

The Role of Complement in the Pathogenesis and Treatment of Myasthenia Gravis


Abstract
1. Introduction
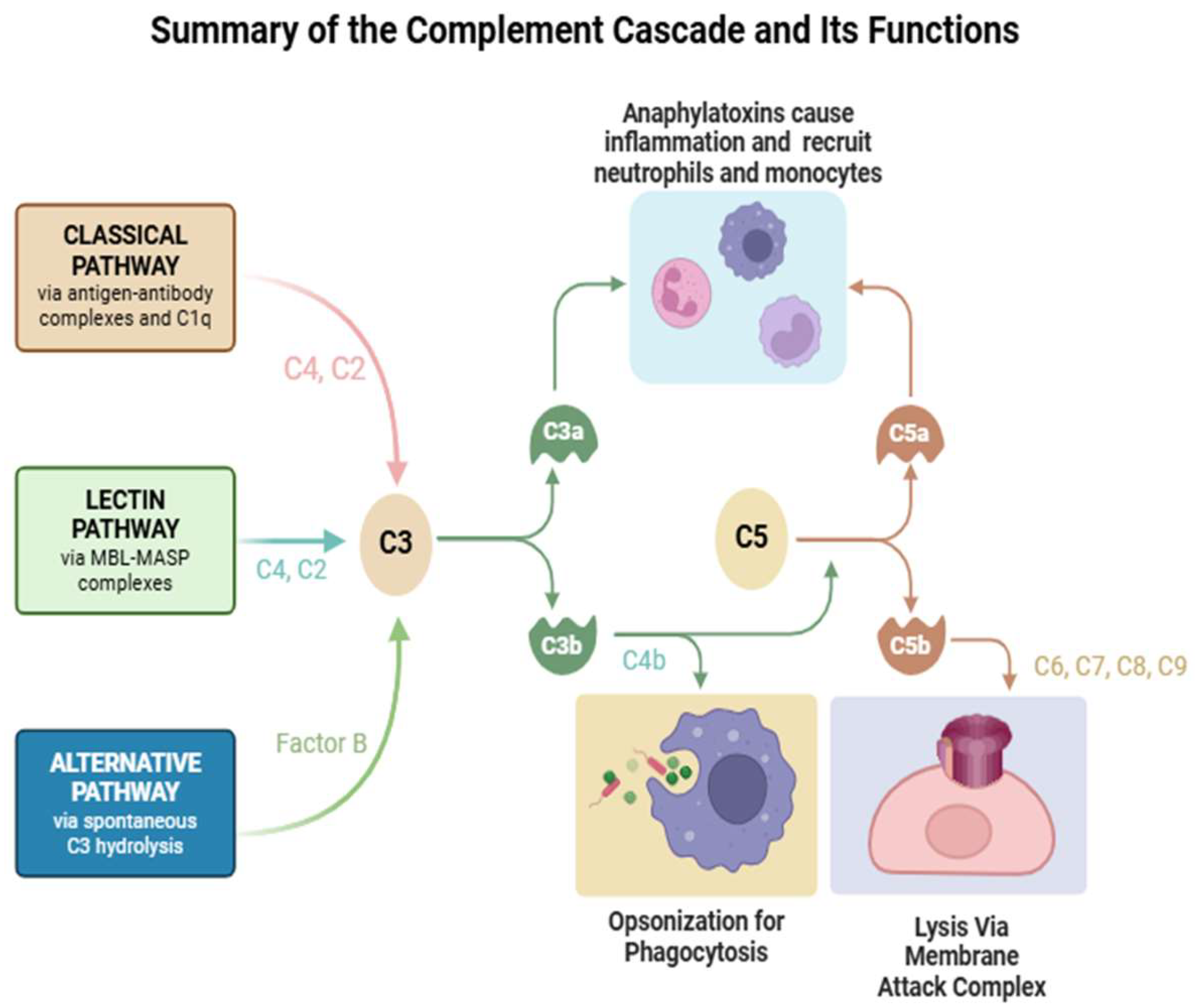
2. Complement Pathways and Components
3. Complement-Related Diseases
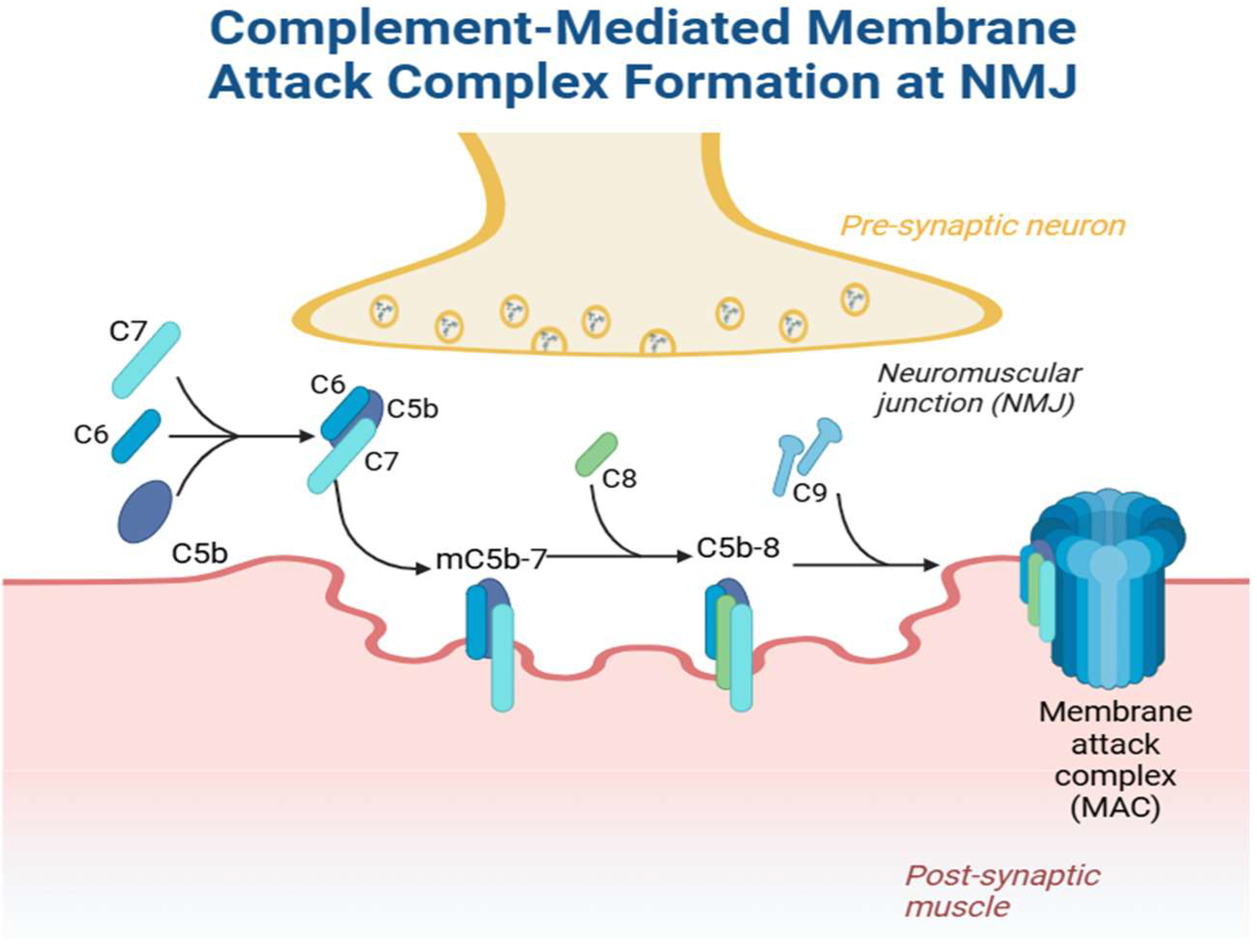
4. The Role of Complement in Myasthenia Gravis
4.1. Preclinical Data
4.2. Clinical Data
4.2.1. Pathophysiology
4.2.2. Complement as Biomarkers
5. Clinical Use of Complement Inhibitors
5.1. Currently Available Therapies
5.2. Emerging Therapies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AChR | Acetylcholine receptor |
| EAMG | Experimental animal model of myasthenia gravis |
| FDA | Food and Drug Administration |
| LRP4 | Low-density lipoprotein receptor-related protein 4 |
| MBL | Mannan-binding lectin |
| MG | Myasthenia gravis |
| MG-ADL | Myasthenia Gravis Activities of Daily Living |
| MuSK | Muscle-specific tyrosine kinase |
| PTMG | Passive transfer models of myasthenia gravis |
| QMG | Quantitative Myasthenia Gravis |
| SLE | Systemic lupus erythematosus |
References
- Dresser, L.; Wlodarski, R.; Rezania, K.; Soliven, B. Myasthenia Gravis: Epidemiology, Pathophysiology and Clinical Manifestations. J. Clin. Med. 2021, 10, 2235. [Google Scholar] [CrossRef] [PubMed]
- San, P.P.; Jacob, S. Role of complement in myasthenia gravis. Front. Neurol. 2023, 14, 1277596. [Google Scholar] [CrossRef]
- Lazaridis, K.; Tzartos, S.J. Autoantibody Specificities in Myasthenia Gravis; Implications for Improved Diagnostics and Therapeutics. Front. Immunol. 2020, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, N.S.R. Complement and myasthenia gravis. Mol. Immunol. 2022, 151, 11–18. [Google Scholar] [CrossRef]
- Okada, K.; Inoue, A.; Okada, M.; Murata, Y.; Kakuta, S.; Jigami, T.; Kubo, S.; Shiraishi, H.; Eguchi, K.; Motomura, M.; et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 2006, 312, 1802–1805. [Google Scholar] [CrossRef] [PubMed]
- Chuquisana, O.; Stascheit, F.; Keller, C.W.; Pučić-Baković, M.; Patenaude, A.-M.; Lauc, G.; Tzartos, S.; Wiendl, H.; Willcox, N.; Meisel, A.; et al. Functional Signature of LRP4 Antibodies in Myasthenia Gravis. Neurol. Neuroimmunol. Neuroinflamm. 2024, 11, e200220. [Google Scholar] [CrossRef]
- Rivner, M.H.; Quarles, B.M.; Pan, J.X.; Yu, Z.; Howard, J.F., Jr.; Corse, A.; Dimachkie, M.M.; Jackson, C.; Vu, T.; Small, G.; et al. Clinical features of LRP4/agrin-antibody-positive myasthenia gravis: A multicenter study. Muscle Nerve 2020, 62, 333–343. [Google Scholar] [CrossRef]
- Dalakas, M.C. Role of complement, anti-complement therapeutics, and other targeted immunotherapies in myasthenia gravis. Expert. Rev. Clin. Immunol. 2022, 18, 691–701. [Google Scholar] [CrossRef]
- Kusner, L.L.; Kaminski, H.J.; Soltys, J. Effect of complement and its regulation on myasthenia gravis pathogenesis. Expert. Rev. Clin. Immunol. 2008, 4, 43–52. [Google Scholar] [CrossRef]
- Basta, M.; Illa, I.; Dalakas, M.C. Increased in vitro uptake of the complement C3b in the serum of patients with Guillain-Barré syndrome, myasthenia gravis and dermatomyositis. J. Neuroimmunol. 1996, 71, 227–229. [Google Scholar] [CrossRef]
