
Investigating the Impact of Delivery Routes for Exon Skipping Therapies in the CNS of DMD Mouse Models
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Antisense Oligonucleotides
2.2. Biodistribution Analysis in Tissue Lysates
2.3. Biodistribution Analysis In Situ
2.4. Exon 51 Skipping Analysis
2.5. Statistical Analysis
3. Results
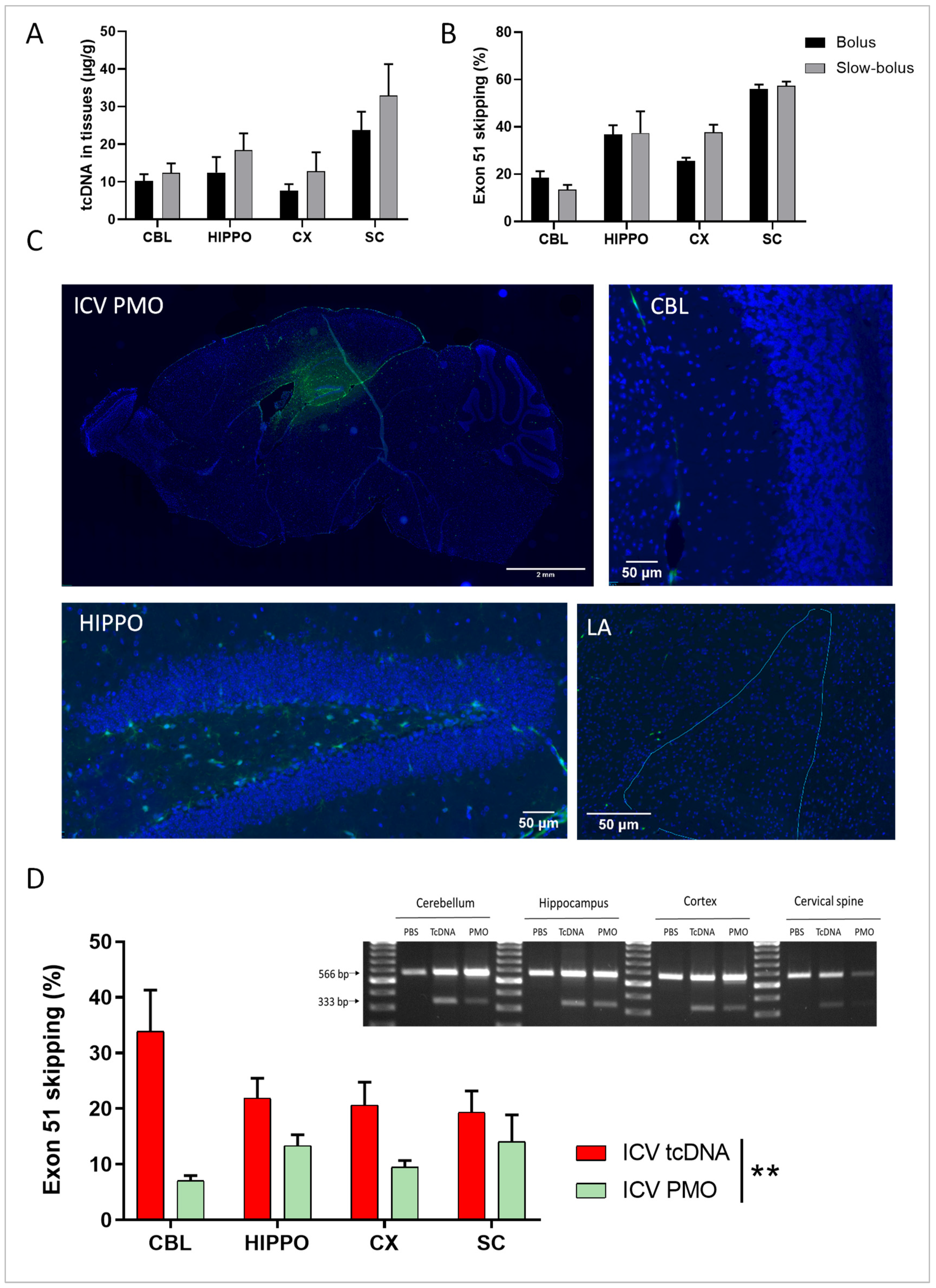
3.1. Single ICV ASO Delivery Optimization
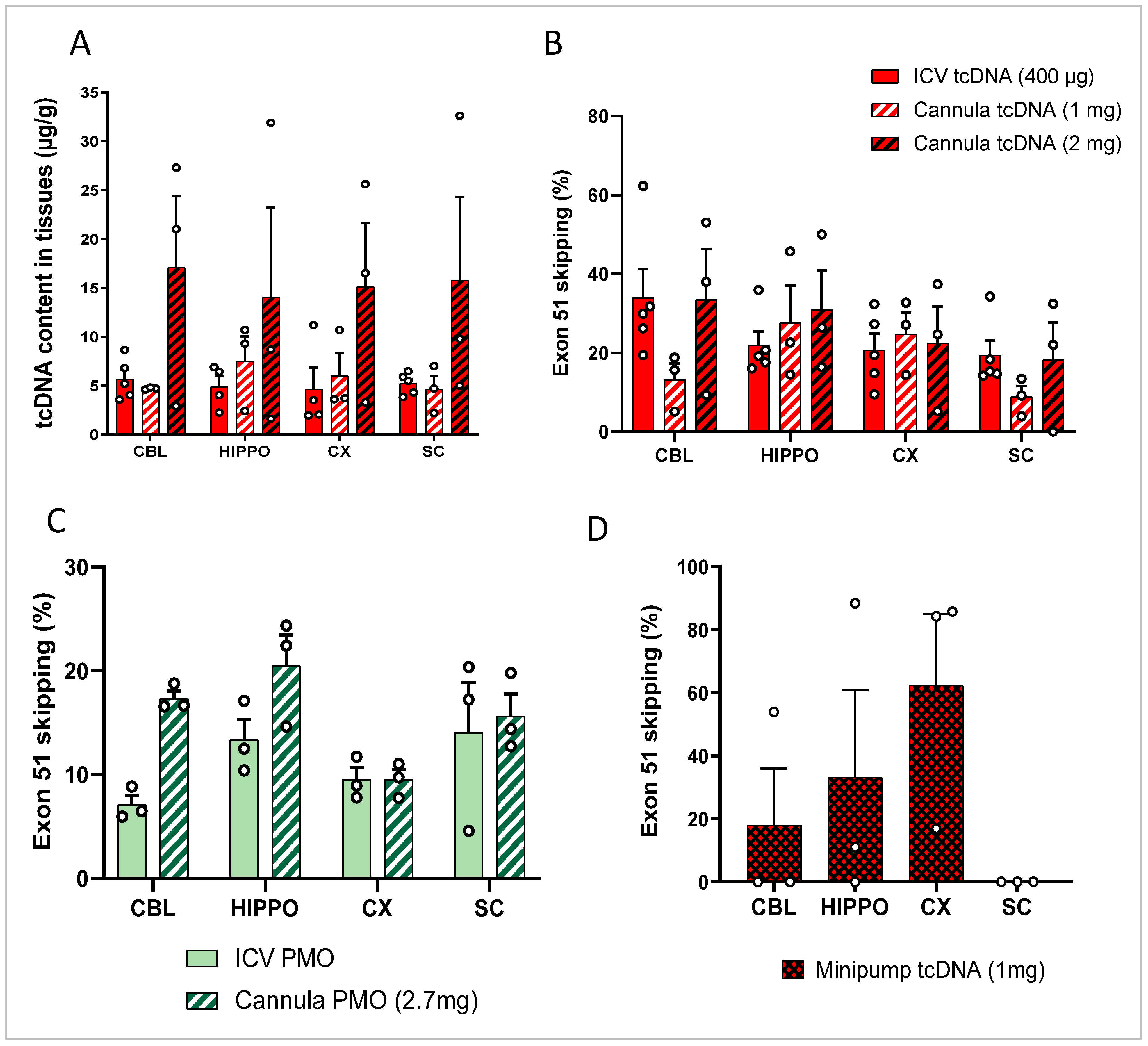
3.2. Repeated ICV ASO Delivery
3.3. ICV Delivery of ASO Using Osmotic Pumps
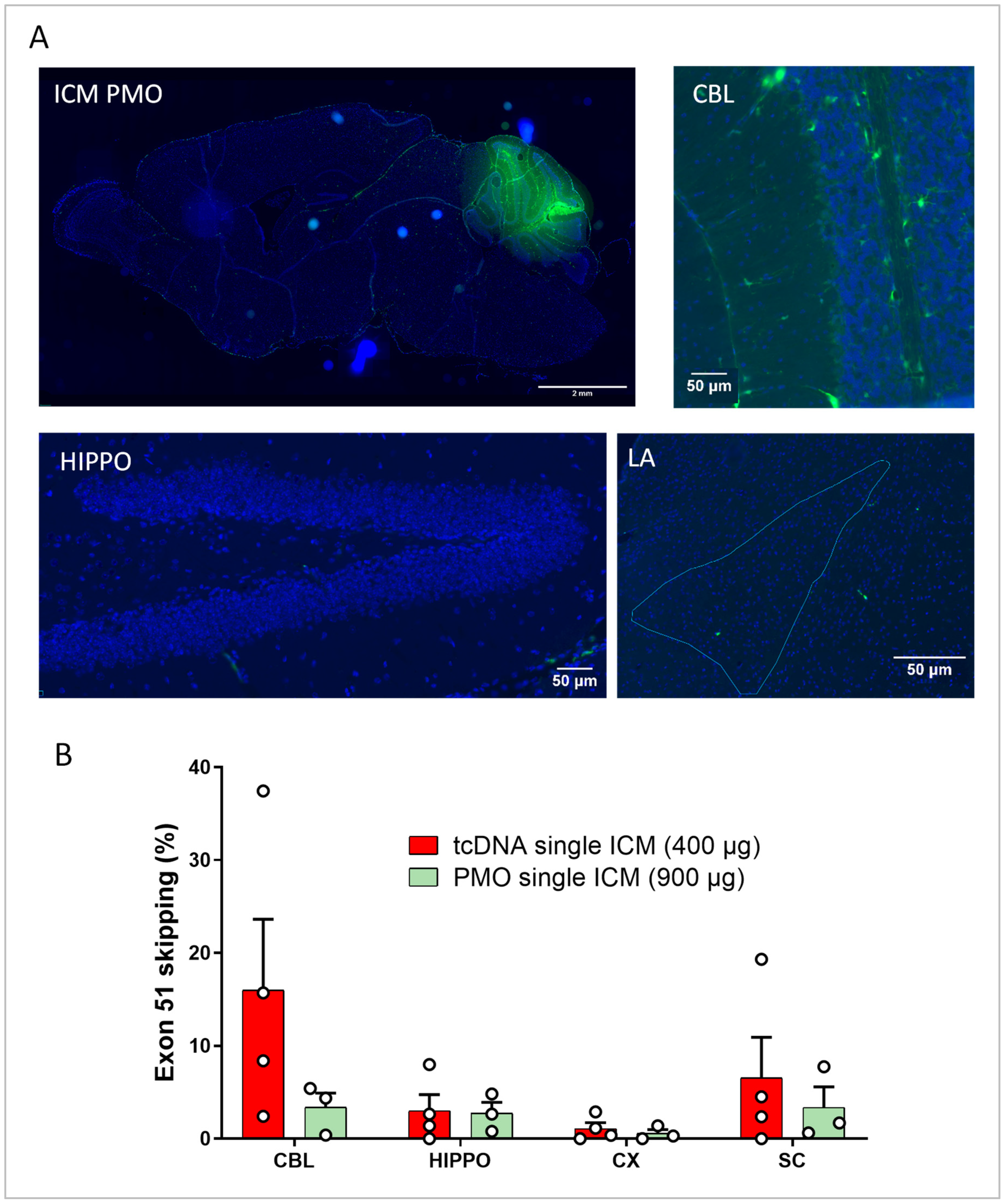
3.4. Intra-Cisterna Magna Delivery of ASOs
3.5. Repeated ICM ASO Administration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilar, M.; Belenky, A.; Smisek, D.L.; Bourque, A.; Cohen, A.S. Kinetics of Phosphorothioate Oligonucleotide Metabolism in Biological Fluids. Nucleic Acids Res. 1997, 25, 3615–3620. [Google Scholar] [CrossRef] [PubMed]
- Griepenburg, J.C.; Rapp, T.L.; Carroll, P.J.; Eberwine, J.; Dmochowski, I.J. Ruthenium-Caged Antisense Morpholinos for Regulating Gene Expression in Zebrafish Embryos. Chem. Sci. 2015, 6, 2342–2346. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Wengel, J.; Maiti, S. LNA-Modified Oligonucleotides Effectively Drive Intramolecular-Stable Hairpin to Intermolecular-Duplex State. Biochem. Biophys. Res. Commun. 