
Current State and Future Directions in the Diagnosis of Amyotrophic Lateral Sclerosis

 , , ,
, , , 
Abstract
1. Introduction
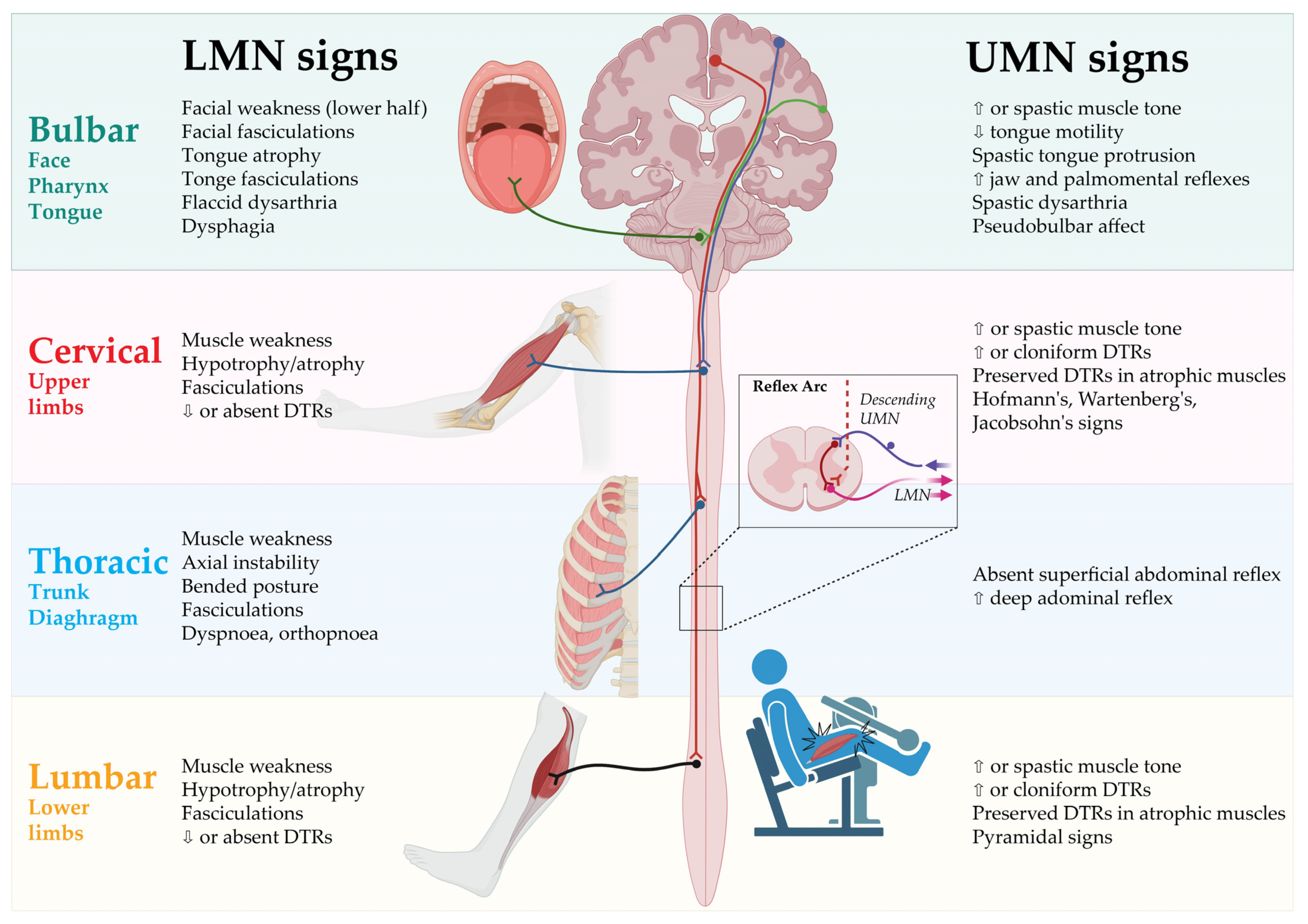
2. Clinical Presentation
2.1. Spinal-Onset ALS
2.2. Bulbar-Onset ALS
2.3. Progressive Muscular Atrophy
2.4. Primary Lateral Sclerosis
2.5. Flail-Arm-Syndrome
2.6. Flail-Leg-Syndrome
2.7. Axial or Respiratory-Onset ALS
2.8. Hemiplegic ALS (Mill’s Syndrome)
3. Diagnostic Criteria
3.1. The El Escorial Criteria (1994) and Revised El Escorial Criteria (2000)
3.2. The Awaji Criteria (2008)
3.3. The Gold Coast Criteria (2020)
4. Clinical Assessments of Disease Severity and Progression
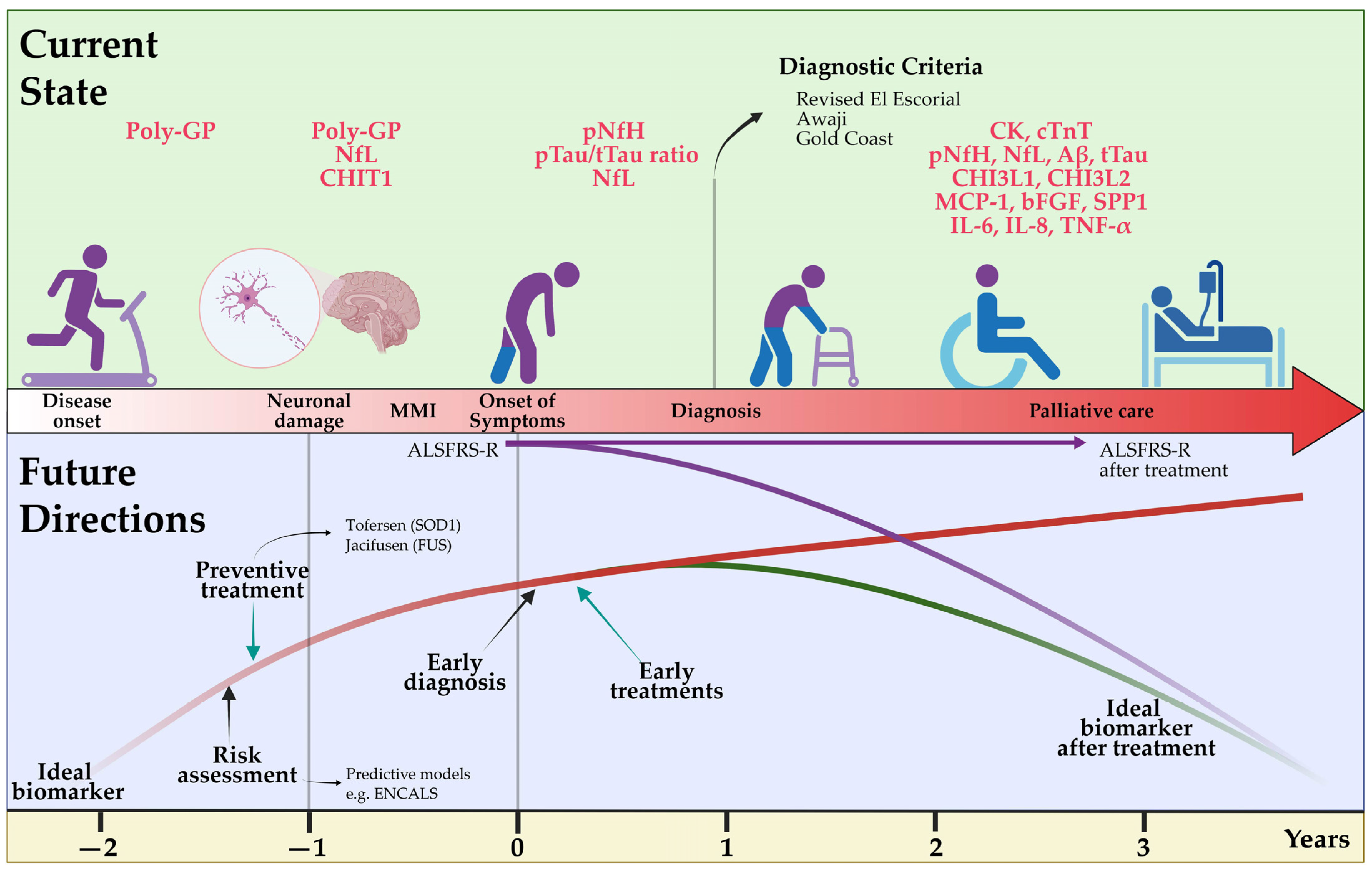
5. Pre-Symptomatic ALS
6. Cognitive and Behavioral Assessment
7. Technical Diagnostic Tools
8. Genetic Testing
- Exclusion of genetic disorders mimicking ALS, such as spinal and bulbar muscular atrophy (SBMA) which is caused by a polyglutamine expansion in the androgen receptor (AR) gene and can be mistaken for LMN-predominant ALS [81]. Other examples include, but are not limited to, adult-onset spinal muscular atrophy, a number of hereditary spastic paraparesis (HSP) subtypes and adult polyglucosan body disease (APBD) [82];
- Identification of patients who are eligible for trials targeting specific mutated genes, such as SOD1, C9orf72, Ataxin 2 (ATXN2) and FUS [83]. For patients with SOD1 mutations, tofersen, an antisense oligonucleotide reducing SOD1 protein synthesis, was shown to reduce neurofilament light chain levels in plasma and is already available in many countries in an early access program [84,85];
- Counselling of family members concerning predictive testing. Predictive testing always needs thorough counselling, especially when there are no measures to prevent the disease, as is currently the case for ALS [86,87]. However, family members may benefit from the knowledge gained from genetic testing, either because they are relieved when they do not harbor the mutation or because they are able to plan ahead for specific life decisions. This does also include preimplantation genetic diagnosis if a desire to have children is present [88]. Also, the exemplary ATLAS trial is already trying to closely follow up asymptomatic carriers of disease-causing mutations to initiate therapy on the basis of biomarker-defined phenoconversion and before the advent of clinical symptoms for an optimal disease-modifying effect [89]. Approaches like these will likely be applied more often as more gene-specific therapies become available;
- Identification of genetic mutations may help in the prediction of the clinical course of the disease, which is of importance for patient and caregiver counselling (see prediction models section).