- Chamberlain-Banoub, J.; Neal, J.W.; Mizuno, M.; Harris, C.L.; Morgan, B.P. Complement membrane attack is required for endplate damage and clinical disease in passive experimental myasthenia gravis in Lewis rats. Clin. Exp. Immunol. 2006, 146, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.F., Jr. Myasthenia gravis: The role of complement at the neuromuscular junction. Ann. N. Y. Acad. Sci. 2018, 1412, 113–128. [Google Scholar] [CrossRef]
- Botto, M.; Kirschfink, M.; Macor, P.; Pickering, M.C.; Würzner, R.; Tedesco, F. Complement in human diseases: Lessons from complement deficiencies. Mol. Immunol. 2009, 46, 2774–2783. [Google Scholar] [CrossRef]
- Figueroa, J.E.; Densen, P. Infectious diseases associated with complement deficiencies. Clin. Microbiol. Rev. 1991, 4, 359–395. [Google Scholar] [CrossRef]
- Sanghvi, S.Y.; Schwartz, R.A. Leiner’s disease (erythroderma desquamativum): A review and approach to therapy. Dermatol. Ther. 2021, 34, e14510. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, M.S.; Ballanti, E.; Triggianese, P.; Perricone, R. Vasculitides and the Complement System: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2015, 49, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Lung, T.; Sakem, B.; Risch, L.; Würzner, R.; Colucci, G.; Cerny, A.; Nydegger, U. The complement system in liver diseases: Evidence-based approach and therapeutic options. J. Transl. Autoimmun. 2019, 2, 100017. [Google Scholar] [CrossRef]
- Ballow, M.; Kennedy, T.L., 3rd; Gaudio, K.M.; Siegel, N.J.; McLean, R.H. Serum hemolytic factor D values in children with steroid-responsive idiopathic nephrotic syndrome. J. Pediatr. 1982, 100, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Boccuni, P.; Del Vecchio, L.; Di Noto, R.; Rotoli, B. Glycosyl phosphatidylinositol (GPI)-anchored molecules and the pathogenesis of paroxysmal nocturnal hemoglobinuria. Crit. Rev. Oncol. Hematol. 2000, 33, 25–43. [Google Scholar] [CrossRef]
- Mantegazza, R.; Cordiglieri, C.; Consonni, A.; Baggi, F. Animal models of myasthenia gravis: Utility and limitations. Int. J. Gen. Med. 2016, 9, 53–64. [Google Scholar] [CrossRef]
- Lindstrom, J. Experimental autoimmune myasthenia gravis. J. Neurol. Neurosurg. Psychiatry 1980, 43, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Link, H.; Xiao, B.-G. Rat models as tool to develop new immunotherapies. Immunol. Rev. 2001, 184, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Kusner, L.L.; Losen, M.; Vincent, A.; Lindstrom, J.; Tzartos, S.; Lazaridis, K.; Martinez-Martinez, P. Guidelines for pre-clinical assessment of the acetylcholine receptor–specific passive transfer myasthenia gravis model-Recommendations for methods and experimental designs. Exp. Neurol. 2015, 270, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Patrick, J.; Lindstrom, J. Autoimmune response to acetylcholine receptor. Science 1973, 180, 871–872. [Google Scholar] [CrossRef]
- Losen, M.; Martinez-Martinez, P.; Molenaar, P.C.; Lazaridis, K.; Tzartos, S.; Brenner, T.; Duan, R.S.; Luo, J.; Lindstrom, J.; Kusner, L. Standardization of the experimental autoimmune myasthenia gravis (EAMG) model by immunization of rats with Torpedo californica acetylcholine receptors–Recommendations for methods and experimental designs. Exp. Neurol. 2015, 270, 18–28. [Google Scholar] [CrossRef]