2007, 352, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Knudson, C.M.; Campbell, K.P.; Kunkel, L.M. Subcellular Fractionation of Dystrophin to the Triads of Skeletal Muscle. Nature 1987, 330, 754–758. [Google Scholar] [CrossRef]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A Systematic Review and Meta-Analysis on the Epidemiology of Duchenne and Becker Muscular Dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Janson, A.A.M.; Heemskerk, J.A.; De Winter, C.L.; Van Ommen, G.-J.B.; Van Deutekom, J.C.T. Therapeutic Modulation of DMD Splicing by Blocking Exonic Splicing Enhancer Sites with Antisense Oligonucleotides. Ann. N. Y. Acad. Sci. 2006, 1082, 74–76. [Google Scholar] [CrossRef]
- Dunckley, M.G.; Manoharan, M.; Villiet, P.; Eperon, I.C.; Dickson, G. Modification of Splicing in the Dystrophin Gene in Cultured Mdx Muscle Cells by Antisense Oligoribonucleotides. Hum Mol. Genet. 1998, 7, 1083–1090. [Google Scholar] [CrossRef]
- Ferlini, A.; Goyenvalle, A.; Muntoni, F. RNA-Targeted Drugs for Neuromuscular Diseases. Science 2021, 371, 29–31. [Google Scholar] [CrossRef]
- Hendriksen, R.G.F.; Vles, J.S.H.; Aalbers, M.W.; Chin, R.F.M.; Hendriksen, J.G.M. Brain-Related Comorbidities in Boys and Men with Duchenne Muscular Dystrophy: A Descriptive Study. Eur. J. Paediatr. Neurol. 2018, 22, 488–497. [Google Scholar] [CrossRef]
