000374759 | Predicted involvement in amino acid transmembrane transport | Down | N-ter region | [27,39] | ||
| ENST00000374759_N | Novel transcript | Upregulated | Exon 2 | N-ter region | [27] | |||
| Truncated 5′ end | ENST00000288439 | Down | N-ter region | [27,39] | ||||
| ENST00000288439_N | Novel transcript | Upregulated | N-ter regionAnd exon 2 | [27] | ||||
| FN1 | Fibronectin I | Cell adhesion and migration | ||||||
| EDA (extra type III domain A) | ENST00000354785.11 | Important for fibroblast activation | Upregulated | Type III domain A | [30] |
2.3. Inflammaging
2.3.1. Innate Immunity and Fibrocytes
2.3.2. Adaptative Immunity
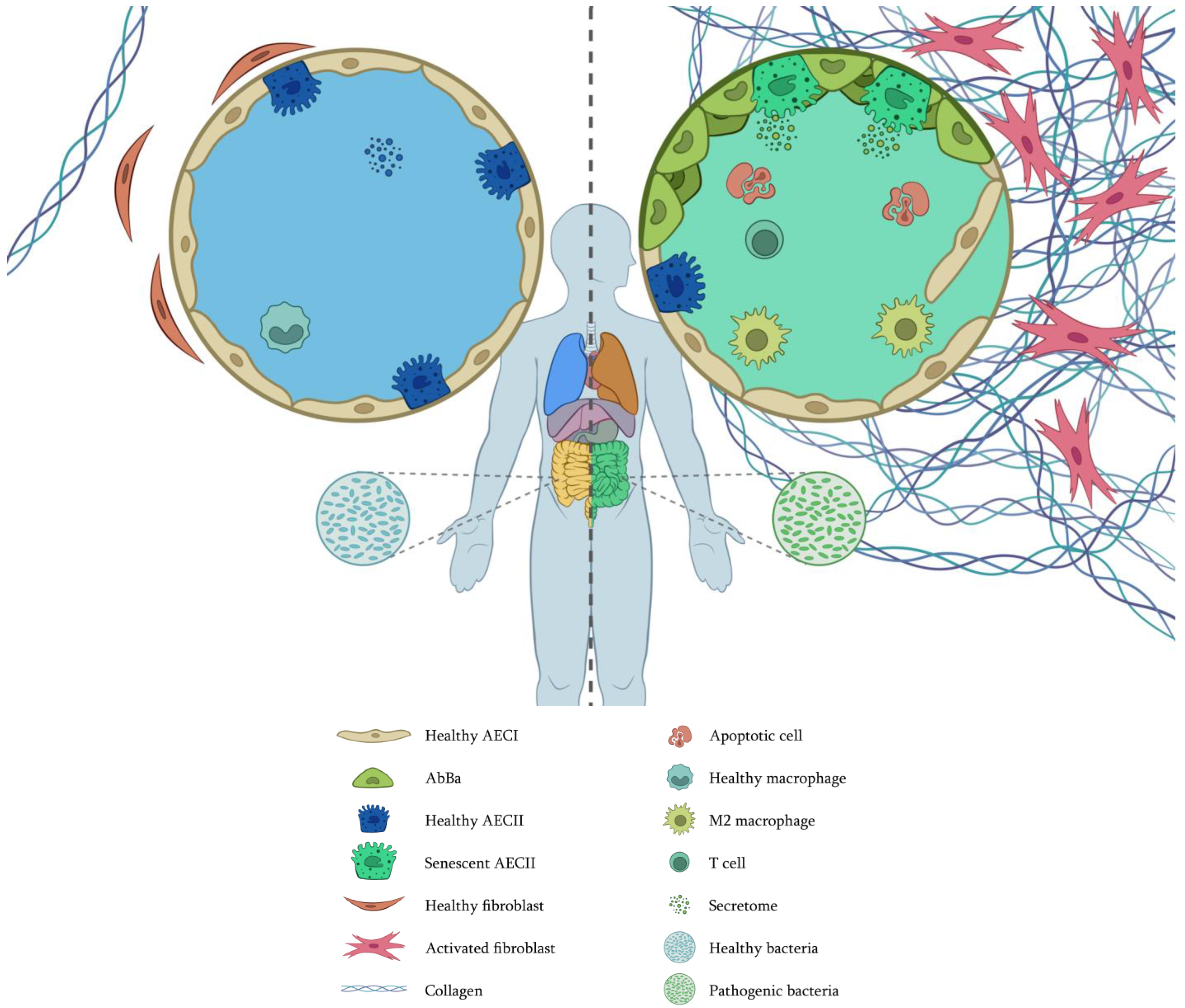
2.4. Dysbiosis
2.5. Disabled Macroautophagy
3. Classic Hallmarks
3.1. Loss of Proteostasis
3.1.1. Decline in Protein Quality Control Systems
3.1.2. Unfolded Protein Response in the ER
3.2. Stem Cell Exhaustion
3.3. Altered Intercellular Communication
3.4. Epigenetic Alterations
3.5. Deregulated Nutrient Sensing
3.6. Cell Senescence
3.7. Other Hallmarks
4. Translational Research and Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Selman, M.; King, T.E., Jr.; Pardo, A.; American Thoracic Society; European Respiratory Society; American College of Chest Physicians. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Selman, M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 755–772. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Schmauck-Medina, T.; Molière, A.; Lautrup, S.; Zhang, J.; Chlopicki, S.; Madsen, H.B.; Cao, S.; Soendenbroe, C.; Mansell, E.; Vestergaard, M.B.; et al. New hallmarks of ageing: A 2022 Copenhagen ageing meeting summary. Aging 2022, 14, 6829–6839. [Google Scholar] [CrossRef]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikström, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Investig. 2014, 124, 1622–1635. [Google Scholar] [CrossRef]
- Nizamoglu, M.; Alleblas, F.; Koster, T.; Borghuis, T.; Vonk, J.M.; Thomas, M.J.; White, E.S.; Watson, C.K.; Timens, W.; El Kasmi, K.C.; et al. Three dimensional fibrotic extracellular matrix directs microenvironment fiber remodeling by fibroblasts. Acta Biomater. 2024, 177, 118–131. [Google Scholar] [CrossRef]
- Hewitt, R.J.; Puttur, F.; Gaboriau, D.C.A.; Fercoq, F.; Fresquet, M.; Traves, W.J.; Yates, L.L.; Walker, S.A.; Molyneaux, P.L.; Kemp, S.V.; et al. Lung extracellular matrix modulates KRT5+ basal cell activity in pulmonary fibrosis. Nat. Commun. 2023, 14, 6039. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Fibroageing: An ageing pathological feature driven by dysregulated extracellular matrix-cell mechanobiology. Ageing Res. Rev. 2021, 70, 101393. [Google Scholar] [CrossRef]
- Mebratu, Y.A.; Soni, S.; Rosas, L.; Rojas, M.; Horowitz, J.C.; Nho, R. The aged extracellular matrix and the profibrotic role of senescence-associated secretory phenotype. Am. J. Physiol. Physiol. 2023, 325, C565–C579. [Google Scholar] [CrossRef] [PubMed]
- Ngassie, M.L.K.; De Vries, M.; Borghuis, T.; Timens, W.; Sin, D.D.; Nickle, D.; Joubert, P.; Horvatovich, P.; Marko-Varga, G.; Teske, J.J.; et al. Age-associated differences in the human lung extracellular matrix. Am. J. Physiol. Lung Cell Mol. Physiol. 2023, 324, L799–L814. [Google Scholar] [CrossRef] [PubMed]
- Martín-Vicente, P.; López-Martínez, C.; López-Alonso, I.; Exojo-Ramírez, S.M.; Duarte-Herrera, I.D.; Amado-Rodríguez, L.; Ordoñez, I.; Cuesta-Llavona, E.; Gómez, J.; Campo, N.; et al. Mechanical Stretch Induces Senescence of Lung Epithelial Cells and Drives Fibroblast Activation by Paracrine Mechanisms. Am. J. Respir. Cell Mol. Biol. 2024, 72, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef]
- Decaris, M.L.; Schaub, J.R.; Chen, C.; Cha, J.; Lee, G.G.; Rexhepaj, M.; Ho, S.S.; Rao, V.; Marlow, M.M.; Kotak, P.; et al. Dual inhibition of αvβ6 and αvβ1 reduces fibrogenesis in lung tissue explants from patients with IPF. Respir. Res. 2021, 22, 265. [Google Scholar] [CrossRef]
- Lancaster, L.; Cottin, V.; Ramaswamy, M.; Wuyts, W.A.; Jenkins, R.G.; Scholand, M.B.; Kreuter, M.; Valenzuela, C.; Ryerson, C.J.; Goldin, J.; et al. Bexotegrast in Patients with Idiopathic Pulmonary Fibrosis: The INTEGRIS-IPF Clinical Trial. Am. J. Respir. Crit. Care Med. 2024, 210, 424–434. [Google Scholar] [CrossRef]
- Campbell, M.G.; Cormier, A.; Ito, S.; Seed, R.I.; Bondesson, A.J.; Lou, J.; Marks, J.D.; Baron, J.L.; Cheng, Y.; Nishimura, S.L. Cryo-EM Reveals Integrin-Mediated TGF-β Activation without Release from Latent TGF-β. Cell 2020, 180, 490–501.e16. [Google Scholar] [CrossRef]
- Phogat, S.; Thiam, F.; Al Yazeedi, S.; Abokor, F.A.; Osei, E.T. 3D in vitro hydrogel models to study the human lung extracellular matrix and fibroblast function. Respir. Res. 2023, 24, 242. [Google Scholar] [CrossRef]
- Duarte, A.C.; Costa, E.C.; Filipe, H.A.; Saraiva, S.M.; Jacinto, T.; Miguel, S.P.; Ribeiro, M.P.; Coutinho, P. Animal-derived products in science and current alternatives. Mater. Sci. Eng. C 2023, 151, 213428. [Google Scholar] [CrossRef]
- Kaemmerer, E.; Melchels, F.P.; Holzapfel, B.M.; Meckel, T.; Hutmacher, D.W.; Loessner, D. Gelatine methacrylamide-based hydrogels: An alternative three-dimensional cancer cell culture system. Acta Biomater. 2014, 10, 2551–2562. [Google Scholar] [CrossRef]
- Caliari, S.R.; Burdick, J.A. A practical guide to hydrogels for cell culture. Nat. Methods 2016, 13, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, N.; Orzechowska, B.; Awsiuk, K.; Rysz, J.; Tymetska, S.; Raczkowska, J. Cell-Specific Response of NSIP- and IPF-Derived Fibroblasts to the Modification of the Elasticity, Biological Properties, and 3D Architecture of the Substrate. Int. J. Mol. Sci. 2022, 23, 14714. [Google Scholar] [CrossRef] [PubMed]
- Machahua, C.; Vicens-Zygmunt, V.; Ríos-Martín, J.; Llatjós, R.; Escobar-Campuzano, I.; Molina-Molina, M.; Montes-Worboys, A. Collagen 3D matrices as a model for the study of cell behavior in pulmonary fibrosis. Exp. Lung Res. 2022, 48, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global Methylation Patterns in Idiopathic Pulmonary Fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef]
- Li, X.; Manley, J.L. New Talents for an Old Acquaintance: The SR Protein Splicing Factor ASF/SF2 Functions in the Maintenance of Genome Stability. Cell Cycle 2005, 4, 1706–1708. [Google Scholar] [CrossRef]