- Viegas, S.; Jacobson, L.; Waters, P.; Cossins, J.; Jacob, S.; Leite, M.I.; Webster, R.; Vincent, A. Passive and active immunization models of MuSK-Ab positive myasthenia: Electrophysiological evidence for pre and postsynaptic defects. Exp. Neurol. 2012, 234, 506–512. [Google Scholar] [CrossRef]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.R.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef]
- Duncan, A.R.; Winter, G. The binding site for C1q on IgG. Nature 1988, 332, 738–740. [Google Scholar] [CrossRef]
- Lubbers, R.; Oostindie, S.C.; Dijkstra, D.J.; Parren, P.W.H.I.; Verheul, M.K.; Abendstein, L.; Sharp, T.H.; de Ru, A.; Janssen, G.M.C.; van Veelen, P.A.; et al. Carbamylation reduces the capacity of IgG for hexamerization and complement activation. Clin. Exp. Immunol. 2020, 200, 1–11. [Google Scholar] [CrossRef]
- Oskam, N.; Damelang, T.; Streutker, M.; Ooijevaar-de Heer, P.; Nouta, J.; Koeleman, C.; Van Coillie, J.; Wuhrer, M.; Vidarsson, G.; Rispens, T. Factors affecting IgG4-mediated complement activation. Front. Immunol. 2023, 14, 1087532. [Google Scholar] [CrossRef]
- Dekkers, G.; Treffers, L.; Plomp, R.; Bentlage, A.E.H.; de Boer, M.; Koeleman, C.A.M.; Lissenberg-Thunnissen, S.N.; Visser, R.; Brouwer, M.; Mok, J.Y.; et al. Decoding the Human Immunoglobulin G-Glycan Repertoire Reveals a Spectrum of Fc-Receptor- and Complement-Mediated-Effector Activities. Front. Immunol. 2017, 8, 877. [Google Scholar] [CrossRef] [PubMed]
- Chinello, C.; de Haan, N.; Capitoli, G.; Trezzi, B.; Radice, A.; Pagani, L.; Criscuolo, L.; Signorini, S.; Galimberti, S.; Sinico, R.A.; et al. Definition of IgG Subclass-Specific Glycopatterns in Idiopathic Membranous Nephropathy: Aberrant IgG Glycoforms in Blood. Int. J. Mol. Sci. 2022, 23, 4664. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Wormald, M.R.; Rudd, P.M.; Fischer, P.B.; Dwek, R.A.; Sim, R.B. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat. Med. 1995, 1, 237–243. [Google Scholar] [CrossRef]
- Vakrakou, A.G.; Karachaliou, E.; Chroni, E.; Zouvelou, V.; Tzanetakos, D.; Salakou, S.; Papadopoulou, M.; Tzartos, S.; Voumvourakis, K.; Kilidireas, C.; et al. Immunotherapies in MuSK-positive Myasthenia Gravis; an IgG4 antibody-mediated disease. Front. Immunol. 2023, 14, 1212757. [Google Scholar] [CrossRef] [PubMed]
- Preston, D.; Shapiro, B. Electromyography and Neuromuscular Disorders: Clinical-Electrodiagnostic-Ultrasound Correlations, Fourth Edition. J. Clin. Neurophysiol. 2021, 38, e19. [Google Scholar] [CrossRef]
- Serra, A.; Ruff, R.L.; Leigh, R.J. Neuromuscular transmission failure in myasthenia gravis: Decrement of safety factor and susceptibility of extraocular muscles. Ann. N. Y. Acad. Sci. 2012, 1275, 129–135. [Google Scholar] [CrossRef]
- Ruff, R.L.; Lennon, V.A. End-plate voltage-gated sodium channels are lost in clinical and experimental myasthenia gravis. Ann. Neurol. 1998, 43, 370–379. [Google Scholar] [CrossRef]
- Ruff, R.L.; Lennon, V.A. How myasthenia gravis alters the safety factor for neuromuscular transmission. J. Neuroimmunol. 2008, 201–202, 13–20. [Google Scholar] [CrossRef]
- Vilquin, J.-T.; Bayer, A.C.; Le Panse, R.; Berrih-Aknin, S. The Muscle Is Not a Passive Target in Myasthenia Gravis. Front. Neurol. 2019, 10, 1343. [Google Scholar] [CrossRef]