- Ricotti, V.; Mandy, W.P.L.; Scoto, M.; Pane, M.; Deconinck, N.; Messina, S.; Mercuri, E.; Skuse, D.H.; Muntoni, F. Neurodevelopmental, Emotional, and Behavioural Problems in Duchenne Muscular Dystrophy in Relation to Underlying Dystrophin Gene Mutations. Dev. Med. Child Neurol. 2016, 58, 77–84. [Google Scholar] [CrossRef]
- Araki, E.; Nakamura, K.; Nakao, K.; Kameya, S.; Kobayashi, O.; Nonaka, I.; Kobayashi, T.; Katsuki, M. Targeted Disruption of Exon 52 in the Mouse Dystrophin Gene Induced Muscle Degeneration Similar to That Observed in Duchenne Muscular Dystrophy. Biochem. Biophys. Res. Commun. 1997, 238, 492–497. [Google Scholar] [CrossRef]
- Colombo, P.; Nobile, M.; Tesei, A.; Civati, F.; Gandossini, S.; Mani, E.; Molteni, M.; Bresolin, N.; D’Angelo, G. Assessing Mental Health in Boys with Duchenne Muscular Dystrophy: Emotional, Behavioural and Neurodevelopmental Profile in an Italian Clinical Sample. Eur. J. Paediatr. Neurol. 2017, 21, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J.; Betts, G.A.; Maroulis, S.; Gilissen, C.; Pedersen, R.L.; Mowat, D.R.; Johnston, H.M.; Buckley, M.F. Dystrophin Gene Mutation Location and the Risk of Cognitive Impairment in Duchenne Muscular Dystrophy. PLoS ONE 2010, 5, e8803. [Google Scholar] [CrossRef] [PubMed]