- Chen, K.-J.; Li, Q.; Weng, C.-M.; Duan, Z.-X.; Zhang, D.-D.; Chen, Z.-Q.; Chen, J.; Wang, J.-M. Bleomycin-enhanced alternative splicing of fibroblast growth factor receptor 2 induces epithelial to mesenchymal transition in lung fibrosis. Biosci. Rep. 2018, 38, BSR20180445. [Google Scholar] [CrossRef]
- Deng, N.; Sanchez, C.G.; Lasky, J.A.; Zhu, D. Detecting Splicing Variants in Idiopathic Pulmonary Fibrosis from Non-Differentially Expressed Genes. PLoS ONE 2013, 8, e68352. [Google Scholar] [CrossRef]
- Nance, T.; Smith, K.S.; Anaya, V.; Richardson, R.; Ho, L.; Pala, M.; Mostafavi, S.; Battle, A.; Feghali-Bostwick, C.; Rosen, G.; et al. Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS ONE 2014, 9, e97550. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Iwamoto, H.; Mazur, W.; Miura, S.; Sakamoto, S.; Horimasu, Y.; Masuda, T.; Miyamoto, S.; Nakashima, T.; Ohshimo, S.; et al. Reduced endogenous secretory RAGE in blood and bronchoalveolar lavage fluid is associated with poor prognosis in idiopathic pulmonary fibrosis. Respir. Res. 2020, 21, 145. [Google Scholar] [CrossRef]
- Muro, A.F.; Moretti, F.A.; Moore, B.B.; Yan, M.; Atrasz, R.G.; Wilke, C.A.; Flaherty, K.R.; Martinez, F.J.; Tsui, J.L.; Sheppard, D.; et al. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 638–645. [Google Scholar] [CrossRef]
- Kasper, M.; Günthert, U.; Dall, P.; Kayser, K.; Schuh, D.; Haroske, G.; Müller, M. Distinct expression patterns of CD44 isoforms during human lung development and in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 1995, 13, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, X.; Hung, C.; Bahudhanapati, H.; Tan, J.; Kass, D.J.; Zhang, Y. The relaxin family peptide receptor 1 (RXFP1): An emerging player in human health and disease. Mol. Genet. Genom. Med. 2020, 8, e1194. [Google Scholar] [CrossRef] [PubMed]
- Nollet, M.; Bachelier, R.; Joshkon, A.; Traboulsi, W.; Mahieux, A.; Moyon, A.; Muller, A.; Somasundaram, I.; Simoncini, S.; Peiretti, F.; et al. Involvement of Multiple Variants of Soluble CD146 in Systemic Sclerosis: Identification of a Novel Profibrotic Factor. Arthritis Rheumatol. 2022, 74, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.J.; Guillen-Guio, B.; Croot, E.; Kraven, L.M.; Moss, S.; Stewart, I.; Jenkins, R.G.; Wain, L.V. Genetic overlap between idiopathic pulmonary fibrosis and COVID-19. Eur. Respir. J. 2022, 60, 2103132. [Google Scholar] [CrossRef]
- Nakanishi, T.; Willett, J.; Farjoun, Y.; Allen, R.J.; Guillen-Guio, B.; Adra, D.; Zhou, S.; Richards, J.B. Alternative splicing in lung influences COVID-19 severity and respiratory diseases. Nat. Commun. 2023, 14, 6198. [Google Scholar] [CrossRef]
- Brinkman, B.M. Splice variants as cancer biomarkers. Clin. Biochem. 2004, 37, 584–594. [Google Scholar] [CrossRef]
- Giblin, S.P.; Midwood, K.S. Tenascin-C: Form versus function. Cell Adhes. Migr. 2014, 9, 48–82. [Google Scholar] [CrossRef]
- Torres-Machorro, A.L.; Becerril, C.; Hernández-Plata, E.; Luis-García, E.R.; Maldonado, M.; Herrera, I.; Negreros, M.; Hernández-Sánchez, F.; Mendoza-Milla, C.; Gaxiola, M.; et al. Altered expression pattern of immune response-related genes and isoforms in hypersensitivity pneumonitis lung fibroblasts. Sci. Rep. 2024, 14, 24002. [Google Scholar] [CrossRef]
- Harrison, P.W.; Amode, M.R.; Austine-Orimoloye, O.; Azov, A.G.; Barba, M.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2024. Nucleic Acids Res. 2024, 52, D891–D899. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.; Brett, J.; Yan, S.; Wang, F.; Pan, Y.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Coutu, D.L.; Wu, J.H.; Monette, A.; Rivard, G.; Blostein, M.D.; Galipeau, J. Periostin, a member of a novel family of vitamin K-dependent proteins, is expressed by mesenchymal stromal cells. J. Biol. Chem. 2008, 283, 17991–18001. [Google Scholar] [CrossRef] [PubMed]
- Hakelius, M.; Saiepour, D.; Göransson, H.; Rubin, K.; Gerdin, B.; Nowinski, D. Differential Gene Regulation in Fibroblasts in Co-culture with Keratinocytes and Head and Neck SCC Cells. Anticancer Res. 2015, 35, 3253–3265. [Google Scholar] [PubMed]
- Gardina, P.J.; A Clark, T.; Shimada, B.; Staples, M.K.; Yang, Q.; Veitch, J.; Schweitzer, A.; Awad, T.; Sugnet, C.; Dee, S.; et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genom. 2006, 7, 325. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R. Interactions of TOM1L1 with the Multivesicular Body Sorting Machinery. J. Biol. Chem. 2005, 280, 9258–9264. [Google Scholar] [CrossRef]
- Mezzadra, R.; Sun, C.; Jae, L.T.; Gomez-Eerland, R.; De Vries, E.; Wu, W.; Logtenberg, M.E.W.; Slagter, M.; Rozeman, E.A.; Hofland, I.; et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017, 549, 106–110. [Google Scholar] [CrossRef]
- Koch, J.; Pranjic, K.; Huber, A.; Ellinger, A.; Hartig, A.; Kragler, F.; Brocard, C. PEX11 family members are membrane elongation factors that coordinate peroxisome proliferation and maintenance. J. Cell Sci. 2010, 123, 3389–3400. [Google Scholar] [CrossRef]
- Saavedra, D.; Añé-Kourí, A.L.; Barzilai, N.; Caruso, C.; Cho, K.-H.; Fontana, L.; Franceschi, C.; Frasca, D.; Ledón, N.; Niedernhofer, L.J.; et al. Aging and chronic inflammation: Highlights from a multidisciplinary workshop. Immun. Ageing 2023, 20, 25. [Google Scholar] [CrossRef]
- Chilosi, M.; Carloni, A.; Rossi, A.; Poletti, V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl. Res. 2013, 162, 156–173. [Google Scholar] [CrossRef]
- Murray, M.A.; Chotirmall, S.H. The Impact of Immunosenescence on Pulmonary Disease. Mediat. Inflamm. 2015, 2015, 692546. [Google Scholar] [CrossRef]
- Bringardner, B.D.; Baran, C.P.; Eubank, T.D.; Marsh, C.B. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2008, 10, 287–302. [Google Scholar] [CrossRef]
- Chen, H.; Xia, Z.; Qing, B.; Wang, W.; Gu, L.; Chen, Y.; Wang, J.; Yuan, Y. Analysis of necroptosis-related prognostic genes and immune infiltration in idiopathic pulmonary fibrosis. Front. Immunol. 2023, 14, 1119139. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Choe, E.J.; Jung, S.Y.; Ko, J.; Kim, D.-W.; Lee, J.H. A study on the prevalence and prognosis of progressive pulmonary fibrosis: A retrospective observational study. Medicine 2024, 103, e38226. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Martinez, F.J.; Brown, K.K.; Costabel, U.; Cottin, V.; Wells, A.U.; Lancaster, L.; Gibson, K.F.; Haddad, T.; Agarwal, P.; et al. CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: A phase 2 trial of carlumab. Eur. Respir. J. 2015, 46, 1740–1750. [Google Scholar] [CrossRef] [PubMed]