9. Diagnostic and Prognostic Value of Fluid Biomarkers
9.1. Neurofilaments
9.2. Inflammatory Biomarkers
9.3. Chitinase, Tau Protein, TDP-43, Creatine Kinase and Other Fluid Biomarkers
9.4. Frontiers in Fluid Biomarkers
10. Predictive Models
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALSFRS-R | Revised Amyotrophic Lateral Sclerosis Functional Rating Scale |
| ADM | abductor digiti minimi |
| ALS | amyotrophic lateral sclerosis |
| ALS-bci | ALS with cognitive and behavioral impairment |
| ALS-bi | ALS with behavioral impairment |
| ALS-ci | ALS with cognitive impariment |
| APB | abductor pollicis brevis |
| APBD | adult polyglucosan body disease |
| AR | androgen receptor |
| ASO | antisense oligonucleotide |
| ATXN2 | Ataxin 2 |
| Aβ | Amyloid β |
| bFGF | basic fibroblast growth factor |
| C9orf72 | chromosome 9 open reading frame 72 |
| CD83 | cluster of differentiation 83 |
| CHI3L1 | chitinase-3-like protein 1 |
| CHI3L2 | chitinase-3-like protein 2 |
| CHIT1 | chitotriosidase 1 |
| CJD | Creutzfeld-Jakob disease |
| CK | creatine kinase |
| CLP | chitinase-like proteins |
| CSA | cross-sectional areas |
| CSF | cerebrospinal fluid |
| CST | corticospinal tract |
| cTnT | cardiac troponin T |
| CXCL1 | chemokine (C-X-C motif) ligand 1 |
| CXCL2 | chemokine (C-X-C motif) ligand 2 |
| DTI | diffusion tensor imaging |
| DTR | deep tendon reflex |
| ECAS | Edinburgh Cognitive and Behavioral ALS Screen |
| ED | electrodiagnostic |
| EMG | electromyography |
| FAB | Frontal Assessment Battery |
| FAS | flail-arm-syndrome |
| FDG-PET | [18F]Fluorodeoxyglucose-PET |
| FDI | first dorsal interosseus |
| FLS | flail-leg-syndrome |
| FOSB | FBJ murine osteosarcoma viral oncogene homolog B |
| FTD | Frontotemporal dementia |
| FUS | fused-in sarcoma |
| GWAS | Genome-wide association studies |
| HRE | hexanucleotide (G4C2)n repeat expansion |
| HSP | hereditary spastic paraparesis |
| IL-18 | interleukin-18 |
| IL-8 | interleukin-8 |
| iPSCs | induced pluripotent stem cells |
| LMN | lower motor neurons |
| MCP-1 | monocyte chemoattractant protein-1 |
| MiToS | Milano Torino Staging System |
| MMI | mild motor impairment |
| MMN | multifocal motor neuropathy |
| MMSE | Mini Mental State Assessment |
| MND | motor neuron disease |
| MoCa | Montreal Cognitive Assessment Test |
| MRI | magnetic resonance imaging |
| MS | multiple sclerosis |
| Nf | neurofilaments |
| NfL | neurofilament light chain |
| NGS | next-generation sequencing |
| PET | positron emission tomography |
| PLS | primary lateral sclerosis |
| PMA | progressive muscular atrophy |
| pNfH | phosphorylated neurofilament heavy chain |
| Poly-GP | arginine containing dipeptide repeat polymers |
| PRS | polygenic risk scores |
| pTau | phosphorylated Tau |
| ROADS | Rasch overall ALS disability scale |
| sAPPβ | soluble amyloid precursor protein |
| SBMA | spinal and bulbar muscular atrophy |
| SMA | spinal muscular atrophy |
| SOCS3 | suppressor of cytokine signaling 3 |
| SOD1 | superoxide dismutase 1 |
| SPP1 | secreted Phosphoprotein 1 |
| TARDBP | TAR DNA binding protein |
| TDP-43 | transactive response DNA binding protein 43 |
| TNF-α | tumor necrosis factor |
| TSPO | 18pkD translocator protein |
| t-Tau | total Tau |
| UMN | upper motor neurons |
| Vn | vagus nerve |
References
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Recent Advances in the Diagnosis and Prognosis of Amyotrophic Lateral Sclerosis. Lancet Neurol. 2022, 21, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic Lateral Sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Štětkářová, I.; Ehler, E. Diagnostics of Amyotrophic Lateral Sclerosis: Up to Date. Diagnostics 2021, 11, 231. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.; Morren, J.A.; Pioro, E.P. Time to Diagnosis and Factors Affecting Diagnostic Delay in Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2020, 417, 117054. [Google Scholar] [CrossRef]
- Weise, D.; Menze, I.; Metelmann, M.C.F.; Woost, T.B.; Classen, J.; Otto Pelz, J. Multimodal Assessment of Autonomic Dysfunction in Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2022, 29, 715–723. [Google Scholar] [CrossRef]
- Sharma, R.; Hicks, S.; Berna, C.M.; Kennard, C.; Talbot, K.; Turner, M.R. Oculomotor Dysfunction in Amyotrophic Lateral Sclerosis: A Comprehensive Review. Arch. Neurol. 2011, 68, 857–861. [Google Scholar] [CrossRef]
- Walhout, R.; Verstraete, E.; van den Heuvel, M.P.; Veldink, J.H.; van den Berg, L.H. Patterns of Symptom Development in Patients with Motor Neuron Disease. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 21–28. [Google Scholar] [CrossRef]
- Ramamoorthy, D.; Severson, K.; Ghosh, S.; Sachs, K.; Glass, J.D.; Fournier, C.N.; Herrington, T.M.; Berry, J.D.; Ng, K.; Fraenkel, E. Identifying Patterns in Amyotrophic Lateral Sclerosis Progression from Sparse Longitudinal Data. Nat. Comput. Sci. 2022, 2, 605–616. [Google Scholar] [CrossRef]
- Brooks, B.R. El Escorial World Federation of Neurology Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 1994, 124, 96–107. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O.; Kiernan, M.C.; Chiò, A.; Rix-Brooks, B.; van den Berg, L.H. Amyotrophic Lateral Sclerosis: Moving towards a New Classification System. Lancet Neurol. 2016, 15, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- Longinetti, E.; Fang, F. Epidemiology of Amyotrophic Lateral Sclerosis: An Update of Recent Literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef]
- Wilbourn, A.J. The “Split Hand Syndrome”. Muscle Nerve 2000, 23, 138. [Google Scholar] [CrossRef]
- Turner, M.R.; Wicks, P.; Brownstein, C.A.; Massagli, M.P.; Toronjo, M.; Talbot, K.; Al-Chalabi, A. Concordance between Site of Onset and Limb Dominance in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 853–854. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Jin, J.; Chen, Q.; Kang, L.; Jia, R.; Qin, X.; Liu, X.; Dang, Y.; Dang, J. Dissociated Lower Limb Muscle Involvement in Amyotrophic Lateral Sclerosis and Its Differential Diagnosis Value. Sci. Rep. 2019, 9, 17786. [Google Scholar] [CrossRef]
- Swash, M. Why Are Upper Motor Neuron Signs Difficult to Elicit in Amyotrophic Lateral Sclerosis? J. Neurol. Neurosurg. Psychiatry 2012, 83, 659–662. [Google Scholar] [CrossRef]
- Huynh, W.; Simon, N.G.; Grosskreutz, J.; Turner, M.R.; Vucic, S.; Kiernan, M.C. Assessment of the Upper Motor Neuron in Amyotrophic Lateral Sclerosis. Clin. Neurophysiol. 2016, 127, 2643–2660. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Vidovic, M.; Aust, E.; Hermann, A.; Günther, R. The Palmomental Reflex in Amyotrophic Lateral Sclerosis—A Clinical Sign of Executive or Motor Dysfunction? Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 588–591. [Google Scholar] [CrossRef]