- Saoudi, A.; Zarrouki, F.; Sebrié, C.; Izabelle, C.; Goyenvalle, A.; Vaillend, C. Emotional Behavior and Brain Anatomy of the Mdx52 Mouse Model of Duchenne Muscular Dystrophy. Dis. Model Mech. 2021, 14, dmm049028. [Google Scholar] [CrossRef] [PubMed]
- Boursereau, R.; Donadieu, A.; Dabertrand, F.; Dubayle, D.; Morel, J.-L. Blood Brain Barrier Precludes the Cerebral Arteries to Intravenously-Injected Antisense Oligonucleotide. Eur. J. Pharmacol. 2015, 747, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.; Rosch, J.; Putnam, D. Concepts, Technologies, and Practices for Drug Delivery Past the Blood–Brain Barrier to the Central Nervous System. J. Control. Release 2016, 240, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.K.; Bjornsson, C.S.; Dymecki, S.M.; Gilbertson, R.J.; Holtzman, D.M.; Monuki, E.S. The Choroid Plexus and Cerebrospinal Fluid: Emerging Roles in Development, Disease, and Therapy. J. Neurosci. 2013, 33, 17553–17559. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2019; Available online: https://www.elsevier.com/books/paxinos-and-franklins-the-mouse-brain-in-stereotaxic-coordinates-compact/franklin/978-0-12-816159-3 (accessed on 14 October 2022).