- Fraser, E.; Denney, L.; Antanaviciute, A.; Blirando, K.; Vuppusetty, C.; Zheng, Y.; Repapi, E.; Iotchkova, V.; Taylor, S.; Ashley, N.; et al. Multi-Modal Characterization of Monocytes in Idiopathic Pulmonary Fibrosis Reveals a Primed Type I Interferon Immune Phenotype. Front. Immunol. 2021, 12, 623430. [Google Scholar] [CrossRef]
- Andersson-Sjöland, A.; de Alba, C.G.; Nihlberg, K.; Becerril, C.; Ramírez, R.; Pardo, A.; Westergren-Thorsson, G.; Selman, M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 2129–2140. [Google Scholar] [CrossRef]
- Heukels, P.; van Hulst, J.A.C.; van Nimwegen, M.; Boorsma, C.E.; Melgert, B.N.; Toorn, L.M.v.D.; Boomars, K.A.T.; Wijsenbeek, M.S.; Hoogsteden, H.; von der Thüsen, J.H.; et al. Fibrocytes are increased in lung and peripheral blood of patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 90. [Google Scholar] [CrossRef]
- de Alba, C.G.; Buendia-Roldán, I.; Salgado, A.; Becerril, C.; Ramírez, R.; González, Y.; Checa, M.; Navarro, C.; Ruiz, V.; Pardo, A.; et al. Fibrocytes contribute to inflammation and fibrosis in chronic hypersensitivity pneumonitis through paracrine effects. Am. J. Respir. Crit. Care Med. 2015, 191, 427–436. [Google Scholar] [CrossRef]
- Gomez-Manjarres, D.C.; Axell-House, D.B.; Patel, D.C.; Odackal, J.; Yu, V.; Burdick, M.D.; Mehrad, B. Sirolimus suppresses circulating fibrocytes in idiopathic pulmonary fibrosis in a randomized controlled crossover trial. J. Clin. Investig. 2023, 8, e166901. [Google Scholar] [CrossRef]
- Wang, Q.-R.; Liu, S.-S.; Min, J.-L.; Yin, M.; Zhang, Y.; Zhang, Y.; Tang, X.-N.; Li, X.; Liu, S.-S. CCL17 drives fibroblast activation in the progression of pulmonary fibrosis by enhancing the TGF-β/Smad signaling. Biochem. Pharmacol. 2023, 210, 115475. [Google Scholar] [CrossRef]
- Stewart, I.D.; Nanji, H.; Figueredo, G.; Fahy, W.A.; Maher, T.M.; Ask, A.J.; Maharaj, S.; Ask, K.; Kolb, M.; Jenkins, G.R. Circulating fibrocytes are not disease-specific prognosticators in idiopathic pulmonary fibrosis. Eur. Respir. J. 2021, 58, 2100172. [Google Scholar] [CrossRef]
- Guo, W.; Guo, T.; Zhou, Q.; Long, Y.; Luo, M.; Shen, Q.; Duan, W.; Ouyang, X.; Peng, H. Cofilin-1 promotes fibrocyte differentiation and contributes to pulmonary fibrosis. Biochem. Biophys. Res. Commun. 2021, 565, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-Y.; Lu, Y.-T.; Chen, S.-Y.; Hsu, C.-F.; Lu, Y.-C.; Wang, C.-Y.; Huang, K.-L. Targeting pathogenic macrophages by the application of SHP-1 agonists reduces inflammation and alleviates pulmonary fibrosis. Cell Death Dis. 2023, 14, 352. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, C.; Xie, Y.; Li, X. Down-regulation of CYTL1 attenuates bleomycin-induced pulmonary fibrosis in mice by inhibiting M2 macrophage polarization via the TGF-β/CCN2 axis. Clin. Exp. Pharmacol. Physiol. 2024, 51, e13913. [Google Scholar] [CrossRef]
- Veeraraghavan, S.; Latsi, P.; Wells, A.; Pantelidis, P.; Nicholson, A.; Colby, T.; Haslam, P.; Renzoni, E.; du Bois, R. BAL findings in idiopathic nonspecific interstitial pneumonia and usual interstitial pneumonia. Eur. Respir. J. 2003, 22, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-C.; Chen, N.-J.; Chen, H.-P.; Yu, W.-K.; Su, V.Y.-F.; Chen, H.; Wu, H.-H.; Yang, K.-Y. Nintedanib Reduces Neutrophil Chemotaxis via Activating GRK2 in Bleomycin-Induced Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 4735. [Google Scholar] [CrossRef]
- Polverino, E.; Rosales-Mayor, E.; Dale, G.E.; Dembowsky, K.; Torres, A. The Role of Neutrophil Elastase Inhibitors in Lung Diseases. Chest 2017, 152, 249–262. [Google Scholar] [CrossRef]
- Passalacqua, G.; Mincarini, M.; Colombo, D.; Troisi, G.; Ferrari, M.; Bagnasco, D.; Balbi, F.; Riccio, A.; Canonica, G.W. IL-13 and idiopathic pulmonary fibrosis: Possible links and new therapeutic strategies. Pulm. Pharmacol. Ther. 2017, 45, 95–100. [Google Scholar] [CrossRef]
- Sumida, A.; Hasegawa, Y.; Okamoto, M.; Hashimoto, N.; Imaizumi, K.; Yatsuya, H.; Yokoi, T.; Takagi, K.; Shimokata, K.; Kawabe, T. TH1/TH2 immune response in lung fibroblasts in interstitial lung disease. Arch. Med. Res. 2008, 39, 503–510. [Google Scholar] [CrossRef]
- Deng, K.M.; Yang, X.S.; Luo, Q.; She, Y.X.; Yu, Q.Y.; Tang, X.X. Deleterious Role of Th9 Cells in Pulmonary Fibrosis. Cells 2021, 10, 3209. [Google Scholar] [CrossRef]
- Tao, C.; Xian, H.; Nian-Yu, Z.; Jia-Cui, S.; Dong, W.; Hui-Ping, L. C-type lectin Mincle initiates IL-17-mediated inflammation in acute exacerbations of idiopathic pulmonary fibrosis. Biomed. Pharmacother. 2023, 159, 114253. [Google Scholar] [CrossRef]
- Lei, L.; Zhao, C.; Qin, F.; He, Z.-Y.; Wang, X.; Zhong, X.-N. Th17 cells and IL-17 promote the skin and lung inflammation and fibrosis process in a bleomycin-induced murine model of systemic sclerosis. Clin. Exp. Rheumatol. 2016, 34, 14–22. [Google Scholar] [PubMed]
- A Papiris, S.; Kollintza, A.; Karatza, M.; Manali, E.D.; Sotiropoulou, C.; Milic-Emili, J.; Roussos, C.; Daniil, Z. CD8+ T lymphocytes in bronchoalveolar lavage in idiopathic pulmonary fibrosis. J. Inflamm. 2007, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Jin, C.; Li, D.; Wang, Y.; Zheng, S.; Feng, Q.; Shi, N.; Kong, W.; Ma, X.; Wang, J. Single-cell transcriptomics reveals CD8+ T cell structure and developmental trajectories in idiopathic pulmonary fibrosis. Mol. Immunol. 2024, 172, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.-Y.; Lou, Y.; Qin, Y.-C.; Lin, W.; Liang, B.-B.; Sooranna, S.R.; Ma, Y.-L.; Zhou, S.-F. Novel kinase 1 regulates CD8+T cells as a potential therapeutic mechanism for idiopathic pulmonary fibrosis. Int. J. Med. Sci. 2024, 21, 1079–1090. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.; Chen, S.; Jiang, J. Unveiling the role of copper metabolism and STEAP2 in idiopathic pulmonary fibrosis molecular landscape. J. Cell. Mol. Med. 2024, 28, e18414. [Google Scholar] [CrossRef]
- Ran, Z.; Mu, B.-R.; Zhu, T.; Zhang, Y.; Luo, J.-X.; Yang, X.; Li, B.; Wang, D.-M.; Lu, M.-H. Predicting biomarkers related to idiopathic pulmonary fibrosis: Robust ranking aggregation analysis and animal experiment verification. Int. Immunopharmacol. 2024, 139, 112766. [Google Scholar] [CrossRef]
- Hata, K.; Yanagihara, T.; Matsubara, K.; Kunimura, K.; Suzuki, K.; Tsubouchi, K.; Eto, D.; Ando, H.; Uehara, M.; Ikegame, S.; et al. Mass cytometry identifies characteristic immune cell subsets in bronchoalveolar lavage fluid from interstitial lung diseases. Front. Immunol. 2023, 14, 1145814. [Google Scholar] [CrossRef]
