
Fecal Supernatants from Patients with Crohn’s Disease Induce Inflammatory Alterations in M2 Macrophages and Fibroblasts

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects and Ethical Statement
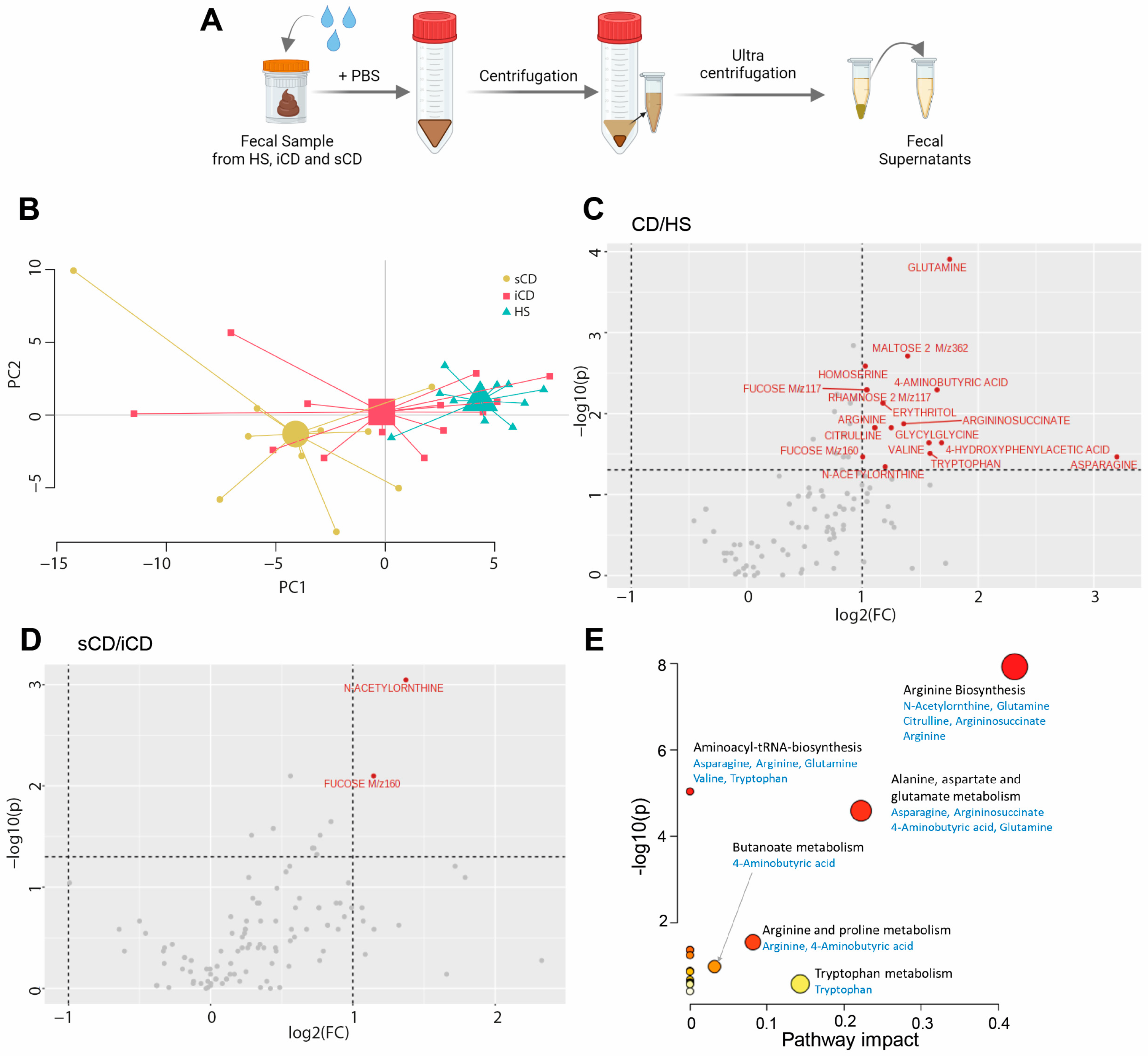
2.2. Preparation of Fecal Supernatants
2.3. Metabolomic Analysis of Fecal Supernatants
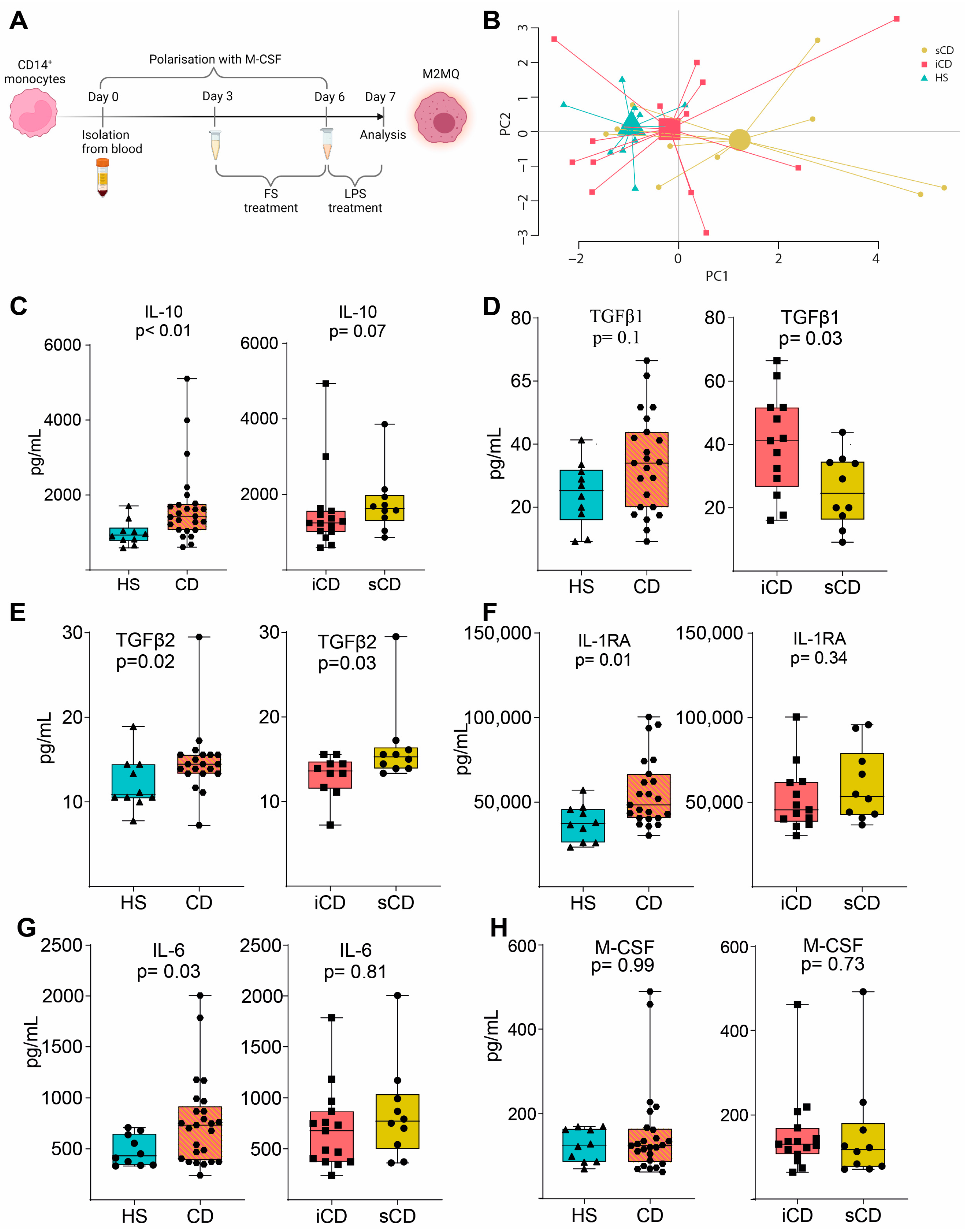
2.4. Peripheral Blood CD14+ Monocyte Isolation
2.5. M2MQ Polarization and FS Conditioning
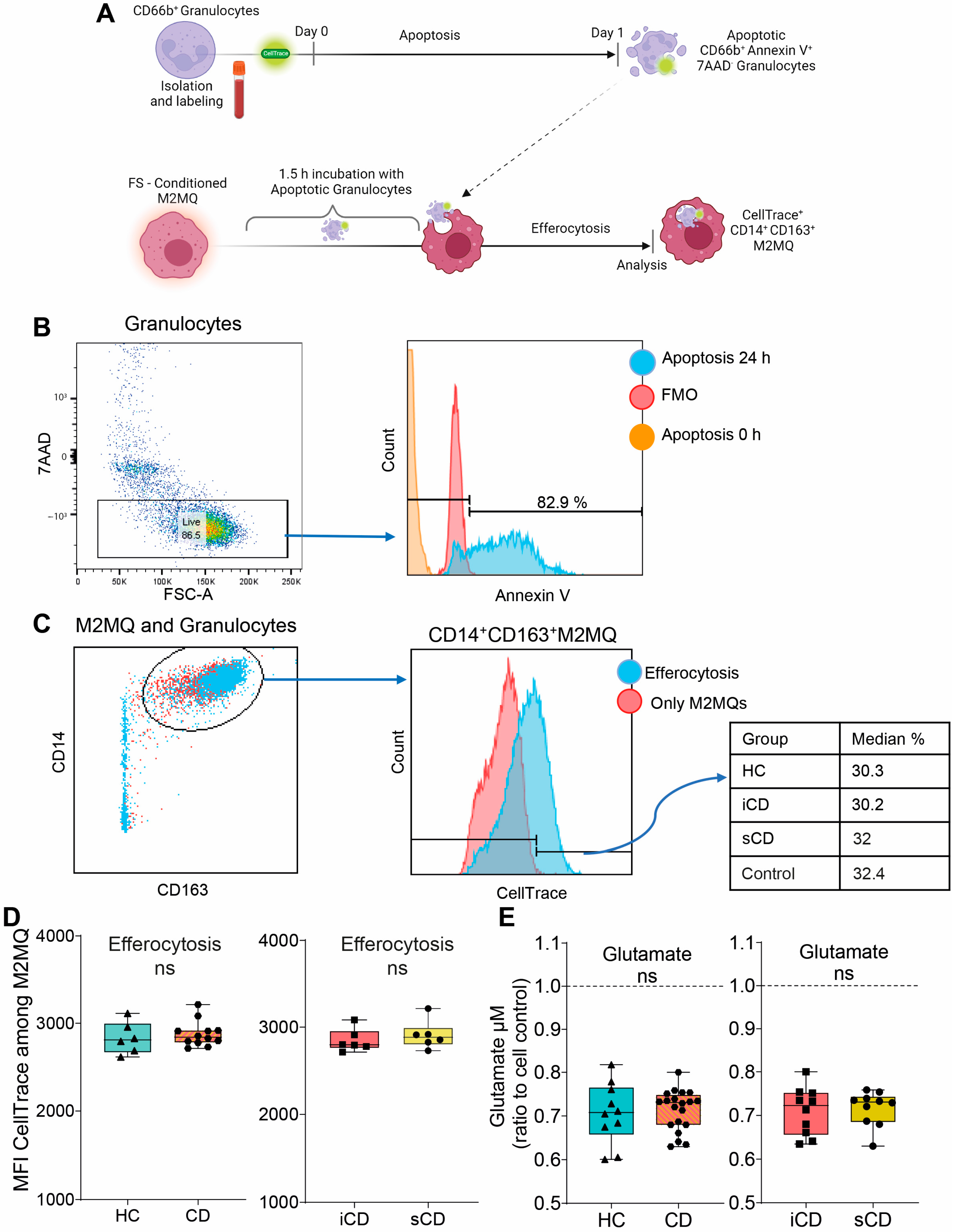
2.6. Peripheral Blood CD66b+ Granulocyte Isolation
2.7. Efferocytosis Assay
2.8. Glutamate Assay for Evaluation of Metabolism
2.9. Primary Fibroblast Cell Line Culture
2.10. Fibroblast Assays with Fecal Supernatants or M2MQ Conditioned Media
2.11. Fibrosis Gene Expression Array
2.12. Protein Analyses
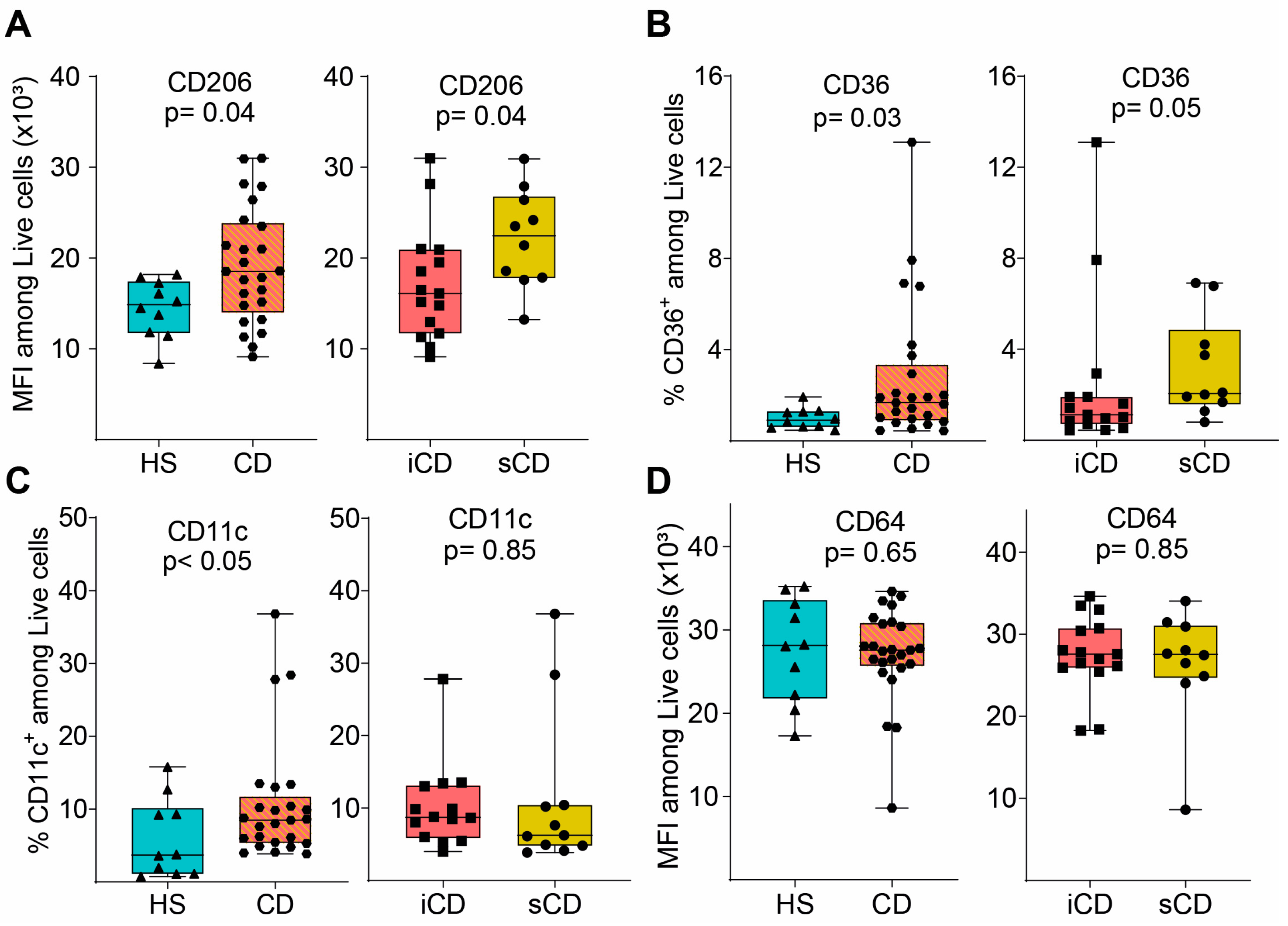
2.13. Flow Cytometry Analyses
2.14. Biostatistics and Data Analysis
3. Results
3.1. Patients with CD Have a Skewed Profile of Fecal-Derived Metabolites
3.2. Fecal Supernatants from Patients with CD Condition Macrophages to Express a Distinct anti-Inflammatory Protein Profile upon LPS Stimulation
3.3. Efferocytotic Function of Macrophages Is Unaltered after Exposure to Fecal Supernatants