- Liewluck, T.; Saperstein, D.S. Progressive Muscular Atrophy. Neurol. Clin. 2015, 33, 761–773. [Google Scholar] [CrossRef]
- Kim, W.-K.; Liu, X.; Sandner, J.; Pasmantier, M.; Andrews, J.; Rowland, L.P.; Mitsumoto, H. Study of 962 Patients Indicates Progressive Muscular Atrophy Is a Form of ALS. Neurology 2009, 73, 1686–1692. [Google Scholar] [CrossRef]
- Turner, M.R.; Barohn, R.J.; Corcia, P.; Fink, J.K.; Harms, M.B.; Kiernan, M.C.; Ravits, J.; Silani, V.; Simmons, Z.; Statland, J.; et al. Primary Lateral Sclerosis: Consensus Diagnostic Criteria. J. Neurol. Neurosurg. Psychiatry 2020, 91, 373–377. [Google Scholar] [CrossRef]
- Chiò, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G.; PARALS study group. Phenotypic Heterogeneity of Amyotrophic Lateral Sclerosis: A Population Based Study. J. Neurol. Neurosurg. Psychiatry 2011, 82, 740–746. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Mathers, S.; Talman, P.; Galtrey, C.; Parkinson, M.H.; Ganesalingam, J.; Willey, E.; Ampong, M.A.; Ellis, C.M.; Shaw, C.E.; et al. Natural History and Clinical Features of the Flail Arm and Flail Leg ALS Variants. Neurology 2009, 72, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.T.; Ellis, C.M.; Al-Chalabi, A.; Leigh, P.N.; Shaw, C.E. Flail Arm Syndrome: A Distinctive Variant of Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 1998, 65, 950–951. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic Lateral Sclerosis: A Clinical Review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- De Carvalho, M.; Pinto, S.; Swash, M. Association of Paraspinal and Diaphragm Denervation in ALS. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2010, 11, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Robberecht, W. The Phenotypic Variability of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef]
- Johnsen, B. Diagnostic Criteria for Amyotrophic Lateral Sclerosis from El Escorial to Gold Coast. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2020, 131, 1962–1963. [Google Scholar] [CrossRef] [PubMed]
- Kee, R.; O’Gorman, C.M. Teaching Video NeuroImages: The Pathologic Deep Abdominal Reflex. Neurology 2017, 89, e245. [Google Scholar] [CrossRef] [PubMed]
- Rajabally, Y.A.; Hbahbih, M.; Abbott, R.J. Hemiplegic ALS: Mills Syndrome. Neurology 2005, 64, 1984–1985. [Google Scholar] [CrossRef] [PubMed]
- Jaiser, S.R.; Mitra, D.; Williams, T.L.; Baker, M.R. Mills’ Syndrome Revisited. J. Neurol. 2019, 266, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Gastaut, J.L.; Bartolomei, F. Mills’ Syndrome: Ascending (or Descending) Progressive Hemiplegia: A Hemiplegic Form of Primary Lateral Sclerosis? J. Neurol. Neurosurg. Psychiatry 1994, 57, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Rutter-Locher, Z.; Turner, M.R.; Leigh, P.N.; Al-Chalabi, A. Analysis of Terms Used for the Diagnosis and Classification of Amyotrophic Lateral Sclerosis and Motor Neuron Disease. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 600–604. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic Criteria for Diagnosis of ALS. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.; Swash, M.; de Carvalho, M. Awaji Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis: A Systematic Review. Arch. Neurol. 2012, 69, 1410–1416. [Google Scholar] [CrossRef]
- Johnsen, B.; Pugdahl, K.; Fuglsang-Frederiksen, A.; Kollewe, K.; Paracka, L.; Dengler, R.; Camdessanché, J.P.; Nix, W.; Liguori, R.; Schofield, I.; et al. Diagnostic Criteria for Amyotrophic Lateral Sclerosis: A Multicentre Study of Inter-Rater Variation and Sensitivity. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2019, 130, 307–314. [Google Scholar] [CrossRef]
- Traynor, B.J.; Codd, M.B.; Corr, B.; Forde, C.; Frost, E.; Hardiman, O.M. Clinical Features of Amyotrophic Lateral Sclerosis According to the El Escorial and Airlie House Diagnostic Criteria: A Population-Based Study. Arch. Neurol. 2000, 57, 1171. [Google Scholar] [CrossRef]
- Shefner, J.M.; Al-Chalabi, A.; Baker, M.R.; Cui, L.-Y.; de Carvalho, M.; Eisen, A.; Grosskreutz, J.; Hardiman, O.; Henderson, R.; Matamala, J.M.; et al. A Proposal for New Diagnostic Criteria for ALS. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2020, 131, 1975–1978. [Google Scholar] [CrossRef]
- Turner, M.R. Diagnosing ALS: The Gold Coast Criteria and the Role of EMG. Pract. Neurol. 2022, 22, 176–178. [Google Scholar] [CrossRef] [PubMed]
- ALS CNTF Treatment Study Phase I–II Group; Brooks, B.R.; Sanjak, M. The Amyotrophic Lateral Sclerosis Functional Rating Scale: Assessment of Activities of Daily Living in Patients With Amyotrophic Lateral Sclerosis. Arch. Neurol. 1996, 53, 141–147. [Google Scholar] [CrossRef]
- Cedarbaum, J.M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A. The ALSFRS-R: A Revised ALS Functional Rating Scale That Incorporates Assessments of Respiratory Function. J. Neurol. Sci. 1999, 169, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.C.; Rojas-Garcia, R.; Scott, K.M.; Scotton, W.; Ellis, C.E.; Burman, R.; Wijesekera, L.; Turner, M.R.; Leigh, P.N.; Shaw, C.E.; et al. A Proposed Staging System for Amyotrophic Lateral Sclerosis. Brain J. Neurol. 2012, 135, 847–852. [Google Scholar] [CrossRef]
- Chiò, A.; Hammond, E.R.; Mora, G.; Bonito, V.; Filippini, G. Development and Evaluation of a Clinical Staging System for Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Fournier, C.N.; Bedlack, R.; Quinn, C.; Russell, J.; Beckwith, D.; Kaminski, K.H.; Tyor, W.; Hertzberg, V.; James, V.; Polak, M.; et al. Development and Validation of the Rasch-Built Overall Amyotrophic Lateral Sclerosis Disability Scale (ROADS). JAMA Neurol. 2020, 77, 480–488. [Google Scholar] [CrossRef]
- Hartmaier, S.L.; Rhodes, T.; Cook, S.F.; Schlusser, C.; Chen, C.; Han, S.; Zach, N.; Murthy, V.; Davé, S. Qualitative Measures That Assess Functional Disability and Quality of Life in ALS. Health Qual. Life Outcomes 2022, 20, 12. [Google Scholar] [CrossRef]
- Benatar, M.; Turner, M.R.; Wuu, J. Defining Pre-Symptomatic Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 303–309. [Google Scholar] [CrossRef]
- Benatar, M.; Granit, V.; Andersen, P.M.; Grignon, A.-L.; McHutchison, C.; Cosentino, S.; Malaspina, A.; Wuu, J. Mild Motor Impairment as Prodromal State in Amyotrophic Lateral Sclerosis: A New Diagnostic Entity. Brain 2022, 145, 3500–3508. [Google Scholar] [CrossRef]