- Fu, C.; Tian, X.; Wu, S.; Chu, X.; Cheng, Y.; Wu, X.; Yang, W. Role of telomere dysfunction and immune infiltration in idiopathic pulmonary fibrosis: New insights from bioinformatics analysis. Front. Genet. 2024, 15, 1447296. [Google Scholar] [CrossRef]
- Ali, M.F.; Egan, A.M.; Shaughnessy, G.F.; Anderson, D.K.; Kottom, T.J.; Dasari, H.; Van Keulen, V.P.; Aubry, M.-C.; Yi, E.S.; Limper, A.H.; et al. Antifibrotics Modify B-Cell–induced Fibroblast Migration and Activation in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 722–733. [Google Scholar] [CrossRef]
- Prêle, C.M.; Miles, T.; Pearce, D.R.; O’Donoghue, R.J.; Grainge, C.; Barrett, L.; Birnie, K.; Lucas, A.D.; Baltic, S.; Ernst, M.; et al. Plasma cell but not CD20-mediated B-cell depletion protects from bleomycin-induced lung fibrosis. Eur. Respir. J. 2022, 60, 2101469. [Google Scholar] [CrossRef]
- Chavez-Galan, L.; Becerril, C.; Ruiz, A.; Ramon-Luing, L.A.; Cisneros, J.; Montaño, M.; Salgado, A.; Ramos, C.; Buendía-Roldán, I.; Pardo, A.; et al. Fibroblasts From Idiopathic Pulmonary Fibrosis Induce Apoptosis and Reduce the Migration Capacity of T Lymphocytes. Front. Immunol. 2022, 13, 820347. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Hou, W.; Zhong, H.; Liu, D. Lung microbiota: Implications and interactions in chronic pulmonary diseases. Front. Cell. Infect. Microbiol. 2024, 14, 1401448. [Google Scholar] [CrossRef] [PubMed]
- Shulgina, L.; Cahn, A.P.; Chilvers, E.R.; Parfrey, H.; Clark, A.B.; Wilson, E.C.F.; Twentyman, O.P.; Davison, A.G.; Curtin, J.J.; Crawford, M.B.; et al. Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: A randomised controlled trial. Thorax 2012, 68, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Anstrom, K.J.; For the CleanUP-IPF Study Team; Noth, I.; Flaherty, K.R.; Edwards, R.H.; Albright, J.; Baucom, A.; Brooks, M.; Clark, A.B.; Clausen, E.S.; et al. Design and rationale of a multi-center, pragmatic, open-label randomized trial of antimicrobial therapy—The study of clinical efficacy of antimicrobial therapy strategy using pragmatic design in Idiopathic Pulmonary Fibrosis (CleanUP-IPF) clinical trial. Respir. Res. 2020, 21, 68. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, M.; Yang, L.; Gao, L.; Sun, W. Potential targeted therapy based on deep insight into the relationship between the pulmonary microbiota and immune regulation in lung fibrosis. Front. Immunol. 2023, 14, 1032355. [Google Scholar] [CrossRef]
- Invernizzi, R.; Wu, B.G.; Barnett, J.; Ghai, P.; Kingston, S.; Hewitt, R.J.; Feary, J.; Li, Y.; Chua, F.; Wu, Z.; et al. The Respiratory Microbiome in Chronic Hypersensitivity Pneumonitis Is Distinct from That of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 339–347. [Google Scholar] [CrossRef]
- Friaza, V.; de la Horra, C.; Rodríguez-Domínguez, M.J.; Martín-Juan, J.; Cantón, R.; Calderón, E.J.; del Campo, R. Metagenomic analysis of bronchoalveolar lavage samples from patients with idiopathic interstitial pneumonia and its antagonic relation with Pneumocystis jirovecii colonization. J. Microbiol. Methods 2010, 82, 98–101. [Google Scholar] [CrossRef]
- Yoon, H.-Y.; Moon, S.-J.; Song, J.W. Lung Tissue Microbiome Is Associated With Clinical Outcomes of Idiopathic Pulmonary Fibrosis. Front. Med. 2021, 8, 744523. [Google Scholar] [CrossRef]
- Han, M.K.; Zhou, Y.; Murray, S.; Tayob, N.; Noth, I.; Lama, V.N.; Moore, B.B.; White, E.S.; Flaherty, K.R.; Huffnagle, G.B.; et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: An analysis of the COMET study. Lancet Respir. Med. 2014, 2, 548–556. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, S.-F.; Espindola, M.S.; Vij, R.; Oldham, J.M.; Huffnagle, G.B.; Erb-Downward, J.R.; Flaherty, K.R.; Moore, B.B.; White, E.S.; et al. Microbes Are Associated with Host Innate Immune Response in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 208–219. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Ashley, S.L.; Gurczynski, S.J.; Xia, M.; Wilke, C.; Falkowski, N.R.; Norman, K.C.; Arnold, K.B.; Huffnagle, G.B.; Salisbury, M.L.; et al. Lung Microbiota Contribute to Pulmonary Inflammation and Disease Progression in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Huffnagle, G.B.; Flaherty, K.R.; White, E.S.; Martinez, F.J.; Erb-Downward, J.R.; Moore, B.B.; O’dwyer, D.N. Radiographic Honeycombing and Altered Lung Microbiota in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Valenzi, E.; Yang, H.; Sembrat, J.C.; Yang, L.; Winters, S.; Nettles, R.; Kass, D.J.; Qin, S.; Wang, X.; Myerburg, M.M.; et al. Topographic heterogeneity of lung microbiota in end-stage idiopathic pulmonary fibrosis: The Microbiome in Lung Explants-2 (MiLEs-2) study. Thorax 2021, 76, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Xue, B.; Ma, J. Causal relationship between gut microbiota and idiopathic pulmonary fibrosis: A two-sample Mendelian randomization. Medicine 2024, 103, e39013. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Yin, Z.; Chen, S.; Lang, J.; Han, L.; Yi, J.; Zhang, L.; Yue, Q.; Tian, W.; Chen, P.; et al. The gut-lung axis: Gut microbiota changes associated with pulmonary fibrosis in mouse models induced by bleomycin. Front. Pharmacol. 2022, 13, 985223. [Google Scholar] [CrossRef]
- Shi, H.; Zhao, T.; Geng, R.; Sun, L.; Fan, H. The associations between gut microbiota and chronic respiratory diseases: A Mendelian randomization study. Front. Microbiol. 2023, 14, 1200937. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, Y.; Cheng, Y.; Qin, H.; Zhao, H. Genetic liability of gut microbiota for idiopathic pulmonary fibrosis and lung function: A two-sample Mendelian randomization study. Front. Cell. Infect. Microbiol. 2024, 14, 1348685. [Google Scholar] [CrossRef]
- Ornatowski, W.; Lu, Q.; Yegambaram, M.; Garcia, A.E.; Zemskov, E.A.; Maltepe, E.; Fineman, J.R.; Wang, T.; Black, S.M. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020, 36, 101679. [Google Scholar] [CrossRef]
- Zhang, Z.; Yue, P.; Lu, T.; Wang, Y.; Wei, Y.; Wei, X. Role of lysosomes in physiological activities, diseases, and therapy. J. Hematol. Oncol. 2021, 14, 79. [Google Scholar] [CrossRef]
- Keller, C.W.; Adamopoulos, I.E.; Lünemann, J.D. Autophagy pathways in autoimmune diseases. J. Autoimmun. 2023, 136, 103030. [Google Scholar] [CrossRef]
- Tao, H.; Lv, Q.; Zhang, J.; Chen, L.; Yang, Y.; Sun, W. Different Levels of Autophagy Activity in Mesenchymal Stem Cells Are Involved in the Progression of Idiopathic Pulmonary Fibrosis. Stem Cells Int. 2024, 2024, 3429565. [Google Scholar] [CrossRef] [PubMed]