- Rose, N.; Holdermann, S.; Callegari, I.; Kim, H.; Fruh, I.; Kappos, L.; Kuhle, J.; Müller, M.; Sanderson, N.S.R.; Derfuss, T. Receptor clustering and pathogenic complement activation in myasthenia gravis depend on synergy between antibodies with multiple subunit specificities. Acta Neuropathol. 2022, 144, 1005–1025. [Google Scholar] [CrossRef]
- Engel, A.G. Morphologic and immunopathologic findings in myasthenia gravis and in congenital myasthenic syndromes. J. Neurol. Neurosurg. Psychiatry 1980, 43, 577–589. [Google Scholar] [CrossRef]
- Nakano, S.; Engel, A.G. Myasthenia gravis: Quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the end-plate in 30 patients. Neurology 1993, 43, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Morsch, M.; Reddel, S.W.; Ghazanfari, N.; Toyka, K.V.; Phillips, W.D. Muscle specific kinase autoantibodies cause synaptic failure through progressive wastage of postsynaptic acetylcholine receptors. Exp. Neurol. 2012, 237, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Klooster, R.; Plomp, J.J.; Huijbers, M.G.; Niks, E.H.; Straasheijm, K.R.; Detmers, F.J.; Hermans, P.W.; Sleijpen, K.; Verrips, A.; Losen, M.; et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 2012, 135, 1081–1101. [Google Scholar] [CrossRef]
- Aguirre, F.; Manin, A.; Fernandez, V.C.; Justo, M.E.; Leoni, J.; Paz, M.L.; Villa, A.M. C3, C5a and anti-acetylcholine receptor antibody as severity biomarkers in myasthenia gravis. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420935697. [Google Scholar] [CrossRef]
- Stascheit, F.; Chuquisana, O.; Keller, C.W.; Ambrose, P.A.; Hoffmann, S.; Gross, C.C.; Lehnerer, S.; Wiendl, H.; Willcox, N.; Meisel, A.; et al. Complement activation profiles in anti-acetylcholine receptor positive myasthenia gravis. Eur. J. Neurol. 2023, 30, 1409–1416. [Google Scholar] [CrossRef]
- Huang, Y.F.; Sandholm, K.; Persson, B.; Nilsson, B.; Punga, A.R. Visualization and characterization of complement activation in acetylcholine receptor antibody seropositive myasthenia gravis. Muscle Nerve 2024, 70, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Nelke, C.; Schroeter, C.B.; Barman, S.; Stascheit, F.; Masanneck, L.; Theissen, L.; Huntemann, N.; Walli, S.; Cengiz, D.; Dobelmann, V.; et al. Identification of disease phenotypes in acetylcholine receptor-antibody myasthenia gravis using proteomics-based consensus clustering. eBioMedicine 2024, 105, 105231. [Google Scholar] [CrossRef]
- Romi, F.; Skeie, G.O.; Vedeler, C.; Aarli, J.A.; Zorzato, F.; Gilhus, N.E. Complement activation by titin and ryanodine receptor autoantibodies in myasthenia gravis. A study of IgG subclasses and clinical correlations. J. Neuroimmunol. 2000, 111, 169–176. [Google Scholar] [CrossRef]
- Tong, T.; Zhang, J.; Jia, L.; Liang, P.; Wang, N. Integrated proteomics and metabolomics analysis reveals hubs protein and network alterations in myasthenia gravis. Aging 2022, 14, 5417–5426. [Google Scholar] [CrossRef]