- Phukan, J.; Pender, N.P.; Hardiman, O. Cognitive Impairment in Amyotrophic Lateral Sclerosis. Lancet Neurol. 2007, 6, 994–1003. [Google Scholar] [CrossRef]
- Phukan, J.; Elamin, M.; Bede, P.; Jordan, N.; Gallagher, L.; Byrne, S.; Lynch, C.; Pender, N.; Hardiman, O. The Syndrome of Cognitive Impairment in Amyotrophic Lateral Sclerosis: A Population-Based Study. J. Neurol. Neurosurg. Psychiatry 2012, 83, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, A.; Iazzolino, B.; Calvo, A.; Moglia, C.; Lopiano, L.; Restagno, G.; Brunetti, M.; Ossola, I.; Lo Presti, A.; Cammarosano, S.; et al. Cognitive Correlates in Amyotrophic Lateral Sclerosis: A Population-Based Study in Italy. J. Neurol. Neurosurg. Psychiatry 2015, 86, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Beeldman, E.; Raaphorst, J.; Klein Twennaar, M.; de Visser, M.; Schmand, B.A.; de Haan, R.J. The Cognitive Profile of ALS: A Systematic Review and Meta-Analysis Update. J. Neurol. Neurosurg. Psychiatry 2016, 87, 611–619. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Strong, M.J.; Grace, G.M.; Freedman, M.; Lomen-Hoerth, C.; Woolley, S.; Goldstein, L.H.; Murphy, J.; Shoesmith, C.; Rosenfeld, J.; Leigh, P.N.; et al. Consensus Criteria for the Diagnosis of Frontotemporal Cognitive and Behavioural Syndromes in Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. 2009, 10, 131–146. [Google Scholar] [CrossRef]
- Strong, M.J.; Abrahams, S.; Goldstein, L.H.; Woolley, S.; Mclaughlin, P.; Snowden, J.; Mioshi, E.; Roberts-South, A.; Benatar, M.; Hortobá Gyi, T.; et al. Amyotrophic Lateral Sclerosis-Frontotemporal Spectrum Disorder (ALS-FTSD): Revised Diagnostic Criteria. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 153–174. [Google Scholar] [CrossRef]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal Lobar Degeneration: A Consensus on Clinical Diagnostic Criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U.; et al. Sensitivity of Revised Diagnostic Criteria for the Behavioural Variant of Frontotemporal Dementia. Brain J. Neurol. 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- Gosselt, I.K.; Nijboer, T.C.W.; Van Es, M.A. An Overview of Screening Instruments for Cognition and Behavior in Patients with ALS: Selecting the Appropriate Tool for Clinical Practice. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 324–336. [Google Scholar] [CrossRef]
- Abrahams, S.; Newton, J.; Niven, E.; Foley, J.; Bak, T.H. Screening for Cognition and Behaviour Changes in ALS. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Vielhaber, S.; Schreiber, F.; Cartwright, M.S. Peripheral Nerve Imaging in Amyotrophic Lateral Sclerosis. Clin. Neurophysiol. 2020, 131, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Loewenbrück, K.F.; Werner, R.; Günther, R.; Dittrich, M.; Klingenberger, R.; Reichmann, H.; Storch, A.; Hermann, A. One Nerve Suffices: A Clinically Guided Nerve Ultrasound Protocol for the Differentiation of Multifocal Motor Neuropathy (MMN) and Amyotrophic Lateral Sclerosis (ALS). J. Neurol. 2021, 268, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Holzapfel, K.; Naumann, M. Ultrasound Detection of Vagus Nerve Atrophy in Bulbar Amyotrophic Lateral Sclerosis. J. Neuroimaging 2020, 30, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, M.; Bakola, E.; Papapostolou, A.; Stefanou, M.I.; Moschovos, C.; Salakou, S.; Zis, P.; Zouvelou, V.; Kimiskidis, V.K.; Chroni, E.; et al. Autonomic Dysfunction in Amyotrophic Lateral Sclerosis: A Neurophysiological and Neurosonology Study. J. Neuroimaging Off. J. Am. Soc. Neuroimaging 2022, 32, 710–719. [Google Scholar] [CrossRef]
- Lichtenstein, T.; Sprenger, A.; Weiss, K.; Große Hokamp, N.; Maintz, D.; Schlamann, M.; Fink, G.R.; Lehmann, H.C.; Henning, T.D. MRI DTI and PDFF as Biomarkers for Lower Motor Neuron Degeneration in ALS. Front. Neurosci. 2021, 15, 682126. [Google Scholar] [CrossRef]
- Simon, N.G.; Lagopoulos, J.; Paling, S.; Pfluger, C.; Park, S.B.; Howells, J.; Gallagher, T.; Kliot, M.; Henderson, R.D.; Vucic, S.; et al. Peripheral Nerve Diffusion Tensor Imaging as a Measure of Disease Progression in ALS. J. Neurol. 2017, 264, 882–890. [Google Scholar] [CrossRef]
- Baek, S.-H.; Park, J.; Kim, Y.H.; Seok, H.Y.; Oh, K.-W.; Kim, H.-J.; Kwon, Y.-J.; Sim, Y.; Tae, W.-S.; Kim, S.H.; et al. Usefulness of Diffusion Tensor Imaging Findings as Biomarkers for Amyotrophic Lateral Sclerosis. Sci. Rep. 2020, 10, 5199. [Google Scholar] [CrossRef] [PubMed]
- Kassubek, J.; Müller, H.-P. Advanced Neuroimaging Approaches in Amyotrophic Lateral Sclerosis: Refining the Clinical Diagnosis. Expert Rev. Neurother. 2020, 20, 237–249. [Google Scholar] [CrossRef]
- Kocar, T.D.; Behler, A.; Ludolph, A.C.; Müller, H.-P.; Kassubek, J. Multiparametric Microstructural MRI and Machine Learning Classification Yields High Diagnostic Accuracy in Amyotrophic Lateral Sclerosis: Proof of Concept. Front. Neurol. 2021, 12, 745475. [Google Scholar] [CrossRef]
- Rajagopalan, V.; Pioro, E.P. Comparing Brain Structural MRI and Metabolic FDG-PET Changes in Patients with ALS-FTD: “the Chicken or the Egg?” Question. J. Neurol. Neurosurg. Psychiatry 2015, 86, 952–958. [Google Scholar] [CrossRef]
- Van Laere, K.; Vanhee, A.; Verschueren, J.; De Coster, L.; Driesen, A.; Dupont, P.; Robberecht, W.; Van Damme, P. Value of 18fluorodeoxyglucose-Positron-Emission Tomography in Amyotrophic Lateral Sclerosis: A Prospective Study. JAMA Neurol. 2014, 71, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Cistaro, A.; Campi, C.; Calvo, A.; Caponnetto, C.; Nobili, F.M.; Fania, P.; Beltrametti, M.C.; Moglia, C.; Novi, G.; et al. A PET/CT Approach to Spinal Cord Metabolism in Amyotrophic Lateral Sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Hatakeyama, T.; Sato, K.; Fukui, Y.; Hishikawa, N.; Ohta, Y.; Nishiyama, Y.; Kawai, N.; Tamiya, T.; Abe, K. Flow-Metabolism Uncoupling in the Cervical Spinal Cord of ALS Patients. Neurol. Sci. 2017, 38, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Alshikho, M.J.; Zürcher, N.R.; Loggia, M.L.; Cernasov, P.; Reynolds, B.; Pijanowski, O.; Chonde, D.B.; Izquierdo Garcia, D.; Mainero, C.; Catana, C.; et al. Integrated Magnetic Resonance Imaging and [11C]-PBR28 Positron Emission Tomographic Imaging in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2018, 83, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Masson, G.L. Genetics of Amyotrophic Lateral Sclerosis: A Review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Gibson, S.B.; Downie, J.M.; Tsetsou, S.; Feusier, J.E.; Figueroa, K.P.; Bromberg, M.B.; Jorde, L.B.; Pulst, S.M. The Evolving Genetic Risk for Sporadic ALS. Neurology 2017, 89, 226–233. [Google Scholar] [CrossRef]