- Romero, Y.; Bueno, M.; Ramirez, R.; Álvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. mTORC 1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Chen, Y.-Y.; Hsiao, C.-M.; Pan, M.-H.; Wang, B., Jr.; Chen, Y.-C.; Ho, C.-T.; Huang, K.-C.; Chen, R.-J. Induction of Autophagy by Pterostilbene Contributes to the Prevention of Renal Fibrosis via Attenuating NLRP3 Inflammasome Activation and Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2020, 8, 436. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Lv, Z.-D.; Huang, W.-D.; Wei, S.-C.; Liu, X.-M.; Song, W.-D. LncRNA TUG1 promotes pulmonary fibrosis progression via up-regulating CDC27 and activating PI3K/Akt/mTOR pathway. Epigenetics 2023, 18, 2195305. [Google Scholar] [CrossRef]
- Qi, J.; Wu, Y.; Guo, Z.; Zhu, S.; Xiong, J.; Hu, F.; Liang, X.; Ye, X. Fibroblast growth factor 21 alleviates idiopathic pulmonary fibrosis by inhibiting PI3K-AKT-mTOR signaling and stimulating autophagy. Int. J. Biol. Macromol. 2024, 273, 132896. [Google Scholar] [CrossRef]
- Yue, Y.-L.; Zhang, M.-Y.; Liu, J.-Y.; Fang, L.-J.; Qu, Y.-Q. The role of autophagy in idiopathic pulmonary fibrosis: From mechanisms to therapies. Ther. Adv. Respir. Dis. 2022, 16, 17534666221140972. [Google Scholar] [CrossRef]
- Zheng, D.; Guo, J.; Liang, Z.; Jin, Y.; Ding, Y.; Liu, J.; Qi, C.; Shi, K.; Xie, L.; Zhu, M.; et al. Supramolecular Nanofibers Ameliorate Bleomycin-Induced Pulmonary Fibrosis by Restoring Autophagy. Adv. Sci. 2024, 11, e2401327. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted protein degradation: Mechanisms, strategies and application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar] [CrossRef]
- Jayaraj, G.G.; Hipp, M.S.; Hartl, F.U. Functional Modules of the Proteostasis Network. Cold Spring Harb. Perspect. Biol. 2020, 12, a033951. [Google Scholar] [CrossRef]
- Margulis, B.; Tsimokha, A.; Zubova, S.; Guzhova, I. Molecular Chaperones and Proteolytic Machineries Regulate Protein Homeostasis in Aging Cells. Cells 2020, 9, 1308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, X.; Huang, W.; Ge, X. The role of heat shock proteins in the regulation of fibrotic diseases. Biomed. Pharmacother. 2021, 135, 111067. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Dang, Z.; Xu, S.; Chong, S. Heat shock protein 47 effects on hepatic stellate cell-associated receptors in hepatic fibrosis of Schistosoma japonicum-infected mice. Biol. Chem. 2017, 398, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Morry, J.; Ngamcherdtrakul, W.; Gu, S.; Goodyear, S.M.; Castro, D.J.; Reda, M.M.; Sangvanich, T.; Yantasee, W. Dermal delivery of HSP47 siRNA with NOX4-modulating mesoporous silica-based nanoparticles for treating fibrosis. Biomaterials 2015, 66, 41–52. [Google Scholar] [CrossRef]
- Ruigrok, M.J.R.; El Amasi, K.E.M.; Leeming, D.J.; Sand, J.M.B.; Frijlink, H.W.; Hinrichs, W.L.J.; Olinga, P. Silencing Heat Shock Protein 47 (HSP47) in Fibrogenic Precision-Cut Lung Slices: A Surprising Lack of Effects on Fibrogenesis? Front. Med. 2021, 8, 607962. [Google Scholar] [CrossRef]
- Namba, T.; Tanaka, K.-I.; Hoshino, T.; Azuma, A.; Mizushima, T. Suppression of Expression of Heat Shock Protein 70 by Gefitinib and Its Contribution to Pulmonary Fibrosis. PLoS ONE 2011, 6, e27296. [Google Scholar] [CrossRef]
- Zhou, R.; Jin, C.; Jiao, L.; Zhang, S.; Tian, M.; Liu, J.; Yang, S.; Yao, W.; Zhou, F. Geranylgeranylacetone, an inducer of heat shock protein 70, attenuates pulmonary fibrosis via inhibiting NF-κB/NOX4/ROS signalling pathway in vitro and in vivo. Chem. Interact. 2023, 382, 110603. [Google Scholar] [CrossRef]
- Somogyvári, M.; Khatatneh, S.; Sőti, C. Hsp90: From Cellular to Organismal Proteostasis. Cells 2022, 11, 2479. [Google Scholar] [CrossRef]
- Mohammed, O.A.; Abdel-Reheim, M.A.; Saleh, L.A.; Alamri, M.M.S.; Alfaifi, J.; Adam, M.I.E.; Farrag, A.A.; AlQahtani, A.A.J.; BinAfif, W.F.; Hashish, A.A.; et al. Alvespimycin Exhibits Potential Anti-TGF-β Signaling in the Setting of a Proteasome Activator in Rats with Bleomycin-Induced Pulmonary Fibrosis: A Promising Novel Approach. Pharmaceuticals 2023, 16, 1123. [Google Scholar] [CrossRef]
- Lee, J.Y.; Reyes, N.S.; Ravishankar, S.; Zhou, M.; Krasilnikov, M.; Ringler, C.; Pohan, G.; Wilson, C.; Ang, K.K.-H.; Wolters, P.J.; et al. An in vivo screening platform identifies senolytic compounds that target p16INK4a+ fibroblasts in lung fibrosis. J. Clin. Investig. 2024, 134, e173371. [Google Scholar] [CrossRef]
- Plantier, L.; Cazes, A.; Dinh-Xuan, A.-T.; Bancal, C.; Marchand-Adam, S.; Crestani, B. Physiology of the lung in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 170062. [Google Scholar] [CrossRef] [PubMed]
- Hillary, R.F.; FitzGerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.C.; Meier, S.E.; Ingram, A.L.; Abisambra, J.F. PERK-opathies: An Endoplasmic Reticulum Stress Mechanism Underlying Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Dobrinskikh, E.; Hennessy, C.E.; Kurche, J.S.; Kim, E.; Estrella, A.M.; Cardwell, J.; Yang, I.V.; Schwartz, D.A. Epithelial Endoplasmic Reticulum Stress Enhances the Risk of Muc5b-associated Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2023, 68, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Kurche, J.S.; Stancil, I.T.; Michalski, J.E.; Yang, I.V.; Schwartz, D.A. Dysregulated Cell–Cell Communication Characterizes Pulmonary Fibrosis. Cells 2022, 11, 3319. [Google Scholar] [CrossRef]
- Mekhael, O.; Revill, S.D.; I Hayat, A.; Cass, S.P.; MacDonald, K.; Vierhout, M.; Ayoub, A.; Reihani, A.; Padwal, M.; Imani, J.; et al. Myeloid-specific deletion of activating transcription factor 6 alpha increases CD11b+ macrophage subpopulations and aggravates lung fibrosis. Immunol. Cell Biol. 2023, 101, 412–427. [Google Scholar] [CrossRef]
- Wang, Z.; Feng, F.; He, H.; Wu, Q.; Gu, C.; Hrovat, J.; Peng, W.; Xu, Y.; Han, D.; Yang, P.; et al. Citrus alkaline extracts prevent endoplasmic reticulum stress in type II alveolar epithelial cells to ameliorate pulmonary fibrosis via the ATF3/PINK1 pathway. Phytomedicine 2021, 89, 153599. [Google Scholar] [CrossRef]
- Luo, R.; Wei, Y.; Chen, P.; Zhang, J.; Wang, L.; Wang, W.; Wang, P.; Tian, W. Mesenchymal Stem Cells Inhibit Epithelial-to-Mesenchymal Transition by Modulating the IRE1α Branch of the Endoplasmic Reticulum Stress Response. Stem Cells Int. 2023, 2023, 4483776. [Google Scholar] [CrossRef]