- Jin, L.; He, D.; Zeng, Q.; Tan, S.; Shi, J.; Liu, Y.; Zou, Z.; Song, J.; Yan, C.; Huan, X.; et al. Eculizumab in thymoma-associated myasthenia gravis: A real-world cohort study. Ther. Adv. Neurol. Disord. 2024, 17, 17562864241309431. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, Y.; Wang, Z.; Shi, Y.; Chen, Y.; Chen, Y. The impact of immune markers on thymectomy prognosis in thymoma-myasthenia gravis. J. Thorac. Dis. 2024, 16, 6634–6643. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Peng, Y.; Yang, H. Serological diagnosis of myasthenia gravis and its clinical significance. Ann. Transl. Med. 2023, 11, 290. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Li, H.; Guo, S. Eculizumab treatment for myasthenia gravis subgroups: 2021 update. J. Neuroimmunol. 2022, 362, 577767. [Google Scholar] [CrossRef]
- Thomas, T.C.; Rollins, S.A.; Rother, R.P.; Giannoni, M.A.; Hartman, S.L.; Elliott, E.A.; Nye, S.H.; Matis, L.A.; Squinto, S.P.; Evans, M.J. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol. Immunol. 1996, 33, 1389–1401. [Google Scholar] [CrossRef]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef]
- Howard, J.F., Jr.; Utsugisawa, K.; Benatar, M.; Murai, H.; Barohn, R.J.; Illa, I.; Jacob, S.; Vissing, J.; Burns, T.M.; Kissel, J.T.; et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): A phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017, 16, 976–986. [Google Scholar] [CrossRef]
- Dhillon, S. Eculizumab: A Review in Generalized Myasthenia Gravis. Drugs 2018, 78, 367–376. [Google Scholar] [CrossRef]
- Suh, J.; Clarke, V.; Amato, A.A.; Guidon, A.C. Safety and outcomes of eculizumab for acetylcholine receptor-positive generalized myasthenia gravis in clinical practice. Muscle Nerve 2022, 66, 348–353. [Google Scholar] [CrossRef]
- Lathia, C.; Gao, X.; Kassir, N.; Mouksassi, S.; Jayaraman, B.; Marier, J.F.; Wang, J.; Bedrosian, C.L. Population pharmacokinetic and pharmacodynamic analysis of eculizumab to support phase 3 dosing regimen in patients with refractory generalized myasthenia gravis. In Clinical Pharmacology & Therapeutics; Wiley-Blackwell: Hoboken, NJ, USA, 2015. [Google Scholar]
- Monteleone, J.P.R.; Gao, X.; Kleijn, H.J.; Bellanti, F.; Pelto, R. Eculizumab Pharmacokinetics and Pharmacodynamics in Patients with Generalized Myasthenia Gravis. Front. Neurol. 2021, 12, 696385. [Google Scholar] [CrossRef]
- Murai, H.; Uzawa, A.; Suzuki, Y.; Imai, T.; Shiraishi, H.; Suzuki, H.; Okumura, M.; O’Brien, F.; Wang, J.J.; Fujita, K.P.; et al. Long-term efficacy and safety of eculizumab in Japanese patients with generalized myasthenia gravis: A subgroup analysis of the REGAIN open-label extension study. J. Neurol. Sci. 2019, 407, 116419. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kulasekararaj, A.G. Ravulizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Expert. Opin. Biol. Ther. 2020, 20, 227–237. [Google Scholar] [CrossRef]
- Sheridan, D.; Yu, Z.X.; Zhang, Y.; Patel, R.; Sun, F.; Lasaro, M.A.; Bouchard, K.; Andrien, B.; Marozsan, A.; Wang, Y.; et al. Design and preclinical characterization of ALXN1210: A novel anti-C5 antibody with extended duration of action. PLoS ONE 2018, 13, e0195909. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Wiendl, H.; Katsuno, M.; Reddel, S.W.; Howard, J.F., Jr. Ravulizumab in Myasthenia Gravis: A Review of the Current Evidence. Neuropsychiatr. Dis. Treat. 2023, 19, 2639–2655. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Ortiz, S.; Katsuno, M.; Annane, D.; Mantegazza, R.; Beasley, K.N.; Aguzzi, R.; Howard, J.F., Jr. Ravulizumab pharmacokinetics and pharmacodynamics in patients with generalized myasthenia gravis. J. Neurol. 2023, 270, 3129–3137. [Google Scholar] [CrossRef]