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An Antisense Oligonucleotide against SOD1 Delivered Intrathecally for Patients with SOD1 Familial Amyotrophic Lateral Sclerosis: A Phase 1, Randomised, First-in-Man Study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef]
- Madan, E.; Carrié, S.; Donado, C.; Lobo, K.; Souris, M.; Laine, R.; Beers, E.; Cornelissen, L.; Darras, B.T.; Koka, A.; et al. Nusinersen for Patients with Spinal Muscular Atrophy: 1415 Doses via an Interdisciplinary Institutional Approach. Pediatr. Neurol. 2022, 132, 33–40. [Google Scholar] [CrossRef]
- Kennedy, Z.; Gilbert, J.W.; Godinho, B.M.D.C. Intrathecal Delivery of Therapeutic Oligonucleotides for Potent Modulation of Gene Expression in the Central Nervous System. Methods Mol. Biol. 2022, 2434, 345–353. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of Infantile-Onset Spinal Muscular Atrophy with Nusinersen: A Phase 2, Open-Label, Dose-Escalation Study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.M.; Rozenberg, A.; Gray, S.J. Comparison of High-Dose Intracisterna Magna and Lumbar Puncture Intrathecal Delivery of AAV9 in Mice to Treat Neuropathies. Brain Res. 2020, 1739, 146832. [Google Scholar] [CrossRef] [PubMed]
- Relizani, K.; Echevarría, L.; Zarrouki, F.; Gastaldi, C.; Dambrune, C.; Aupy, P.; Haeberli, A.; Komisarski, M.; Tensorer, T.; Larcher, T.; et al. Palmitic Acid Conjugation Enhances Potency of Tricyclo-DNA Splice Switching Oligonucleotides. Nucleic Acids Res. 2022, 50, 17–34. [Google Scholar] [CrossRef]
- Zarrouki, F.; Relizani, K.; Bizot, F.; Tensorer, T.; Garcia, L.; Vaillend, C.; Goyenvalle, A. Partial Restoration of Brain Dystrophin and Behavioral Deficits by Exon Skipping in the Muscular Dystrophy X-Linked (Mdx) Mouse. Ann. Neurol. 2022, 92, 213–229. [Google Scholar] [CrossRef]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and Mutations: One Gene, Several Proteins, Multiple Phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Bremmer-Bout, M.; Aartsma-Rus, A.; de Meijer, E.J.; Kaman, W.E.; Janson, A.A.M.; Vossen, R.H.A.M.; van Ommen, G.-J.B.; den Dunnen, J.T.; van Deutekom, J.C.T. Targeted Exon Skipping in Transgenic HDMD Mice: A Model for Direct Preclinical Screening of Human-Specific Antisense Oligonucleotides. Mol. Ther. 2004, 10, 232–240. [Google Scholar] [CrossRef]
- Veltrop, M.; van Vliet, L.; Hulsker, M.; Claassens, J.; Brouwers, C.; Breukel, C.; van der Kaa, J.; Linssen, M.M.; den Dunnen, J.T.; Verbeek, S.; et al. A Dystrophic Duchenne Mouse Model for Testing Human Antisense Oligonucleotides. PLoS ONE 2018, 13, e0193289. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Chun, S.J.; Norris, D.A.; Hung, G.; Lee, S.; Matson, J.; Fey, R.A.; Gaus, H.; Hua, Y.; Grundy, J.S.; et al. Pharmacology of a Central Nervous System Delivered 2′-O-Methoxyethyl–Modified Survival of Motor Neuron Splicing Oligonucleotide in Mice and Nonhuman Primates. J. Pharmacol. Exp. Ther. 2014, 350, 46–55. [Google Scholar] [CrossRef]
- Simon, M.J.; Iliff, J.J. Regulation of Cerebrospinal Fluid (CSF) Flow in Neurodegenerative, Neurovascular and Neuroinflammatory Disease. Biochim. Biophys. Acta 2016, 1862, 442–451. [Google Scholar] [CrossRef]