- Komatsu, S.; Fan, L.; Idell, S.; Shetty, S.; Ikebe, M. Caveolin-1-Derived Peptide Reduces ER Stress and Enhances Gelatinolytic Activity in IPF Fibroblasts. Int. J. Mol. Sci. 2022, 23, 3316. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, H.; Ding, S.F.X.; Lv, W.; Jiang, Y.; Liang, Y.; Xu, H.; Dai, J. PGC-1α regulates endoplasmic reticulum stress in IPF-derived fibroblasts. Int. Immunopharmacol. 2024, 138, 112514. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, X.; Lin, Y.; Zeng, Y. Roles of airway basal stem cells in lung homeostasis and regenerative medicine. Respir. Res. 2022, 23, 122. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Castillo, J.A.; Pérez, D.B.; Ntokou, A.; Seeger, W.; Morty, R.E.; Ahlbrecht, K. Understanding alveolarization to induce lung regeneration. Respir. Res. 2018, 19, 148. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Tan, C.; Zhang, J. Alveolar Epithelial Type 2 Cell Dysfunction in Idiopathic Pulmonary Fibrosis. Lung 2022, 200, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef]
- Mi, L.; Hu, J.; Li, N.; Gao, J.; Huo, R.; Peng, X.; Zhang, N.; Liu, Y.; Zhao, H.; Liu, R.; et al. The Mechanism of Stem Cell Aging. Stem Cell Rev. Rep. 2022, 18, 1281–1293. [Google Scholar] [CrossRef]
- Pouikli, A.; Tessarz, P. Epigenetic alterations in stem cell ageing—A promising target for age-reversing interventions? Brief. Funct. Genom. 2021, 21, 35–42. [Google Scholar] [CrossRef]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Franzén, L.; Lindvall, M.O.; Hühn, M.; Ptasinski, V.; Setyo, L.; Keith, B.P.; Collin, A.; Oag, S.; Volckaert, T.; Borde, A.; et al. Mapping spatially resolved transcriptomes in human and mouse pulmonary fibrosis. Nat. Genet. 2024, 56, 1725–1736. [Google Scholar] [CrossRef]
- Kadota, T.; Yoshioka, Y.; Fujita, Y.; Araya, J.; Minagawa, S.; Hara, H.; Miyamoto, A.; Suzuki, S.; Fujimori, S.; Kohno, T.; et al. Extracellular Vesicles from Fibroblasts Induce Epithelial-Cell Senescence in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 623–636. [Google Scholar] [CrossRef]
- Elliot, S.; Catanuto, P.; Pereira-Simon, S.; Xia, X.; Shahzeidi, S.; Roberts, E.; Ludlow, J.; Hamdan, S.; Daunert, S.; Parra, J.; et al. Urine-derived exosomes from individuals with IPF carry pro-fibrotic cargo. eLife 2022, 11, e79543. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, Y.; Yue, R.; Peng, X.; Yu, H.; Huang, X. Exosomes derived from hypoxia-induced alveolar epithelial cells stimulate interstitial pulmonary fibrosis through a HOTAIRM1-dependent mechanism. Mod. Pathol. 2022, 102, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Song, W.; Gao, Y.; Zheng, X.; Wang, F.; Zhang, Z.; Zen, K.; Liang, H.; Yan, X. Exosomal circRNAs in the plasma serve as novel biomarkers for IPF diagnosis and progression prediction. J. Transl. Med. 2024, 22, 264. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Fujimoto, S.; Miyamoto, A.; Kaneko, R.; Kadota, T.; Watanabe, N.; Kizawa, R.; Kawamoto, H.; Watanabe, J.; Utsumi, H.; et al. Fibroblast-derived Extracellular Vesicles Induce Lung Cancer Progression in the Idiopathic Pulmonary Fibrosis Microenvironment. Am. J. Respir. Cell Mol. Biol. 2023, 69, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Hayek, H.; Rehbini, O.; Kosmider, B.; Brandt, T.; Chatila, W.; Marchetti, N.; Criner, G.J.; Bolla, S.; Kishore, R.; Bowler, R.P.; et al. The Regulation of FASN by Exosomal miR-143-5p and miR-342-5p in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2023, 70, 259–282. [Google Scholar] [CrossRef]
- Feng, Z.; Jing, Z.; Li, Q.; Chu, L.; Jiang, Y.X.; Zhang, X.; Yan, L.; Liu, Y.; Jiang, J.; Xu, P.; et al. Exosomal STIMATE derived from type II alveolar epithelial cells controls metabolic reprogramming of tissue-resident alveolar macrophages. Theranostics 2023, 13, 991–1009. [Google Scholar] [CrossRef]
- Kadota, T.; Fujita, Y.; Araya, J.; Watanabe, N.; Fujimoto, S.; Kawamoto, H.; Minagawa, S.; Hara, H.; Ohtsuka, T.; Yamamoto, Y.; et al. Human bronchial epithelial cell-derived extracellular vesicle therapy for pulmonary fibrosis via inhibition of TGF-β-WNT crosstalk. J. Extracell. Vesicles 2021, 10, e12124. [Google Scholar] [CrossRef]
- Zhu, L.; Xu, Y.; Wang, J.; Zhang, Y.; Zhou, J.; Wu, H. Mesenchymal stem cells-derived exosomes carrying microRNA-30b confer protection against pulmonary fibrosis by downregulating Runx1 via Spred. Mol. Genet. Genom. 2024, 299, 33. [Google Scholar] [CrossRef]
- Guiot, J.; Cambier, M.; Boeckx, A.; Henket, M.; Nivelles, O.; Gester, F.; Louis, E.; Malaise, M.; Dequiedt, F.; Louis, R.; et al. Macrophage-derived exosomes attenuate fibrosis in airway epithelial cells through delivery of antifibrotic miR-142-3p. Thorax 2020, 75, 870–881. [Google Scholar] [CrossRef]
- Fujita, Y. Extracellular vesicles in idiopathic pulmonary fibrosis: Pathogenesis and therapeutics. Inflamm. Regen. 2022, 42, 23. [Google Scholar] [CrossRef]
- Wang, K.; Liu, H.; Hu, Q.; Wang, L.; Liu, J.; Zheng, Z.; Zhang, W.; Ren, J.; Zhu, F.; Liu, G.-H. Epigenetic regulation of aging: Implications for interventions of aging and diseases. Signal Transduct. Target. Ther. 2022, 7, 374. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, F.; Malavolta, M.; Calabrese, R.; Guastafierro, T.; Bacalini, M.G.; Reale, A.; Franceschi, C.; Capri, M.; Hervonen, A.; Hurme, M.; et al. Age-dependent expression of DNMT1 and DNMT3B in PBMCs from a large European population enrolled in the MARK-AGE study. Aging Cell 2016, 15, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Lopatina, N.; Haskell, J.F.; Andrews, L.G.; Poole, J.C.; Saldanha, S.; Tollefsbol, T. Differential maintenance and de novo methylating activity by three DNA methyltransferases in aging and immortalized fibroblasts. J. Cell. Biochem. 2001, 84, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-H.; Chen, J.A.; Sayed, F.; Ward, M.E.; Gao, F.; Nguyen, T.A.; Krabbe, G.; Sohn, P.D.; Lo, I.; Minami, S.; et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 2015, 35, 807–818. [Google Scholar] [CrossRef]
- Djeghloul, D.; Kuranda, K.; Kuzniak, I.; Barbieri, D.; Naguibneva, I.; Choisy, C.; Bories, J.-C.; Dosquet, C.; Pla, M.; Vanneaux, V.; et al. Age-Associated Decrease of the Histone Methyltransferase SUV39H1 in HSC Perturbs Heterochromatin and B Lymphoid Differentiation. Stem Cell Rep. 2016, 6, 970–984. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, Y.; Hu, Y.; An, J.; Li, L.; Wang, Y.; Zhang, X. Decreased expression of m6A demethylase FTO in ovarian aging. Arch. Gynecol. Obstet. 2021, 303, 1363–1369. [Google Scholar] [CrossRef]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic Predictor of Age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef]