- Grassano, M.; Calvo, A.; Moglia, C.; Sbaiz, L.; Brunetti, M.; Barberis, M.; Casale, F.; Manera, U.; Vasta, R.; Canosa, A.; et al. Systematic Evaluation of Genetic Mutations in ALS: A Population-Based Study. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1190–1193. [Google Scholar] [CrossRef]
- Vajda, A.; McLaughlin, R.L.; Heverin, M.; Thorpe, O.; Abrahams, S.; Al-Chalabi, A.; Hardiman, O. Genetic Testing in ALS: A Survey of Current Practices. Neurology 2017, 88, 991–999. [Google Scholar] [CrossRef]
- Al Khleifat, A.; Iacoangeli, A.; van Vugt, J.J.F.A.; Bowles, H.; Moisse, M.; Zwamborn, R.A.J.; van der Spek, R.A.A.; Shatunov, A.; Cooper-Knock, J.; Topp, S.; et al. Structural Variation Analysis of 6,500 Whole Genome Sequences in Amyotrophic Lateral Sclerosis. Npj Genomic Med. 2022, 7, 1–8. [Google Scholar] [CrossRef]
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural Variant Calling: The Long and the Short of It. Genome Biol. 2019, 20, 246. [Google Scholar] [CrossRef]
- Arnold, F.J.; Merry, D.E. Molecular Mechanisms and Therapeutics for SBMA/Kennedy’s Disease. Neurother. J. Am. Soc. Exp. Neurother. 2019, 16, 928–947. [Google Scholar] [CrossRef]
- Hansel, A.; Dorst, J.; Rosenbohm, A.; Hübers, A.; Ludolph, A.C. ALS Mimics. Neurol. Int. Open 2018, 02, E60–E71. [Google Scholar] [CrossRef]
- Amado, D.A.; Davidson, B.L. Gene Therapy for ALS: A Review. Mol. Ther. 2021, 29, 3345–3358. [Google Scholar] [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef]
- Evans, J.P.; Skrzynia, C.; Burke, W. The Complexities of Predictive Genetic Testing. BMJ 2001, 322, 1052–1056. [Google Scholar] [CrossRef]
- Oliveri, S.; Ferrari, F.; Manfrinati, A.; Pravettoni, G. A Systematic Review of the Psychological Implications of Genetic Testing: A Comparative Analysis Among Cardiovascular, Neurodegenerative and Cancer Diseases. Front. Genet. 2018, 9, 624. [Google Scholar] [CrossRef]
- Sciorio, R.; Aiello, R.; Irollo, A.M. Review: Preimplantation Genetic Diagnosis (PGD) as a Reproductive Option in Patients with Neurodegenerative Disorders. Reprod. Biol. 2021, 21, 100468. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Bucelli, R.C.; Andrews, J.A.; Otto, M.; Farahany, N.A.; Harrington, E.A.; Chen, W.; Mitchell, A.A.; et al. Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: The ATLAS Study. Neurotherapeutics 2022, 19, 1248–1258. [Google Scholar] [CrossRef]
- Pecoraro, V.; Mandrioli, J.; Carone, C.; Chiò, A.; Traynor, B.J.; Trenti, T. The NGS Technology for the Identification of Genes Associated with the ALS. A Systematic Review. Eur. J. Clin. Invest. 2020, 50, e13228. [Google Scholar] [CrossRef]
- Mehta, P.R.; Iacoangeli, A.; Opie-Martin, S.; van Vugt, J.J.F.A.; Al Khleifat, A.; Bredin, A.; Ossher, L.; Andersen, P.M.; Hardiman, O.; Mehta, A.R.; et al. The Impact of Age on Genetic Testing Decisions in Amyotrophic Lateral Sclerosis. Brain 2022, 145, 4440–4447. [Google Scholar] [CrossRef]
- Roggenbuck, J.; Quick, A.; Kolb, S.J. Genetic Testing and Genetic Counseling for Amyotrophic Lateral Sclerosis: An Update for Clinicians. Genet. Med. 2017, 19, 267–274. [Google Scholar] [CrossRef]
- Van Rheenen, W.; van der Spek, R.A.A.; Bakker, M.K.; van Vugt, J.J.F.A.; Hop, P.J.; Zwamborn, R.A.J.; de Klein, N.; Westra, H.-J.; Bakker, O.B.; Deelen, P.; et al. Common and Rare Variant Association Analyses in Amyotrophic Lateral Sclerosis Identify 15 Risk Loci with Distinct Genetic Architectures and Neuron-Specific Biology. Nat. Genet. 2021, 53, 1636–1648. [Google Scholar] [CrossRef]
- Placek, K.; Benatar, M.; Wuu, J.; Rampersaud, E.; Hennessy, L.; Van Deerlin, V.M.; Grossman, M.; Irwin, D.J.; Elman, L.; McCluskey, L.; et al. Machine Learning Suggests Polygenic Risk for Cognitive Dysfunction in Amyotrophic Lateral Sclerosis. EMBO Mol. Med. 2021, 13, e12595. [Google Scholar] [CrossRef]
- Dehestani, M.; Liu, H.; Sreelatha, A.A.K.; Schulte, C.; Bansal, V.; Gasser, T. Mitochondrial and Autophagy-Lysosomal Pathway Polygenic Risk Scores Predict Parkinson’s Disease. Mol. Cell. Neurosci. 2022, 121, 103751. [Google Scholar] [CrossRef]
- Bacioglu, M.; Maia, L.F.; Preische, O.; Schelle, J.; Apel, A.; Kaeser, S.A.; Schweighauser, M.; Eninger, T.; Lambert, M.; Pilotto, A.; et al. Neurofilament Light Chain in Blood and CSF as Marker of Disease Progression in Mouse Models and in Neurodegenerative Diseases. Neuron 2016, 91, 494–496. [Google Scholar] [CrossRef]
- Gafson, A.R.; Barthélemy, N.R.; Bomont, P.; Carare, R.O.; Durham, H.D.; Julien, J.-P.; Kuhle, J.; Leppert, D.; Nixon, R.A.; Weller, R.O.; et al. Neurofilaments: Neurobiological Foundations for Biomarker Applications. Brain 2020, 143, 1975–1998. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as Biomarkers in Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Lombardi, V.; Malaspina, A. Neurofilament Light: A Candidate Biomarker of Presymptomatic Amyotrophic Lateral Sclerosis and Phenoconversion. Ann. Neurol. 2018, 84, 130–139. [Google Scholar] [CrossRef]
- Poesen, K.; De Schaepdryver, M.; Stubendorff, B.; Gille, B.; Muckova, P.; Wendler, S.; Prell, T.; Ringer, T.M.; Rhode, H.; Stevens, O.; et al. Neurofilament Markers for ALS Correlate with Extent of Upper and Lower Motor Neuron Disease. Neurology 2017, 88, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Vacchiano, V.; Mastrangelo, A.; Zenesini, C.; Masullo, M.; Quadalti, C.; Avoni, P.; Polischi, B.; Cherici, A.; Capellari, S.; Salvi, F.; et al. Plasma and CSF Neurofilament Light Chain in Amyotrophic Lateral Sclerosis: A Cross-Sectional and Longitudinal Study. Front. Aging Neurosci. 2021, 13, 753242. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhu, Y.; Hsiao-Nakamoto, J.; Tang, X.; Dugas, J.C.; Moscovitch-Lopatin, M.; Glass, J.D.; Brown, R.H.; Ladha, S.S.; Lacomis, D.; et al. Longitudinal Biomarkers in Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Oeckl, P.; Jardel, C.; Salachas, F.; Lamari, F.; Andersen, P.M.; Bowser, R.; de Carvalho, M.; Costa, J.; van Damme, P.; Gray, E.; et al. Multicenter Validation of CSF Neurofilaments as Diagnostic Biomarkers for ALS. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Qin, X.; Chang, X.; Wang, H.; Guo, J.; Zhang, W. Neurofilament Markers in Serum and Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis. J. Cell. Mol. Med. 2022, 26, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Feneberg, E.; Weishaupt, J.; Brettschneider, J.; Tumani, H.; Andersen, P.M.; von Arnim, C.A.F.; Böhm, S.; Kassubek, J.; Kubisch, C.; et al. Neurofilaments in the Diagnosis of Motoneuron Diseases: A Prospective Study on 455 Patients. J. Neurol. Neurosurg. Psychiatry 2016, 87, 12–20. [Google Scholar] [CrossRef]