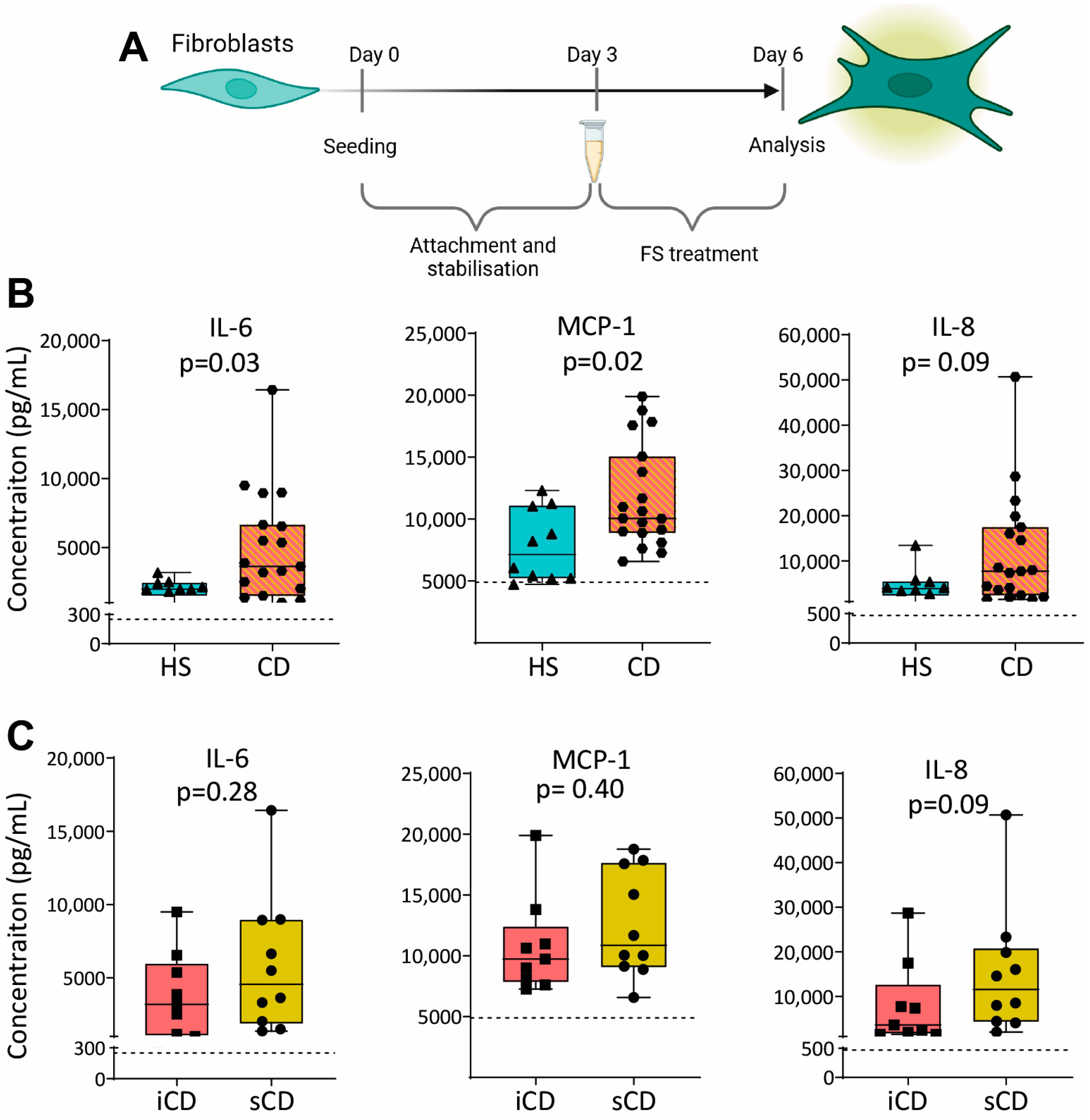
3.4. Fecal Supernatants from Patients with CD Induce Pro-Inflammatory Protein Expression in Fibroblasts
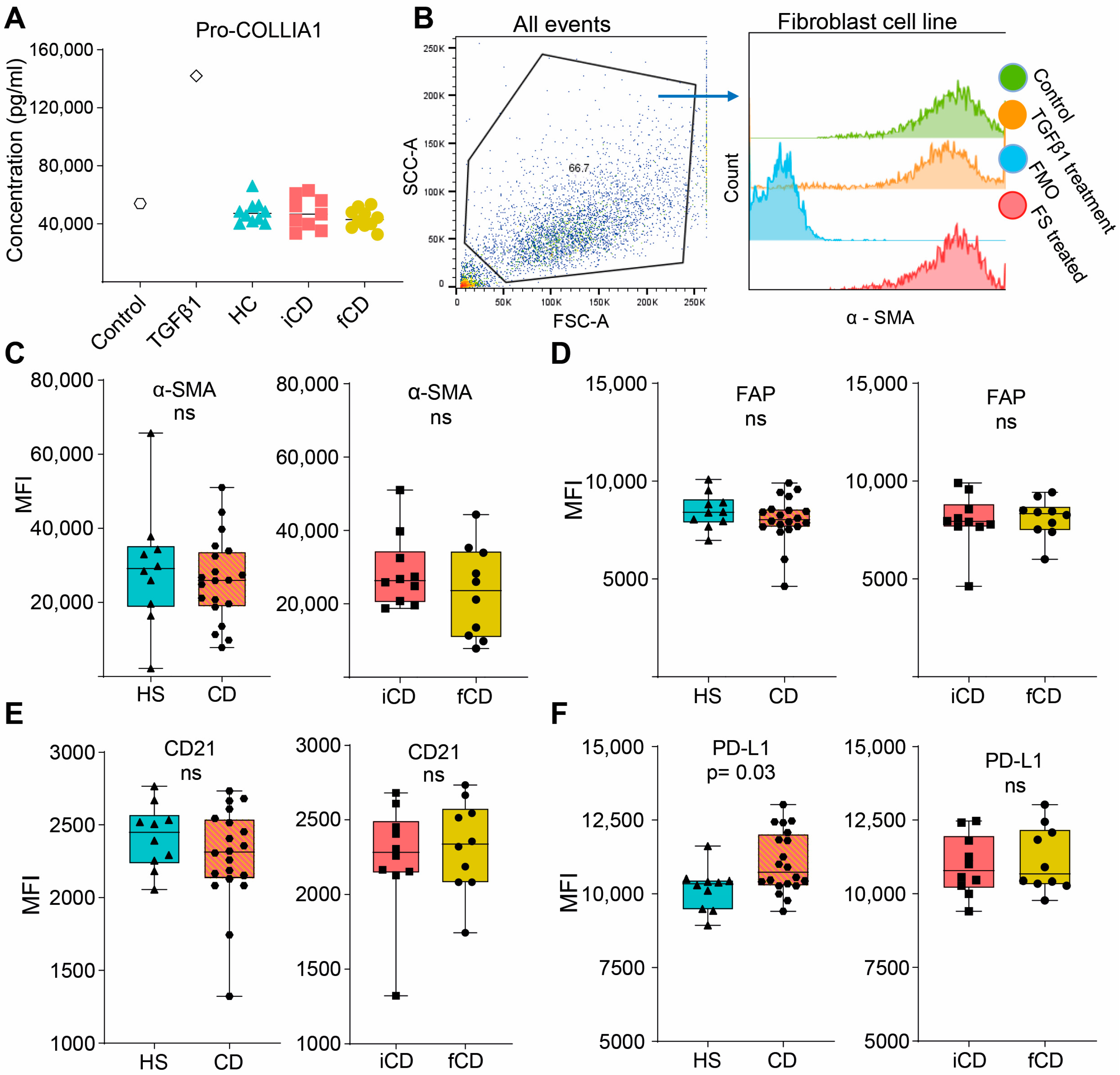
3.5. Fecal Supernatants Do Not Induce Fibrosis-Related Alterations in Fibroblasts
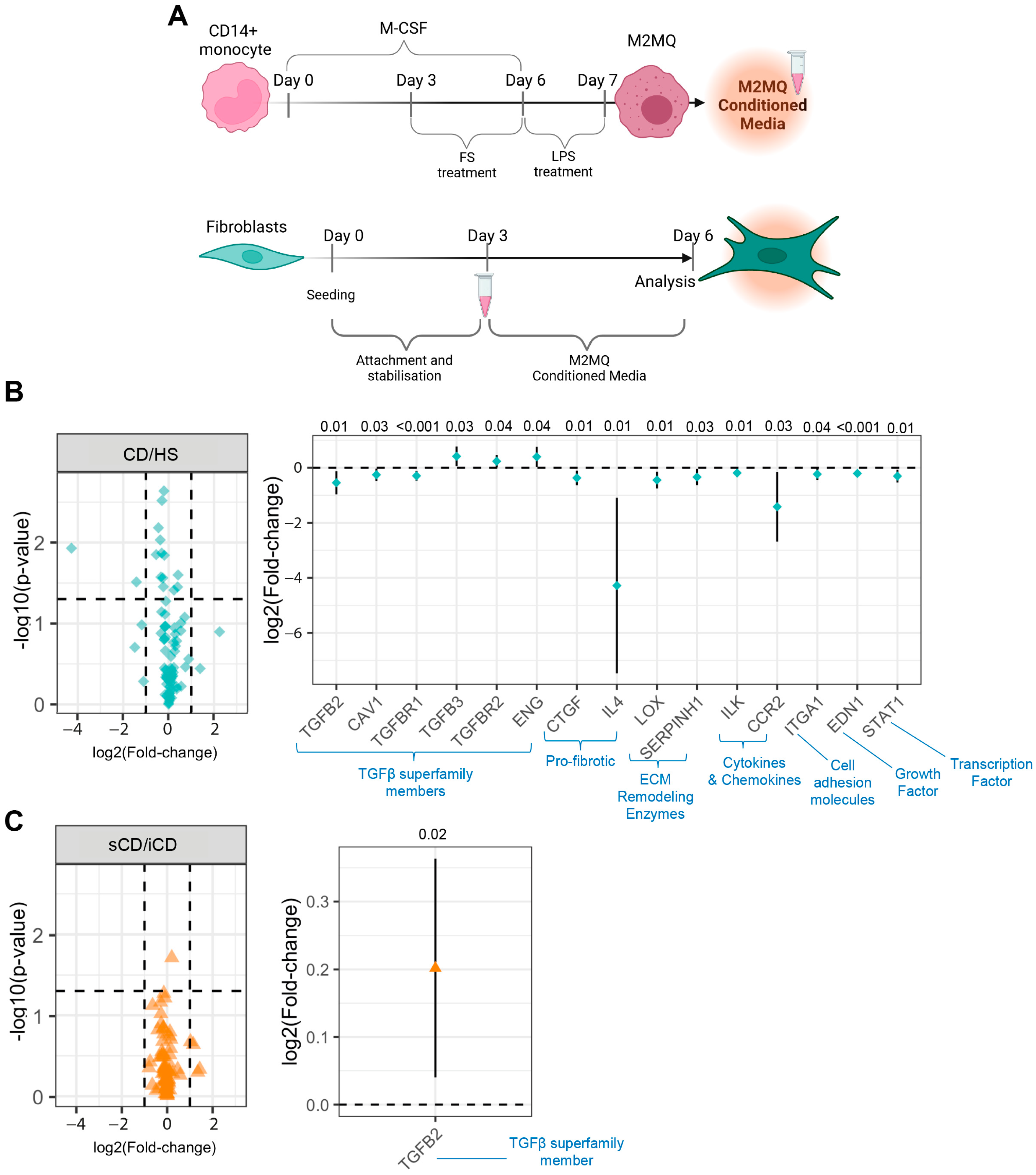
3.6. Conditioned Media from M2MQs Treated with FS from Patients with CD Induce Alterations of TGFβ Superfamily-Related Genes and IL-4 in Fibroblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burisch, J.; Kiudelis, G.; Kupcinskas, L.; Kievit, H.A.L.; Andersen, K.W.; Andersen, V.; Salupere, R.; Pedersen, N.; Kjeldsen, J.; D’incà, R.; et al. Natural disease course of Crohn’s disease during the first 5 years after diagnosis in a European population-based inception cohort: An Epi-IBD study. Gut 2019, 68, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.; Catesson, A.; Griffin, J.L.; Holmes, E.; Williams, H.R.T. Metabolomic Analysis in Inflammatory Bowel Disease: A Systematic Review. J. Crohn’s Colitis 2021, 15, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Galazzo, G.; I