- Doorenweerd, N.; Mahfouz, A.; van Putten, M.; Kaliyaperumal, R.; T’ Hoen, P.A.C.; Hendriksen, J.G.M.; Aartsma-Rus, A.M.; Verschuuren, J.J.G.M.; Niks, E.H.; Reinders, M.J.T.; et al. Timing and Localization of Human Dystrophin Isoform Expression Provide Insights into the Cognitive Phenotype of Duchenne Muscular Dystrophy. Sci. Rep. 2017, 7, 12575. [Google Scholar] [CrossRef]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep Drives Metabolite Clearance from the Adult Brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Ries, M.; Decker, Y.; Müller, A.; Riner, C.; Bücker, A.; Fassbender, K.; Detmar, M.; Proulx, S.T. Rapid Lymphatic Efflux Limits Cerebrospinal Fluid Flow to the Brain. Acta Neuropathol. 2019, 137, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Toonen, L.J.A.; Casaca-Carreira, J.; Pellisé-Tintoré, M.; Mei, H.; Temel, Y.; Jahanshahi, A.; van Roon-Mom, W.M.C. Intracerebroventricular Administration of a 2′-O-Methyl Phosphorothioate Antisense Oligonucleotide Results in Activation of the Innate Immune System in Mouse Brain. Nucleic Acid Ther. 2018, 28, 63–73. [Google Scholar] [CrossRef]
- Moazami, M.; Rembestsy-Brown, J.; Wang, F.; Krishnamurthy, P.M.; Brown, R.; Watts, J.K. Quantifying and Mitigating Motor Phenotypes Induced by Antisense Oligonucleotides in the Central Nervous System. BioRxiv 2021. [Google Scholar] [CrossRef]
- Hagedorn, P.H.; Brown, J.M.; Easton, A.; Pierdomenico, M.; Jones, K.; Olson, R.E.; Mercer, S.E.; Li, D.; Loy, J.; Høg, A.M.; et al. Acute Neurotoxicity of Antisense Oligonucleotides After Intracerebroventricular Injection into Mouse Brain Can Be Predicted from Sequence Features. Nucleic Acid Ther. 2022, 32, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Jimenez-Mallebrera, C.; van Roon, W.; Sewing, S.; Krieg, A.M.; Arechavala-Gomeza, V.; Andersson, P. Considerations in the Preclinical Assessment of the Safety of Antisense Oligonucleotides. Nucleic Acid Ther. 2023, 33, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Xavier, A.L.R.; Hauglund, N.L.; von Holstein-Rathlou, S.; Li, Q.; Sanggaard, S.; Lou, N.; Lundgaard, I.; Nedergaard, M. Cannula Implantation into the Cisterna Magna of Rodents. J. Vis. Exp. 2018, 57378. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saoudi, A.; Fergus, C.; Gileadi, T.; Montanaro, F.; Morgan, J.E.; Kelly, V.P.; Tensorer, T.; Garcia, L.; Vaillend, C.; Muntoni, F.; et al. Investigating the Impact of Delivery Routes for Exon Skipping Therapies in the CNS of DMD Mouse Models. Cells 2023, 12, 908. https://doi.org/10.3390/cells12060908
Saoudi A, Fergus C, Gileadi T, Montanaro F, Morgan JE, Kelly VP, Tensorer T, Garcia L, Vaillend C, Muntoni F, et al. Investigating the Impact of Delivery Routes for Exon Skipping Therapies in the CNS of DMD Mouse Models. Cells. 2023; 12(6):908. https://doi.org/10.3390/cells12060908
Chicago/Turabian StyleSaoudi, Amel, Claire Fergus, Talia Gileadi, Federica Montanaro, Jennifer E. Morgan, Vincent P. Kelly, Thomas Tensorer, Luis Garcia, Cyrille Vaillend, Francesco Muntoni, and et al. 2023. "Investigating the Impact of Delivery Routes for Exon Skipping Therapies in the CNS of DMD Mouse Models" Cells 12, no. 6: 908. https://doi.org/10.3390/cells12060908
APA StyleSaoudi, A., Fergus, C., Gileadi, T., Montanaro, F., Morgan, J. E., Kelly, V. P., Tensorer, T., Garcia, L., Vaillend, C., Muntoni, F., & Goyenvalle, A. (2023). Investigating the Impact of Delivery Routes for Exon Skipping Therapies in the CNS of DMD Mouse Models. Cells, 12(6), 908. https://doi.org/10.3390/cells12060908