- Behzadi, A.; Pujol-Calderón, F.; Tjust, A.E.; Wuolikainen, A.; Höglund, K.; Forsberg, K.; Portelius, E.; Blennow, K.; Zetterberg, H.; Andersen, P.M. Neurofilaments Can Differentiate ALS Subgroups and ALS from Common Diagnostic Mimics. Sci. Rep. 2021, 11, 22128. [Google Scholar] [CrossRef]
- Zecca, C.; Dell’Abate, M.T.; Pasculli, G.; Capozzo, R.; Barone, R.; Arima, S.; Pollice, A.; Brescia, V.; Tortelli, R.; Logroscino, G. Role of Plasma Phosphorylated Neurofilament Heavy Chain (PNfH) in Amyotrophic Lateral Sclerosis. J. Cell. Mol. Med. 2022, 26, 3608–3615. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Lee, A.; Murphy, A.; Chang, J.; Zipf, A.; Cudkowicz, M.; Atassi, N. Diagnostic Timelines and Delays in Diagnosing Amyotrophic Lateral Sclerosis (ALS). Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 453–456. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Lombardi, V.; Jeromin, A.; Bowser, R.; Andersen, P.M.; Malaspina, A. Neurofilaments in Pre-Symptomatic ALS and the Impact of Genotype. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 538–548. [Google Scholar] [CrossRef]
- De Schaepdryver, M.; Masrori, P.; Van Damme, P.; Poesen, K. Effect of Neurofilament Analysis on the Diagnostic Delay in Amyotrophic Lateral Sclerosis. CNS Neurosci. Ther. 2023, 29, 70–77. [Google Scholar] [CrossRef]
- Verde, F.; Steinacker, P.; Weishaupt, J.H.; Kassubek, J.; Oeckl, P.; Halbgebauer, S.; Tumani, H.; von Arnim, C.A.F.; Dorst, J.; Feneberg, E.; et al. Neurofilament Light Chain in Serum for the Diagnosis of Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-H.; Petzold, A.; Topping, J.; Allen, K.; Macdonald-Wallis, C.; Clarke, J.; Pearce, N.; Kuhle, J.; Giovannoni, G.; Fratta, P.; et al. Plasma Neurofilament Heavy Chain Levels and Disease Progression in Amyotrophic Lateral Sclerosis: Insights from a Longitudinal Study. J. Neurol. Neurosurg. Psychiatry 2015, 86, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Esselin, F.; De la Cruz, E.; Hirtz, C.; Tiers, L.; Alphandery, S.; Baudesson, L.; Taieb, G.; Camu, W.; Lehmann, S. Repeated Neurofilament Light Chain Measurements Did Not Capture Riluzole Therapeutic Effect in Amyotrophic Lateral Sclerosis Patients. CNS Neurosci. Ther. 2022, 28, 1532–1538. [Google Scholar] [CrossRef]
- Walo-Delgado, P.E.; Sainz de la Maza, S.; Villarrubia, N.; Monreal, E.; Medina, S.; Espiño, M.; Fernández-Velasco, J.I.; Rodríguez-Martín, E.; Roldán, E.; Lourido, D.; et al. Low Serum Neurofilament Light Chain Values Identify Optimal Responders to Dimethyl Fumarate in Multiple Sclerosis Treatment. Sci. Rep. 2021, 11, 9299. [Google Scholar] [CrossRef]
- Darras, B.T.; Crawford, T.O.; Finkel, R.S.; Mercuri, E.; De Vivo, D.C.; Oskoui, M.; Tizzano, E.F.; Ryan, M.M.; Muntoni, F.; Zhao, G.; et al. Neurofilament as a Potential Biomarker for Spinal Muscular Atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 932–944. [Google Scholar] [CrossRef]
- Beers, D.R.; Appel, S.H. Immune Dysregulation in Amyotrophic Lateral Sclerosis: Mechanisms and Emerging Therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune Reactivity in a Mouse Model of Familial ALS Correlates with Disease Progression. Neurology 2001, 57, 1282–1289. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.M.G.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 Is Required for Proper Macrophage and Microglial Function in Mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of Dendritic Cells, MCP-1, and Activated Microglia/Macrophages in Amyotrophic Lateral Sclerosis Spinal Cord Tissue. Ann. Neurol. 2004, 55, 221–235. [Google Scholar] [CrossRef]
- Sheean, R.K.; McKay, F.C.; Cretney, E.; Bye, C.R.; Perera, N.D.; Tomas, D.; Weston, R.A.; Scheller, K.J.; Djouma, E.; Menon, P.; et al. Association of Regulatory T-Cell Expansion With Progression of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2018, 75, 681–689. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous Regulatory T Lymphocytes Ameliorate Amyotrophic Lateral Sclerosis in Mice and Correlate with Disease Progression in Patients with Amyotrophic Lateral Sclerosis. Brain 2011, 134, 1293–1314. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.P.; Staff, N.P.; Bornschlegl, S.; Butler, G.W.; Maas, M.L.; Kazamel, M.; Zubair, A.; Gastineau, D.A.; Windebank, A.J.; Dietz, A.B. Comprehensive Immune Profiling Reveals Substantial Immune System Alterations in a Subset of Patients with Amyotrophic Lateral Sclerosis. PLoS ONE 2017, 12, e0182002. [Google Scholar] [CrossRef]
- Jin, M.; Günther, R.; Akgün, K.; Hermann, A.; Ziemssen, T. Peripheral Proinflammatory Th1/Th17 Immune Cell Shift Is Linked to Disease Severity in Amyotrophic Lateral Sclerosis. Sci. Rep. 2020, 10, 5941. [Google Scholar] [CrossRef] [PubMed]
- Prado, L.d.G.R.; Rocha, N.P.; de Souza, L.C.; Bicalho, I.C.S.; Gomez, R.S.; Vidigal-Lopes, M.; Braz, N.F.T.; Vieira, É.L.M.; Teixeira, A.L. Longitudinal Assessment of Clinical and Inflammatory Markers in Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2018, 394, 69–74. [Google Scholar] [CrossRef]
- Tortelli, R.; Zecca, C.; Piccininni, M.; Benmahamed, S.; Dell’Abate, M.T.; Barulli, M.R.; Capozzo, R.; Battista, P.; Logroscino, G. Plasma Inflammatory Cytokines Are Elevated in ALS. Front. Neurol. 2020, 11, 552295. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Cao, C.; Qin, X.-Y.; Yu, Y.; Yuan, J.; Zhao, Y.; Cheng, Y. Increased Peripheral Blood Inflammatory Cytokine Levels in Amyotrophic Lateral Sclerosis: A Meta-Analysis Study. Sci. Rep. 2017, 7, 9094. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Hooten, K.G.; Sieglaff, D.H.; Zhang, A.; Kalyana-Sundaram, S.; Traini, C.M.; Halsey, W.S.; Hughes, A.M.; Sathe, G.M.; et al. Characterization of Gene Expression Phenotype in Amyotrophic Lateral Sclerosis Monocytes. JAMA Neurol. 2017, 74, 677–685. [Google Scholar] [CrossRef]
- Italiani, P.; Carlesi, C.; Giungato, P.; Puxeddu, I.; Borroni, B.; Bossù, P.; Migliorini, P.; Siciliano, G.; Boraschi, D. Evaluating the Levels of Interleukin-1 Family Cytokines in Sporadic Amyotrophic Lateral Sclerosis. J. Neuroinflam. 2014, 11, 94. [Google Scholar] [CrossRef]
- Lunetta, C.; Lizio, A.; Gerardi, F.; Tarlarini, C.; Filippi, M.; Riva, N.; Tremolizzo, L.; Diamanti, S.; Dellanoce, C.C.; Mosca, L.; et al. Urinary Neopterin, a New Marker of the Neuroinflammatory Status in Amyotrophic Lateral Sclerosis. J. Neurol. 2020, 267, 3609–3616. [Google Scholar] [CrossRef]