Tedjo, D.; Wintjens, D.S.J.; Savelkoul, P.H.M.; Masclee, A.A.M.; Bodelier, A.G.L.; Pierik, M.J.; A E Jonkers, D.M.; Penders, J. Faecal Microbiota Dynamics and their Relation to Disease Course in Crohn’s Disease. J. Crohn’s Colitis 2019, 13, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Strömbeck, A.; Lasson, A.; Strid, H.; Sundin, J.; Stotzer, P.-O.; Simrén, M.; Magnusson, M.K.; Öhman, L. Fecal microbiota composition is linked to the postoperative disease course in patients with Crohn’s disease. BMC Gastroenterol. 2020, 20, 130. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. Immune regulation by microbiome metabolites. Immunology 2018, 154, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Gorreja, F.; Caër, C.; Rush, S.T.A.; Forsskål, S.K.; Härtlova, A.; Magnusson, M.K.; Lindskog, E.B.; Börjesson, L.G.; Block, M.; Wick, M.J. MEFV and NLRP3 Inflammasome Expression Is Attributed to Immature Macrophages and Correlates with Serum Inflammatory Proteins in Crohn´s Disease Patients. Inflammation 2022, 45, 1631–1650. [Google Scholar] [CrossRef]
- Chapuy, L.; Bsat, M.; Sarkizova, S.; Rubio, M.; Therrien, A.; Wassef, E.; Bouin, M.; Orlicka, K.; Weber, A.; Hacohen, N.; et al. Two distinct colonic CD14+ subsets characterized by single-cell RNA profiling in Crohn’s disease. Mucosal Immunol. 2019, 12, 703–719. [Google Scholar] [CrossRef]