- Trapp, A.; Trapp, A.; Kerepesi, C.; Kerepesi, C.; Gladyshev, V.N.; Gladyshev, V.N. Profiling epigenetic age in single cells. Nat. Aging 2021, 1, 1189–1201. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA Methylation Profile in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535. [Google Scholar] [CrossRef]
- Heyn, H.; Li, N.; Ferreira, H.J.; Moran, S.; Pisano, D.G.; Gomez, A.; Diez, J.; Sanchez-Mut, J.V.; Setien, F.; Carmona, F.J.; et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 2012, 109, 10522–10527. [Google Scholar] [CrossRef]
- Lu, A.T.; Fei, Z.; Haghani, A.; Robeck, T.R.; Zoller, J.A.; Li, C.Z.; Lowe, R.; Yan, Q.; Zhang, J.; Vu, H.; et al. Universal DNA methylation age across mammalian tissues. Nat. Aging 2023, 3, 1144–1166. [Google Scholar] [CrossRef] [PubMed]
- Pokhreal, D.; Crestani, B.; Helou, D.G. Macrophage Implication in IPF: Updates on Immune, Epigenetic, and Metabolic Pathways. Cells 2023, 12, 2193. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Bai, L.; Zhou, J.Q.; Cevallos, R.R.; Sanders, J.R.; Liu, G.; Bernard, K.; Sanders, Y.Y. Epigenetic regulation of IPF fibroblast phenotype by glutaminolysis. Mol. Metab. 2022, 67, 101655. [Google Scholar] [CrossRef]
- Deskata, K.; Malli, F.; Jagirdar, R.; Vavougios, G.D.; Zarogiannis, S.; I Gourgoulianis, K.; Daniil, Z. Evaluation of Sirtuin 1 Levels in Peripheral Blood Mononuclear Cells of Patients With Idiopathic Pulmonary Fibrosis. Cureus 2022, 14, e30862. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Pan, L.; Cheng, Y.; Yang, W.; Wu, X.; Zhu, H.; Hu, M.; Zhang, Y.; Zhang, M. Nintedanib Ameliorates Bleomycin-Induced Pulmonary Fibrosis, Inflammation, Apoptosis, and Oxidative Stress by Modulating PI3K/Akt/mTOR Pathway in Mice. Inflammation 2023, 46, 1531–1542. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, Y.; Li, K.; Huo, Y.; Wang, S.; Dong, S.; Ma, M. Targeting the PI3K/mTOR pathway in idiopathic pulmonary fibrosis: Advances and therapeutic potential. Bioorganic Med. Chem. 2024, 115, 117908. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Cheng, H.; Li, X.; Feng, D.; Yue, S.; Xu, J.; Xie, H.; Luo, Z. Therapeutic Effects of Omentin-1 on Pulmonary Fibrosis by Attenuating Fibroblast Activation via AMP-Activated Protein Kinase Pathway. Biomedicines 2022, 10, 2715. [Google Scholar] [CrossRef]
- Wei, S.; Liu, Y.; Ran, C.; Li, Y.; Tang, B.; Lu, M.; Wang, H. Calpain-1 Up-Regulation Promotes Bleomycin-Induced Pulmonary Fibrosis by Activating Ferroptosis. Am. J. Pathol. 2024, 194, 2272–2289. [Google Scholar] [CrossRef]
- Guo, F.; Xu, F.; Li, S.; Zhang, Y.; Lv, D.; Zheng, L.; Gan, Y.; Zhou, M.; Zhao, K.; Xu, S.; et al. Amifostine ameliorates bleomycin-induced murine pulmonary fibrosis via NAD+/SIRT1/AMPK pathway-mediated effects on mitochondrial function and cellular metabolism. Eur. J. Med. Res. 2024, 29, 68. [Google Scholar] [CrossRef]
- Tirunavalli, S.K.; Andugulapati, S.B. Geneticin ameliorates pulmonary fibrosis by attenuating the TGF-β/Smad via modulating AMPK/SIRT1 signaling. Life Sci. 2024, 346, 122626. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, F.; D’agnano, V.; Mariniello, D.F.; Castaldo, G.; Vitale, M.; Cazzola, M.; Bianco, A.; Scialò, F. Potential role of SIRT-1 and SIRT-3 as biomarkers for the diagnosis and prognosis of idiopathic pulmonary fibrosis. Respir. Res. 2024, 25, 189. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiao, Y.; Wu, Z.; Liu, H.; Li, Y.; Cai, Y.; Wei, W.; Cao, F. The role of quercetin in ameliorating bleomycin-induced pulmonary fibrosis: Insights into autophagy and the SIRT1/AMPK signaling pathway. Mol. Biol. Rep. 2024, 51, 795. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.; Ravichandran, R.; Sanborn, K.; Fleming, T.; Wheatcroft, S.B.; Kearney, M.T.; Tokman, S.; Walia, R.; Smith, M.A.; Flint, D.J.; et al. Loss of IGFBP2 mediates alveolar type 2 cell senescence and promotes lung fibrosis. Cell Rep. Med. 2023, 4, 100945. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, À.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Hamsanathan, S.; Alder, J.K.; Sellares, J.; Rojas, M.; Gurkar, A.U.; Mora, A.L. Cellular Senescence: The Trojan Horse in Chronic Lung Diseases. Am. J. Respir. Cell Mol. Biol. 2019, 61, 21–30. [Google Scholar] [CrossRef]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017, 50, 1602367. [Google Scholar] [CrossRef]
- Yanai, H.; Shteinberg, A.; Porat, Z.; Budovsky, A.; Braiman, A.; Zeische, R.; Fraifeld, V.E. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging 2015, 7, 664–672. [Google Scholar] [CrossRef]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef] [PubMed]
- Blokland, K.E.C.; Waters, D.W.; Schuliga, M.; Read, J.; Pouwels, S.D.; Grainge, C.L.; Jaffar, J.; Westall, G.; Mutsaers, S.E.; Prêle, C.M.; et al. Senescence of IPF Lung Fibroblasts Disrupt Alveolar Epithelial Cell Proliferation and Promote Migration in Wound Healing. Pharmaceutics 2020, 12, 389. [Google Scholar] [CrossRef] [PubMed]
- Blokland, K.E.C.; Habibie, H.; Borghuis, T.; Teitsma, G.J.; Schuliga, M.; Melgert, B.N.; Knight, D.A.; Brandsma, C.-A.; Pouwels, S.D.; Burgess, J.K. Regulation of Cellular Senescence Is Independent from Profibrotic Fibroblast-Deposited ECM. Cells 2021, 10, 1628. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Boatz, J.C.; Shi, J.; Hu, Q.; Li, X.; Zhang, Y.; Königshoff, M.; Kliment, C.R. Loss of ANT1 Increases Fibrosis and Epithelial Cell Senescence in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2023, 69, 556–569. [Google Scholar] [CrossRef]
- Su, W.; Guo, Y.; Wang, Q.; Ma, L.; Zhang, Q.; Zhang, Y.; Geng, Y.; Jin, T.; Guo, J.; Yang, R.; et al. YAP1 inhibits the senescence of alveolar epithelial cells by targeting Prdx3 to alleviate pulmonary fibrosis. Exp. Mol. Med. 2024, 56, 1643–1654. [Google Scholar] [CrossRef]
- Guan, R.; Yuan, L.; Li, J.; Wang, J.; Li, Z.; Cai, Z.; Guo, H.; Fang, Y.; Lin, R.; Liu, W.; et al. Bone morphogenetic protein 4 inhibits pulmonary fibrosis by modulating cellular senescence and mitophagy in lung fibroblasts. Eur. Respir. J. 2022, 60, 2102307. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Zhu, Y.; Prata, L.G.L.; Gerdes, E.O.W.; Netto, J.M.E.; Pirtskhalava, T.; Giorgadze, N.; Tripathi, U.; Inman, C.L.; Johnson, K.O.; Xue, A.; et al. Orally-active, clinically-translatable senolytics restore α-Klotho in mice and humans. EBioMedicine 2022, 77, 103912. [Google Scholar] [CrossRef]