- Vu, T.; Meisel, A.; Mantegazza, R.; Annane, D.; Katsuno, M.; Aguzzi, R.; Enayetallah, A.; Beasley, K.N.; Rampal, N.; Howard, J.F. Terminal Complement Inhibitor Ravulizumab in Generalized Myasthenia Gravis. NEJM Evid. 2022, 1, EVIDoa2100066. [Google Scholar] [CrossRef]
- Meisel, A.; Annane, D.; Vu, T.; Mantegazza, R.; Katsuno, M.; Aguzzi, R.; Frick, G.; Gault, L.; Howard, J.F., Jr. Long-term efficacy and safety of ravulizumab in adults with anti-acetylcholine receptor antibody-positive generalized myasthenia gravis: Results from the phase 3 CHAMPION MG open-label extension. J. Neurol. 2023, 270, 3862–3875. [Google Scholar] [CrossRef]
- Howard, J.F., Jr.; Vu, T.; Mantegazza, R.; Kushlaf, H.; Suzuki, S.; Wiendl, H.; Beasley, K.N.; Liao, S.; Meisel, A.; The CHAMPION MG Study Group. Efficacy of ravulizumab in patients with generalized myasthenia gravis by time from diagnosis: A post hoc subgroup analysis of the CHAMPION MG study. Muscle Nerve 2024, 69, 556–565. [Google Scholar] [CrossRef]
- Zadeh, S.; Price, H.; Drews, R.; Bouffard, M.A.; Young, L.H.; Narayanaswami, P. Novel uses of complement inhibitors in myasthenia gravis-Two case reports. Muscle Nerve 2024, 69, 368–372. [Google Scholar] [CrossRef]
- Howard, J.F., Jr.; Vissing, J.; Gilhus, N.E.; Leite, M.I.; Utsugisawa, K.; Duda, P.W.; Farzaneh-Far, R.; Murai, H.; Wiendl, H. Zilucoplan: An Investigational Complement C5 Inhibitor for the Treatment of Acetylcholine Receptor Autoantibody-Positive Generalized Myasthenia Gravis. Expert. Opin. Investig. Drugs 2021, 30, 483–493. [Google Scholar] [CrossRef]
- Howard, J.F., Jr.; Bresch, S.; Genge, A.; Hewamadduma, C.; Hinton, J.; Hussain, Y.; Juntas-Morales, R.; Kaminski, H.J.; Maniaol, A.; Mantegazza, R.; et al. Safety and efficacy of zilucoplan in patients with generalised myasthenia gravis (RAISE): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Neurol. 2023, 22, 395–406. [Google Scholar] [CrossRef]
- Howard, J.F., Jr.; Bresch, S.; Farmakidis, C.; Freimer, M.; Genge, A.; Hewamadduma, C.; Hinton, J.; Hussain, Y.; Juntas-Morales, R.; Kaminski, H.J.; et al. Long-term safety and efficacy of zilucoplan in patients with generalized myasthenia gravis: Interim analysis of the RAISE-XT open-label extension study. Ther. Adv. Neurol. Disord. 2024, 17, 17562864241243186. [Google Scholar] [CrossRef]
- Weiss, M.D.; Freimer, M.; Leite, M.I.; Maniaol, A.; Utsugisawa, K.; Bloemers, J.; Boroojerdi, B.; Howard, E.; Savic, N.; Howard, J.F., Jr. Improvement of fatigue in generalised myasthenia gravis with zilucoplan. J. Neurol. 2024, 271, 2758–2767. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Nie, X.; Zhu, G.; Qi, W.; Hao, L.; Guo, X. The Efficacy and Safety of Different Targeted Drugs for the Treatment of Generalized Myasthenia Gravis: A Systematic Review and Bayesian Network Meta-analysis. CNS Drugs 2024, 38, 93–104. [Google Scholar] [CrossRef]