- Dharmasiri, S.; Garrido-Martin, E.M.; Harris, R.J.; Bateman, A.C.; E Collins, J.; Cummings, J.R.F.; Sanchez-Elsner, T. Human Intestinal Macrophages Are Involved in the Pathology of Both Ulcerative Colitis and Crohn Disease. Inflamm. Bowel Dis. 2021, 27, 1641–1652. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Zidar, N.; Langner, C.; Jerala, M.; Boštjančič, E.; Drobne, D.; Tomažič, A. Pathology of Fibrosis in Crohn’s Disease—Contribution to Understanding Its Pathogenesis. Front. Med. 2020, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Alam, M.M.; Zhao, X.-F.; Liao, Y.; Shen, J.; Morgan, S.; Huang, T.; Lee, H.; Lee, E.; Huang, Y.; et al. Induction of autophagy in Cx3cr1+ mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis. Mucosal Immunol. 2019, 12, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Hata, K.; Araki, Y.; Fujiyama, Y.; Bamba, T. Interleukin (IL)-4 and IL-17 synergistically stimulate IL-6 secretion in human colonic myofibroblasts. Int. J. Mol. Med. 2002, 10, 631–634. [Google Scholar] [PubMed]
- Friedrich, M.; Pohin, M.; Jackson, M.A.; Korsunsky, I.; Bullers, S.J.; Rue-Albrecht, K.; Christoforidou, Z.; Sathananthan, D.; Thomas, T.; Ravindran, R.; et al. IL-1-driven stromal–neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat. Med. 2021, 27, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Lawrance, I.C.; Maxwell, L.; Doe, W. Inflammation location, but not type, determines the increase in TGF-beta1 and IGF-1 expression and collagen deposition in IBD intestine. Inflamm. Bowel Dis. 2001, 7, 16–26. [Google Scholar] [CrossRef]
- Bendix, M.; Dige, A.; Jørgensen, S.P.; Dahlerup, J.F.; Bibby, B.M.; Deleuran, B.; Agnholt, J. Decrease in Mucosal IL17A, IFNγ and IL10 Expressions in Active Crohn’s Disease Patients Treated with High-Dose Vitamin D Alone or Combined with Infliximab. Nutrients 2020, 12, 3699. [Google Scholar] [CrossRef]
- Bak, N.F.; Bendix, M.; Hald, S.; Reinert, L.; Magnusson, M.K.; Agnholt, J. High-dose vitamin D3 supplementation decreases the number of colonic CD103+ dendritic cells in healthy subjects. Eur. J. Nutr. 2018, 57, 2607–2619. [Google Scholar] [CrossRef]