- Shepheard, S.R.; Karnaros, V.; Benyamin, B.; Schultz, D.W.; Dubowsky, M.; Wuu, J.; Chataway, T.; Malaspina, A.; Benatar, M.; Rogers, M.-L. Urinary Neopterin: A Novel Biomarker of Disease Progression in Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2022, 29, 990–999. [Google Scholar] [CrossRef]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a Neuroprotective to a Neurotoxic Microglial Phenotype in a Mouse Model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Guttenplan, K.A.; Weigel, M.K.; Adler, D.I.; Couthouis, J.; Liddelow, S.A.; Gitler, A.D.; Barres, B.A. Knockout of Reactive Astrocyte Activating Factors Slows Disease Progression in an ALS Mouse Model. Nat. Commun. 2020, 11, 3753. [Google Scholar] [CrossRef]
- Jara, J.H.; Gautam, M.; Kocak, N.; Xie, E.F.; Mao, Q.; Bigio, E.H.; Özdinler, P.H. MCP1-CCR2 and Neuroinflammation in the ALS Motor Cortex with TDP-43 Pathology. J. Neuroinflam. 2019, 16, 196. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, H.; Qin, X.; Tian, M.; Zhang, H. PET Imaging of Reactive Astrocytes in Neurological Disorders. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Distefano, G.; Zorena, K.; Malaguarnera, L. Chitinases and Immunity: Ancestral Molecules with New Functions. Immunobiology 2016, 221, 399–411. [Google Scholar] [CrossRef]
- Steinacker, P.; Verde, F.; Fang, L.; Feneberg, E.; Oeckl, P.; Roeber, S.; Anderl-Straub, S.; Danek, A.; Diehl-Schmid, J.; Fassbender, K.; et al. Chitotriosidase (CHIT1) Is Increased in Microglia and Macrophages in Spinal Cord of Amyotrophic Lateral Sclerosis and Cerebrospinal Fluid Levels Correlate with Disease Severity and Progression. J. Neurol. Neurosurg. Psychiatry 2018, 89, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Bonneh-Barkay, D.; Wang, G.; Starkey, A.; Hamilton, R.L.; Wiley, C.A. In vivo CHI3L1 (YKL-40) Expression in Astrocytes in Acute and Chronic Neurological Diseases. J. Neuroinflam. 2010, 7, 34. [Google Scholar] [CrossRef]
- Pinteac, R.; Montalban, X.; Comabella, M. Chitinases and Chitinase-like Proteins as Biomarkers in Neurologic Disorders. Neurol. Neuroimmunol. Neuroinflam. 2021, 8, e921. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Bampton, A.; Raciborska, D.; Talbot, K.; Turner, M.R. CSF Chitinase Proteins in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1215–1220. [Google Scholar] [CrossRef]
- Gille, B.; De Schaepdryver, M.; Dedeene, L.; Goossens, J.; Claeys, K.G.; Van Den Bosch, L.; Tournoy, J.; Van Damme, P.; Poesen, K. Inflammatory Markers in Cerebrospinal Fluid: Independent Prognostic Biomarkers in Amyotrophic Lateral Sclerosis? J. Neurol. Neurosurg. Psychiatry 2019, 90, 1338–1346. [Google Scholar] [CrossRef]
- Gray, E.; Thompson, A.G.; Wuu, J.; Pelt, J.; Talbot, K.; Benatar, M.; Turner, M.R. CSF Chitinases before and after Symptom Onset in Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1296–1306. [Google Scholar] [CrossRef]
- Agnello, L.; Colletti, T.; Lo Sasso, B.; Vidali, M.; Spataro, R.; Gambino, C.M.; Giglio, R.V.; Piccoli, T.; Bivona, G.; La Bella, V.; et al. Tau Protein as a Diagnostic and Prognostic Biomarker in Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2021, 28, 1868–1875. [Google Scholar] [CrossRef]
- Rusina, R.; Ridzoň, P.; Kulišt’ák, P.; Keller, O.; Bartoš, A.; Buncová, M.; Fialová, L.; Koukolík, F.; Matěj, R. Relationship between ALS and the Degree of Cognitive Impairment, Markers of Neurodegeneration and Predictors for Poor Outcome. A Prospective Study: Risk Factors for Poor Outcome in ALS—A Prospective Study. Eur. J. Neurol. 2010, 17, 23–30. [Google Scholar] [CrossRef]
- Lanznaster, D.; Bejan-Angoulvant, T.; Patin, F.; Andres, C.R.; Vourc’h, P.; Corcia, P.; Blasco, H. Plasma Creatinine and Amyotrophic Lateral Sclerosis Prognosis: A Systematic Review and Meta-Analysis. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Illán-Gala, I.; Alcolea, D.; Montal, V.; Dols-Icardo, O.; Muñoz, L.; de Luna, N.; Turón-Sans, J.; Cortés-Vicente, E.; Sánchez-Saudinós, M.B.; Subirana, A.; et al. CSF SAPPβ, YKL-40, and NfL along the ALS-FTD Spectrum. Neurology 2018, 91, e1619–e1628. [Google Scholar] [CrossRef] [PubMed]
- Majumder, V.; Gregory, J.M.; Barria, M.A.; Green, A.; Pal, S. TDP-43 as a Potential Biomarker for Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. BMC Neurol. 2018, 18, 90. [Google Scholar] [CrossRef]
- Ren, Y.; Li, S.; Chen, S.; Sun, X.; Yang, F.; Wang, H.; Li, M.; Cui, F.; Huang, X. TDP-43 and Phosphorylated TDP-43 Levels in Paired Plasma and CSF Samples in Amyotrophic Lateral Sclerosis. Front. Neurol. 2021, 12, 663637. [Google Scholar] [CrossRef]
- Magen, I.; Yacovzada, N.S.; Yanowski, E.; Coenen-Stass, A.; Grosskreutz, J.; Lu, C.-H.; Greensmith, L.; Malaspina, A.; Fratta, P.; Hornstein, E. Circulating MiR-181 Is a Prognostic Biomarker for Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2021, 24, 1534–1541. [Google Scholar] [CrossRef]
- Ceccanti, M.; Pozzilli, V.; Cambieri, C.; Libonati, L.; Onesti, E.; Frasca, V.; Fiorini, I.; Petrucci, A.; Garibaldi, M.; Palma, E.; et al. Creatine Kinase and Progression Rate in Amyotrophic Lateral Sclerosis. Cells 2020, 9, 1174. [Google Scholar] [CrossRef]
- Castro-Gomez, S.; Radermacher, B.; Tacik, P.; Mirandola, S.R.; Heneka, M.T.; Weydt, P. Teaching an Old Dog New Tricks: Serum Troponin T as a Biomarker in Amyotrophic Lateral Sclerosis. Brain Commun. 2021, 3, fcab274. [Google Scholar] [CrossRef]
- Gong, Z.; Gao, L.; Guo, J.; Lu, Y.; Zang, D. BFGF in the CSF and Serum of SALS Patients. Acta Neurol. Scand. 2015, 132, 171–178. [Google Scholar] [CrossRef]
- Månberg, A.; Skene, N.; Sanders, F.; Trusohamn, M.; Remnestål, J.; Szczepińska, A.; Aksoylu, I.S.; Lönnerberg, P.; Ebarasi, L.; Wouters, S.; et al. Altered Perivascular Fibroblast Activity Precedes ALS Disease Onset. Nat. Med. 2021, 27, 640–646. [Google Scholar] [CrossRef]
- Gendron, T.F.; Chew, J.; Stankowski, J.N.; Hayes, L.R.; Zhang, Y.-J.; Prudencio, M.; Carlomagno, Y.; Daughrity, L.M.; Jansen-West, K.; Perkerson, E.A.; et al. Poly(GP) Proteins Are a Useful Pharmacodynamic Marker for C9ORF72-Associated Amyotrophic Lateral Sclerosis. Sci. Transl. Med. 2017, 9, eaai7866. [Google Scholar] [CrossRef]
- Krishnan, G.; Raitcheva, D.; Bartlett, D.; Prudencio, M.; McKenna-Yasek, D.M.; Douthwright, C.; Oskarsson, B.E.; Ladha, S.; King, O.D.; Barmada, S.J.; et al. Poly(GR) and Poly(GA) in Cerebrospinal Fluid as Potential Biomarkers for C9ORF72-ALS/FTD. Nat. Commun. 2022, 13, 2799. [Google Scholar] [CrossRef]