- Smith, A.G.; Wolfe, G.I.; Habib, A.A.; Qi, C.Z.; Yang, H.; Du, M.; Chen, X.; Gelinas, D.; Brauer, E.; Phillips, G.; et al. Risk-Benefit Analysis of Novel Treatments for Patients with Generalized Myasthenia Gravis. Adv. Ther. 2024, 41, 4628–4647. [Google Scholar] [CrossRef]
- Saccà, F.; Pane, C.; Espinosa, P.E.; Sormani, M.P.; Signori, A. Efficacy of innovative therapies in myasthenia gravis: A systematic review, meta-analysis and network meta-analysis. Eur. J. Neurol. 2023, 30, 3854–3867. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.; Liew, A.; Yeo, S.; Fernström, A.; Barbour, S.; Sperati, C.; Villanueva, R.; Wu, M.-J.; Wang, D.; Borodovsky, A.; et al. Phase 2 Trial of Cemdisiran in Adult Patients with IgA Nephropathy: A Randomized Controlled Trial. Clin. J. Am. Soc. Nephrol. CJASN 2024, 19, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Kusner, L.L.; Yucius, K.; Sengupta, M.; Sprague, A.G.; Desai, D.; Nguyen, T.; Charisse, K.; Kuchimanchi, S.; Kallanthottathil, R.; Fitzgerald, K.; et al. Investigational RNAi Therapeutic Targeting C5 Is Efficacious in Pre-clinical Models of Myasthenia Gravis. Mol. Ther. Methods Clin. Dev. 2019, 13, 484–492. [Google Scholar] [CrossRef]
- Devalaraja-Narashimha, K.; Huang, C.; Cao, M.; Chen, Y.P.; Borodovsky, A.; Olson, W.C.; Morton, L.G.; Retter, M.W. Pharmacokinetics and pharmacodynamics of pozelimab alone or in combination with cemdisiran in non-human primates. PLoS ONE 2022, 17, e0269749. [Google Scholar] [CrossRef]
- Jindal, S.; Pedersen, D.V.; Gera, N.; Chandler, J.; Patel, R.; Neill, A.; Cone, J.; Zhang, Y.; Yuan, C.X.; Millman, E.E.; et al. Characterization of the bispecific VHH antibody gefurulimab (ALXN1720) targeting complement component 5, and designed for low volume subcutaneous administration. Mol. Immunol. 2024, 165, 29–41. [Google Scholar] [CrossRef]
- Lin, K.; Zhang, L.; Kong, M.; Yang, M.; Chen, Y.; Poptic, E.; Hoffner, M.; Xu, J.; Tam, C.; Lin, F. Development of an anti-human complement C6 monoclonal antibody that inhibits the assembly of membrane attack complexes. Blood Adv. 2020, 4, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Lekova, E.; Zelek, W.M.; Gower, D.; Spitzfaden, C.; Osuch, I.H.; John-Morris, E.; Stach, L.; Gormley, D.; Sanderson, A.; Bridges, A.; et al. Discovery of functionally distinct anti-C7 monoclonal antibodies and stratification of anti-nicotinic AChR positive Myasthenia Gravis patients. Front. Immunol. 2022, 13, 968206. [Google Scholar] [CrossRef] [PubMed]
- Würzner, R.; Orren, A.; Potter, P.; Morgan, B.P.; Ponard, D.; Späth, P.; Brai, M.; Schulze, M.; Happe, L.; Götze, O. Functionally active complement proteins C6 and C7 detected in C6- and C7-deficient individuals. Clin. Exp. Immunol. 1991, 83, 430–437. [Google Scholar] [CrossRef]
- Harder, M.J.; Kuhn, N.; Schrezenmeier, H.; Höchsmann, B.; von Zabern, I.; Weinstock, C.; Simmet, T.; Ricklin, D.; Lambris, J.D.; Skerra, A.; et al. Incomplete inhibition by eculizumab: Mechanistic evidence for residual C5 activity during strong complement activation. Blood 2017, 129, 970–980. [Google Scholar] [CrossRef]
- Würzner, R.; Schulze, M.; Happe, L.; Franzke, A.; Bieber, F.A.; Oppermann, M.; Götze, O. Inhibition of terminal complement complex formation and cell lysis by monoclonal antibodies. Complement. Inflamm. 1991, 8, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Jacob, S. Novel Immunotherapies for Myasthenia Gravis. Immunotargets Ther. 2023, 12, 25–45. [Google Scholar] [CrossRef]