- Maasfeh, L.; Härtlova, A.; Isaksson, S.; Sundin, J.; Mavroudis, G.; Savolainen, O.; Strid, H.; Öhman, L.; Magnusson, M.K. Impaired Luminal Control of Intestinal Macrophage Maturation in Patients With Ulcerative Colitis During Remission. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1415–1432. [Google Scholar] [CrossRef]
- Savolainen, O.I.; Sandberg, A.-S.; Ross, A.B. A Simultaneous Metabolic Profiling and Quantitative Multimetabolite Metabolomic Method for Human Plasma Using Gas-Chromatography Tandem Mass Spectrometry. J. Proteome Res. 2016, 15, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Jiye, A.; Trygg, J.; Gullberg, J.; Johansson, A.I.; Jonsson, P.; Antti, H.; Marklund, S.L.; Moritz, T. Extraction and GC/MS analysis of the human blood plasma metabolome. Anal. Chem. 2005, 77, 8086–8094. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 12 September 2023).
- Pang, Z.; Zhou, G.; Ewald, J.; Chang, L.; Hacariz, O.; Basu, N.; Xia, J. Using MetaboAnalyst 5.0 for LC–HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. 2022, 17, 1735–1761. [Google Scholar] [CrossRef]
- Lenth, R. Emmeans: Estimated Marginal Means, Aka Least-Squares Means. R Package Version 1.4.1. 2019. Available online: https://CRAN.R-project.org/package=emmeans (accessed on 12 September 2023).