- Nambiar, A.; Kellogg, D.; Justice, J.; Goros, M.; Gelfond, J.; Pascual, R.; Hashmi, S.; Masternak, M.; Prata, L.; LeBrasseur, N.; et al. Senolytics dasatinib and quercetin in idiopathic pulmonary fibrosis: Results of a phase I, single-blind, single-center, randomized, placebo-controlled pilot trial on feasibility and tolerability. EBioMedicine 2023, 90, 104481. [Google Scholar] [CrossRef]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.Y.; Chen, J.J.-L.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.I.; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. New Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Chen, J.J.-L.; Lancaster, L.; Danoff, S.; Su, S.-C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef]
- Tsang, A.R.; Wyatt, H.D.; Ting, N.S.; Beattie, T.L. hTERT mutations associated with idiopathic pulmonary fibrosis affect telomerase activity, telomere length, and cell growth by distinct mechanisms. Aging Cell 2012, 11, 482–490. [Google Scholar] [CrossRef]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef]
- Cala-Garcia, J.D.; Medina-Rincon, G.J.; Sierra-Salas, P.A.; Rojano, J.; Romero, F. The Role of Mitochondrial Dysfunction in Idiopathic Pulmonary Fibrosis: New Perspectives for a Challenging Disease. Biology 2023, 12, 1237. [Google Scholar] [CrossRef]
- Mercader-Barceló, J.; Martín-Medina, A.; Truyols-Vives, J.; Escarrer-Garau, G.; Elowsson, L.; Montes-Worboys, A.; Río-Bocos, C.; Muncunill-Farreny, J.; Velasco-Roca, J.; Cederberg, A.; et al. Mitochondrial Dysfunction in Lung Resident Mesenchymal Stem Cells from Idiopathic Pulmonary Fibrosis Patients. Cells 2023, 12, 2084. [Google Scholar] [CrossRef]
- Pokharel, M.D.; Garcia-Flores, A.; Marciano, D.; Franco, M.C.; Fineman, J.R.; Aggarwal, S.; Wang, T.; Black, S.M. Mitochondrial network dynamics in pulmonary disease: Bridging the gap between inflammation, oxidative stress, and bioenergetics. Redox Biol. 2024, 70, 103049. [Google Scholar] [CrossRef]
- Heckenbach, I.; Mkrtchyan, G.V.; Ben Ezra, M.; Bakula, D.; Madsen, J.S.; Nielsen, M.H.; Oró, D.; Osborne, B.; Covarrubias, A.J.; Idda, M.L.; et al. Nuclear morphology is a deep learning biomarker of cellular senescence. Nat. Aging 2022, 2, 742–755. [Google Scholar] [CrossRef]
- Rubio, K.; Castillo-Negrete, R.; Barreto, G. Non-coding RNAs and nuclear architecture during epithelial-mesenchymal transition in lung cancer and idiopathic pulmonary fibrosis. Cell. Signal. 2020, 70, 109593. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Pérez, E.R.F.; Costabel, U.; Albera, C.; Lederer, D.J.; Flaherty, K.R.; Ettinger, N.; Perez, R.; Scholand, M.B.; Goldin, J.; et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2020, 8, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Fishman, J.E.; Kim, G.J.; Kyeong, N.Y.; Goldin, J.G.; Glassberg, M.K. Intravenous stem cell dose and changes in quantitative lung fibrosis and DLCO in the AETHER trial: A pilot study. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7568–7572. [Google Scholar] [CrossRef] [PubMed]
- Averyanov, A.; Koroleva, I.; Konoplyannikov, M.; Revkova, V.; Lesnyak, V.; Kalsin, V.; Danilevskaya, O.; Nikitin, A.; Sotnikova, A.; Kotova, S.; et al. First-in-human high-cumulative-dose stem cell therapy in idiopathic pulmonary fibrosis with rapid lung function decline. Stem Cells Transl. Med. 2020, 9, 6–16. [Google Scholar] [CrossRef]


| Sample | Microbiota Increased in IPF | Reference |
|---|---|---|
| BAL | Actinomyces and Veillonella | [86] |
| BAL | Neisseria, Streptococcus, and Actinobacterium sp. | [87] |
| BAL | Streptococcus, Sphingomonas, Clostridium, and Lactobacillus | [88] |
| Bronchoscopy and BAL | Streptococcus and Staphylococcus | [89] |
| BAL | Streptococcus, Prevotella, and Veillonella | [90] |
| Human and murine BAL | Firmicutes | [91] |
| BAL | Prevotella, Veillonella, Porphyomonas, Gemella, and Cronobacter. | [92] |
| Bronchial washing and airway tissue | Streptococcus, Prevotella, and Veillonella | [93] |
| Human gut | Coprococcus 2 | [94] |
| Murine gut | Verrucomicrobiales and Enterobacteriales | [95] |
| Human gut | Blautia and Eisenbergiella | [96] |
| Human gut | Oscillospira and Parasutterella | [97] |
| Role | Cargo | Effect | Reference |
|---|---|---|---|
| Profibrotic | miR-143-5p, miR-342-5p | Downregulation of FASN and ACSL-4 | [145] |
| hsa_circ_0044226 | Associated with acute exacerbations | [143] | |
| hsa_circ_0004099, hsa_circ_0008898 | Proposed diagnostic biomarkers | [143] | |
| HOTAIRM1 | Proliferation and differentiation of fibroblasts | [142] | |
| Low levels of miR-let-7d, miR-29a-5p, and miR-181b-3p and high miR-199a-3p | Increase COL1A1 and α-SMA | [141] | |
| Procancer | mir-19a | Increases proliferation of non-small-cell lung cancer via ZMYND11 and c-Myc | [144] |
| Antifibrotic | STIM-activating enhancer | Controls macrophage differentiation, enhances oxidative phosphorylation, and favors mitochondrial biogenesis | [146] |
| Low miR-30b | Downregulates TNF-α, TβR1, IL-1β, α-SMA, and Collagen 1. | [148] | |
| miR-142-3p | Downregulates TGF-β1, TβR1, COL1A1, and COL3A1 | [149] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Machorro, A.L.; García-Vicente, Á.; Espina-Ordoñez, M.; Luis-García, E.; Negreros, M.; Herrera, I.; Becerril, C.; Toscano, F.; Cisneros, J.; Maldonado, M. Update of Aging Hallmarks in Idiopathic Pulmonary Fibrosis. Cells 2025, 14, 222. https://doi.org/10.3390/cells14030222
Torres-Machorro AL, García-Vicente Á, Espina-Ordoñez M, Luis-García E, Negreros M, Herrera I, Becerril C, Toscano F, Cisneros J, Maldonado M. Update of Aging Hallmarks in Idiopathic Pulmonary Fibrosis. Cells. 2025; 14(3):222. https://doi.org/10.3390/cells14030222
Chicago/Turabian StyleTorres-Machorro, Ana Lilia, Ángeles García-Vicente, Marco Espina-Ordoñez, Erika Luis-García, Miguel Negreros, Iliana Herrera, Carina Becerril, Fernanda Toscano, Jose Cisneros, and Mariel Maldonado. 2025. "Update of Aging Hallmarks in Idiopathic Pulmonary Fibrosis" Cells 14, no. 3: 222. https://doi.org/10.3390/cells14030222
APA StyleTorres-Machorro, A. L., García-Vicente, Á., Espina-Ordoñez, M., Luis-García, E., Negreros, M., Herrera, I., Becerril, C., Toscano, F., Cisneros, J., & Maldonado, M. (2025). Update of Aging Hallmarks in Idiopathic Pulmonary Fibrosis. Cells, 14(3), 222. https://doi.org/10.3390/cells14030222