- Nishimura, J.-i.; Yamamoto, M.; Hayashi, S.; Ohyashiki, K.; Ando, K.; Brodsky, A.L.; Noji, H.; Kitamura, K.; Eto, T.; Takahashi, T.; et al. Genetic Variants in C5 and Poor Response to Eculizumab. N. Engl. J. Med. 2014, 370, 632–639. [Google Scholar] [CrossRef]
- Kulasekararaj, A.; Griffin, M.; Piatek, C.; Shammo, J.; Nishimura, J.-i.; Patriquin, C.; Schrezenmeier, H.; Barcellini, W.; Panse, J.; Gaya, A.; et al. Long-term efficacy and safety of danicopan as add-on therapy to ravulizumab or eculizumab in PNH with significant EVH. Blood 2025, 145, 811–822. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Medication | Mechanism of Action | Targeted pathway | Trials | Evidence |
|---|---|---|---|---|
| Eculizumab | C5 inhibitor, requires arginine in position 855 | Common pathway | REGAIN (Phase 3) | AChR+ with generalized non thymoma associated MG, improve MG ADL scores at 12 weeks, sustained up to 130 weeks |
| Ravulizumab | C5 inhibitor, requires arginine in position 855 | Common pathway | CHAMPION-MG (Phase 3) | AChR+ with generalized non thymoma associated MG, improved MG ADL scores at 12 weeks, sustained up to 60 weeks |
| Zilucoplan | C5 inhibitor, requires arginine in position 855 | Common pathway | RAISE III (Phase 3) | AChR+ with generalized non thymoma associated MG, improved MG ADL scores at 12 weeks, sustained up to 60 weeks |
| Cemdisiran- pzelimab | siRNA targeting synthesis of C5, C5 inhibitor targeting both wild type and mutant R855H | Common pathway | NIMBLE (Phase 3, in progress) | AChR+ and LRP subgroups being studied |
| Gefurulimab | C5 inhibitor targeting both wild type and mutatnt R855H | Common pathway | PREVAIL (Phase 3, in progress) | AChR+ with generalized MG subgroup being studied |
| Vemircopan | Factor D inhibitor | Alternative pathway | NCT05218096 (phase 2 trial in progress) | AChR+ with generalized MG subgroup being studied |
| C6 inhibitors | Inhibition of C6 | Common pathway | Not in trials | N/A |
| C7 inhibitors | Inhibition of C7 | Common pathway | Not in trials | N/A |
| Factor B Inhibitors (IONIS-FB-L-Rx) | Factor B inhibition, antisense oligonucleotide | Alternative pathway | Phase 3 trials for IgA Nephropathy in progress | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez Salazar, A.; Mokhtari, S.; Peguero, E.; Jaffer, M. The Role of Complement in the Pathogenesis and Treatment of Myasthenia Gravis. Cells 2025, 14, 739. https://doi.org/10.3390/cells14100739
Martinez Salazar A, Mokhtari S, Peguero E, Jaffer M. The Role of Complement in the Pathogenesis and Treatment of Myasthenia Gravis. Cells. 2025; 14(10):739. https://doi.org/10.3390/cells14100739
Chicago/Turabian StyleMartinez Salazar, Armando, Sepideh Mokhtari, Edwin Peguero, and Muhammad Jaffer. 2025. "The Role of Complement in the Pathogenesis and Treatment of Myasthenia Gravis" Cells 14, no. 10: 739. https://doi.org/10.3390/cells14100739
APA StyleMartinez Salazar, A., Mokhtari, S., Peguero, E., & Jaffer, M. (2025). The Role of Complement in the Pathogenesis and Treatment of Myasthenia Gravis. Cells, 14(10), 739. https://doi.org/10.3390/cells14100739