- Vu, V.Q. Ggbiplot: A Ggplot2 Based Biplot. R Package Version 0.55. 2016. Available online: http://github.com/vqv/ggbiplot (accessed on 12 September 2023).
- Meelu, P.; Marin, R.; Clish, C.; Zella, G.; Cox, S.; Yajnik, V.; Nguyen, D.D.; Korzenik, J.R. Impaired innate immune function associated with fecal supernatant from crohn’s disease patients. Inflamm. Bowel Dis. 2014, 20, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Scoville, E.A.; Allaman, M.M.; Brown, C.T.; Motley, A.K.; Horst, S.N.; Williams, C.S.; Koyama, T.; Zhao, Z.; Adams, D.W.; Beaulieu, D.B.; et al. Alterations in Lipid, Amino Acid, and Energy Metabolism Distinguish Crohn’s Disease from Ulcerative Colitis and Control Subjects by Serum Metabolomic Profiling. Metabolomics 2018, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mañé, J.; Fernández-Bañares, F.; Ojanguren, I.; Castellá, E.; Bertrán, X.; Bartolí, R.; Alvarez, M.; Gassull, M. Effect of L-arginine on the course of experimental colitis. Clin. Nutr. 2001, 20, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Guo, Y.-C.; Zhou, H.-F.; Yue, T.-T.; Wang, F.-X.; Sun, F.; Wang, W.-Z. Arginine metabolism regulates the pathogenesis of inflammatory bowel disease. Nutr. Rev. 2023, 81, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Gomes, R.G.; de Brito, C.A.A.; Martinelli, V.F.; dos Santos, R.N.; Gomes, F.O.d.S.; Peixoto, C.A.; Crispim, J.O.; Diniz, G.T.N.; Donadi, E.A.; Lucena-Silva, N. HLA-G is expressed in intestinal samples of ulcerative colitis and Crohn’s disease patients and HLA-G5 expression is differentially correlated with TNF and IL-10 cytokine expression. Hum. Immunol. 2018, 79, 477–484. [Google Scholar] [CrossRef]
- Quiros, M.; Nishio, H.; Neumann, P.A.; Siuda, D.; Brazil, J.C.; Azcutia, V.; Hilgarth, R.; O’leary, M.N.; Garcia-Hernandez, V.; Leoni, G.; et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J. Clin. Investig. 2017, 127, 3510–3520. [Google Scholar] [CrossRef]
- Wright, P.B.; McDonald, E.; Bravo-Blas, A.; Baer, H.M.; Heawood, A.; Bain, C.C.; Mowat, A.M.; Clay, S.L.; Robertson, E.V.; Morton, F.; et al. The mannose receptor (CD206) identifies a population of colonic macrophages in health and inflammatory bowel disease. Sci. Rep. 2021, 11, 19616. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, D.; Marin, A.C.; Fernández-Tomé, S.; Montalban-Arques, A.; Carrasco, A.; Tristán, E.; Ortega-Moreno, L.; Mora-Gutiérrez, I.; Díaz-Guerra, A.; Caminero-Fernández, R.; et al. Human intestinal pro-inflammatory CD11chighCCR2+CX3CR1+ macrophages, but not their tolerogenic CD11c−CCR2−CX3CR1− counterparts, are expanded in inflammatory bowel disease. Mucosal Immunol. 2018, 11, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Michlewska, S.; Dransfield, I.; Megson, I.L.; Rossi, A.G. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: Key role for TNF-α. FASEB J. 2009, 23, 844–854. [Google Scholar] [CrossRef]
- Greenberg, M.E.; Sun, M.; Zhang, R.; Febbraio, M.; Silverstein, R.; Hazen, S.L. Oxidized phosphatidylserine–CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 2006, 203, 2613–2625. [Google Scholar] [CrossRef]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.-S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508.e20. [Google Scholar] [CrossRef]
- Catarzi, S.; Favilli, F.; Romagnoli, C.; Marcucci, T.; Picariello, L.; Tonelli, F.; Vincenzini, M.T.; Iantomasi, T. Oxidative state and IL-6 production in intestinal myofibroblasts of Crohn’s disease patients. Inflamm. Bowel Dis. 2011, 17, 1674–1684. [Google Scholar] [CrossRef]
- Acosta-Rodriguez, E.V.; Napolitani, G.; Lanzavecchia, A.; Sallusto, F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17–producing human T helper cells. Nat. Immunol. 2007, 8, 942–949. [Google Scholar] [CrossRef]
- Truffi, M.; Sorrentino, L.; Monieri, M.; Fociani, P.; Mazzucchelli, S.; Bonzini, M.; Zerbi, P.; Sampietro, G.M.; Di Sabatino, A.; Corsi, F. Inhibition of Fibroblast Activation Protein Restores a Balanced Extracellular Matrix and Reduces Fibrosis in Crohn’s Disease Strictures Ex Vivo. Inflamm. Bowel Dis. 2018, 24, 332–345. [Google Scholar] [CrossRef]
- Scharl, M.; Huber, N.; Lang, S.; Fürst, A.; Jehle, E.; Rogler, G. Hallmarks of epithelial to mesenchymal transition are detectable in Crohn’s disease associated intestinal fibrosis. Clin. Transl. Med. 2015, 4, 1. [Google Scholar] [CrossRef]
- Sato, Y.; Mii, A.; Hamazaki, Y.; Fujita, H.; Nakata, H.; Masuda, K.; Nishiyama, S.; Shibuya, S.; Haga, H.; Ogawa, O.; et al. Heterogeneous fibroblasts underlie age-dependent tertiary lymphoid tissues in the kidney. J. Clin. Investig. 2016, 1, e87680. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Liu, X.; Liang, J.; Habiel, D.M.; Vrishika, K.; Coelho, A.L.; Deng, N.; Xie, T.; Wang, Y.; Liu, N.; et al. PD-L1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. J. Clin. Investig. 2019, 4, e125326. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Liu, G.; Xing, J.; Song, H.; Wang, Z. Fecal bacteria from Crohn’s disease patients more potently activated NOD-like receptors and Toll-like receptors in macrophages, in an IL-4-repressible fashion. Microb. Pathog. 2018, 121, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhang, Y.; Xing, J.; Song, X.; Huang, L.; Weng, H.; Wu, X.; Walker, E.; Wang, Z. Fecal bacteria from treatment-naive Crohn’s disease patients can skew helper T cell responses. Exp. Cell Res. 2017, 361, 135–140. [Google Scholar] [CrossRef]
- Holst, L.M.; Iribarren, C.; Sapnara, M.; Savolainen, O.; Törnblom, H.; Wettergren, Y.; Strid, H.; Simrén, M.; Magnusson, M.K.; Öhman, L. Fecal Luminal Factors from Patients with Gastrointestinal Diseases Alter Gene Expression Profiles in Caco-2 Cells and Colonoids. Int. J. Mol. Sci. 2022, 23, 15505. [Google Scholar] [CrossRef]
- Caër, C.; Gorreja, F.; Forsskåhl, S.K.; Brynjolfsson, S.F.; Szeponik, L.; Magnusson, M.K.; Börjesson, L.G.; Block, M.; Bexe-Lindskog, E.; Wick, M.J. TREM-1+ Macrophages Define a Pathogenic Cell Subset in the Intestine of Crohn’s Disease Patients. J. Crohn’s Colitis 2021, 15, 1346–1361. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| iCD (n = 15) | sCD (n = 10) | HS (n = 10) | p-Value 3 | |
|---|---|---|---|---|
| Sex (male/female) | 7/8 | 4/6 | 6/4 | 0.74 4 |
| Age (years) 1 | 31 (20–59) | 28 (20–53) | 27 (21–64) | 0.49 5 |
| Disease duration (years) 1 | 1 (0–8) | 5 (0–24) | NA | 0.04 5 |
| Disease behavior (colonic/ileocolonic/ileocecal) | 5/9/1 | 5/2/3 | NA | 0.10 4 |
| Fecal calprotectin (mg/L) 1 | 700 (190–2600) | 240 (110–6000) | <30 2 | 0.24 5 |
| Blood CRP (mg/L) 1 | 5 (<1–36) | 13 (<1–46) | 1 (<1–7) | 0.19 5 |
| HBI 1 | 7 (5–11) | 7 (5–10) | NA | 0.75 5 |
| CDEIS 1 | 14 (6–30) | 15 (7–49) | NA | 0.30 5 |
| Treatments Azathioprine (yes/no/previously) Infliximab (yes/no/previously) | 2/12/1 2/13/0 | 6/2/2 3/7/0 | NA | 0.01 4 0.30 4 |
| Protein | CD (n = 20) | HS (n = 10) | p-Value 1 | iCD (n = 10) | sCD (n = 10) | p-Value 1 |
|---|---|---|---|---|---|---|
| Pro-COLA1 (ng/mL) 2 | 92 (70–105) | 86 (67–105) | 0.15 | 97 (85–105) | 87 (70–100) | 0.03 |
| Fibronectin (pg/mL) 2 | 323 (272–399) | 322 (296–389) | 0.70 | 324 (273–338) | 322 (272–399) | 0.90 |
| FAP (MFI × 103) 3 | 7.5 (6.7–8.5) | 7.1 (6.2–8.2) | 0.35 | 7.6 (6.7–8.5) | 7.4 (6.7–7.7) | 0.15 |
| PD-L1 (MFI × 103) 3 | 1.6 (1.4–2.1) | 1.6 (1.4–1.9) | 0.90 | 1.6 (1.4–2.1) | 1.6 (1.4–1.9) | 0.70 |
| CD21 (MFI × 103) 3 | 0.48 (0.43–0.59) | 0.49 (0.47–0.53) | 0.40 | 0.47 (0.43–0.52) | 0.50 (0.46–0.59) | 0.01 |
| TGFb1 (MFI × 103) 3 | 1.6 (1.1–2.0) | 1.7 (1.2–1.8) | 0.60 | 1.6 (1.1–2.0) | 1.6 (1.5–2.0) | 0.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorreja, F.; Bendix, M.; Rush, S.T.A.; Maasfeh, L.; Savolainen, O.; Dige, A.; Agnholt, J.; Öhman, L.; Magnusson, M.K. Fecal Supernatants from Patients with Crohn’s Disease Induce Inflammatory Alterations in M2 Macrophages and Fibroblasts. Cells 2024, 13, 60. https://doi.org/10.3390/cells13010060
Gorreja F, Bendix M, Rush STA, Maasfeh L, Savolainen O, Dige A, Agnholt J, Öhman L, Magnusson MK. Fecal Supernatants from Patients with Crohn’s Disease Induce Inflammatory Alterations in M2 Macrophages and Fibroblasts. Cells. 2024; 13(1):60. https://doi.org/10.3390/cells13010060
Chicago/Turabian StyleGorreja, Frida, Mia Bendix, Stephen T. A. Rush, Lujain Maasfeh, Otto Savolainen, Anders Dige, Jorgen Agnholt, Lena Öhman, and Maria K. Magnusson. 2024. "Fecal Supernatants from Patients with Crohn’s Disease Induce Inflammatory Alterations in M2 Macrophages and Fibroblasts" Cells 13, no. 1: 60. https://doi.org/10.3390/cells13010060
APA StyleGorreja, F., Bendix, M., Rush, S. T. A., Maasfeh, L., Savolainen, O., Dige, A., Agnholt, J., Öhman, L., & Magnusson, M. K. (2024). Fecal Supernatants from Patients with Crohn’s Disease Induce Inflammatory Alterations in M2 Macrophages and Fibroblasts. Cells, 13(1), 60. https://doi.org/10.3390/cells13010060