- Ma, X.R.; Prudencio, M.; Koike, Y.; Vatsavayai, S.C.; Kim, G.; Harbinski, F.; Briner, A.; Rodriguez, C.M.; Guo, C.; Akiyama, T.; et al. TDP-43 Represses Cryptic Exon Inclusion in the FTD-ALS Gene UNC13A. Nature 2022, 603, 124–130. [Google Scholar] [CrossRef]
- Holloway, R.G.; Gramling, R.; Kelly, A.G. Estimating and Communicating Prognosis in Advanced Neurologic Disease. Neurology 2013, 80, 764–772. [Google Scholar] [CrossRef]
- Westeneng, H.-J.; Debray, T.P.A.; Visser, A.E.; van Eijk, R.P.A.; Rooney, J.P.K.; Calvo, A.; Martin, S.; McDermott, C.J.; Thompson, A.G.; Pinto, S.; et al. Prognosis for Patients with Amyotrophic Lateral Sclerosis: Development and Validation of a Personalised Prediction Model. Lancet Neurol. 2018, 17, 423–433. [Google Scholar] [CrossRef]
- Kaufmann, P.; Levy, G.; Thompson, J.L.P.; Delbene, M.L.; Battista, V.; Gordon, P.H.; Rowland, L.P.; Levin, B.; Mitsumoto, H. The ALSFRSr Predicts Survival Time in an ALS Clinic Population. Neurology 2005, 64, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Su, X.W.; Simmons, Z.; Mitchell, R.M.; Kong, L.; Stephens, H.E.; Connor, J.R. Biomarker-Based Predictive Models for Prognosis in Amyotrophic Lateral Sclerosis. JAMA Neurol. 2013, 70, 1505–1511. [Google Scholar] [CrossRef]
- Dreger, M.; Steinbach, R.; Gaur, N.; Metzner, K.; Stubendorff, B.; Witte, O.W.; Grosskreutz, J. Cerebrospinal Fluid Neurofilament Light Chain (NfL) Predicts Disease Aggressiveness in Amyotrophic Lateral Sclerosis: An Application of the D50 Disease Progression Model. Front. Neurosci. 2021, 15, 651651. [Google Scholar] [CrossRef]
- Glasmacher, S.A.; Wong, C.; Pearson, I.E.; Pal, S. Survival and Prognostic Factors in C9orf72 Repeat Expansion Carriers: A Systematic Review and Meta-Analysis. JAMA Neurol. 2020, 77, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Kjældgaard, A.-L.; Pilely, K.; Olsen, K.S.; Jessen, A.H.; Lauritsen, A.Ø.; Pedersen, S.W.; Svenstrup, K.; Karlsborg, M.; Thagesen, H.; Blaabjerg, M.; et al. Prediction of Survival in Amyotrophic Lateral Sclerosis: A Nationwide, Danish Cohort Study. BMC Neurol. 2021, 21, 164. [Google Scholar] [CrossRef]
- Van Eenennaam, R.M.; Koppenol, L.S.; Kruithof, W.J.; Kruitwagen-van Reenen, E.T.; Pieters, S.; van Es, M.A.; van den Berg, L.H.; Visser-Meily, J.M.A.; Beelen, A. Discussing Personalized Prognosis Empowers Patients with Amyotrophic Lateral Sclerosis to Regain Control over Their Future: A Qualitative Study. Brain Sci. 2021, 11, 1597. [Google Scholar] [CrossRef]
- Faghri, F.; Brunn, F.; Dadu, A.; Chiò, A.; Calvo, A.; Moglia, C.; Canosa, A.; Manera, U.; Vasta, R.; Palumbo, F.; et al. Identifying and Predicting Amyotrophic Lateral Sclerosis Clinical Subgroups: A Population-Based Machine-Learning Study. Lancet Digit. Health 2022, 4, e359–e369. [Google Scholar] [CrossRef]
- Pancotti, C.; Birolo, G.; Rollo, C.; Sanavia, T.; Di Camillo, B.; Manera, U.; Chiò, A.; Fariselli, P. Deep Learning Methods to Predict Amyotrophic Lateral Sclerosis Disease Progression. Sci. Rep. 2022, 12, 13738. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling Sporadic ALS in IPSC-Derived Motor Neurons Identifies a Potential Therapeutic Agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Tamaki, Y.; Ross, J.P.; Alipour, P.; Castonguay, C.-É.; Li, B.; Catoire, H.; Rochefort, D.; Urushitani, M.; Takahashi, R.; Sonnen, J.A.; et al. Spinal Cord Extracts of Amyotrophic Lateral Sclerosis Spread TDP-43 Pathology in Cerebral Organoids. PLoS Genet. 2023, 19, e1010606. [Google Scholar] [CrossRef]
- Workman, M.J.; Lim, R.G.; Wu, J.; Frank, A.; Ornelas, L.; Panther, L.; Galvez, E.; Perez, D.; Meepe, I.; Lei, S.; et al. Large-Scale Differentiation of IPSC-Derived Motor Neurons from ALS and Control Subjects. Neuron 2023, 111, 1–14. [Google Scholar] [CrossRef]
- Liu, H.; Guan, L.; Deng, M.; Bolund, L.; Kristiansen, K.; Zhang, J.; Luo, Y.; Zhang, Z. Integrative Genetic and Single Cell RNA Sequencing Analysis Provides New Clues to the Amyotrophic Lateral Sclerosis Neurodegeneration. Front. Neurosci. 2023, 17. [Google Scholar] [CrossRef]
- Imamura, K.; Yada, Y.; Izumi, Y.; Morita, M.; Kawata, A.; Arisato, T.; Nagahashi, A.; Enami, T.; Tsukita, K.; Kawakami, H.; et al. Prediction Model of Amyotrophic Lateral Sclerosis by Deep Learning with Patient Induced Pluripotent Stem Cells. Ann. Neurol. 2021, 89, 1226–1233. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinically definite ALS | Clinical or electrophysiological * evidence of UMN and LMN involvement in bulbar region and ≥2 spinal regions |
| or | |
| Clinical or electrophysiological * evidence of UMN and LMN involvement in 3 spinal regions | |
| Clinically probable ALS | Clinical or electrophysiological * evidence of UMN and LMN involvement in ≥2 regions with UMN signs rostral to LMN signs |
| Clinically probable Laboratory-supported ALS ⧫ | Clinical evidence of UMN and LMN involvement in 1 region |
| or | |
| Clinical evidence of isolated UMN involvement in 1 region with electrophysiological evidence of LMN involvement in ≥2 regions | |
| Clinically possible ALS | Clinical or electrophysiological * evidence of UMN and LMN involvement in 1 region |
| or | |
| Evidence of isolated UMN involvement ≥2 regions | |
| or | |
| Evidence of LMN involvement rostral to UMN involvement |
| Progressive motor impairment |
| and |
| Clinical or electrophysiological UMN and LMN involvement in ≥1 region or only LMN involvement in ≥2 regions |
| and |
| Exclusion of other diseases |
| Neurofilament | Cut-Off [pg/mL] | Sensitivity (95% CI) [%] | Specificity (95% CI) [%] | NPV (95% CI) [%] | PPV (95% CI) [%] | ALS vs. | Study |
|---|---|---|---|---|---|---|---|
| NfL | >2200 | 77 (71–82) | 88 (79–94) | 56 (48–65) | 95 (91–98) | ALS mimics | [105] |
| >2200 | - | 85 (79–90) | 75 (69–80) | 87 (81–91) | Other controls | [105] | |
| pNfH | >560 | 83 (78–88) | 80 (70–88) | 62 (52–71) | 93 (88–95) | ALS mimics | [105] |
| >560 | - | 77 (71–83) | 79 (72–84) | 82 (77–86) | Other controls | [105] | |
| NfL | >3819 | 88.4 (78.8–94) | 84.7 (76.8–90.2) | - | - | Disease controls | [100] |
| >2453 | 85.4 | 78 | - | - | ALS mimics | [100] | |
| pNfH | >618 | 94.2 (86–97.7) | 74.8 (66–81.9) | - | - | Disease controls | [100] |
| >768 | 90.7 | 88 | 76 | - | ALS mimics | [100] | |
| NfL | 1431 | 79 (66.1–87.6) | 86.4 (75.7–93.6 | - | - | Controls | [103] |
| pNFH | 568.5 | 78.7 (67.7–87.3) | 93.3 (85.1–97.8) | - | - | Controls | [103] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidovic, M.; Müschen, L.H.; Brakemeier, S.; Machetanz, G.; Naumann, M.; Castro-Gomez, S. Current State and Future Directions in the Diagnosis of Amyotrophic Lateral Sclerosis. Cells 2023, 12, 736. https://doi.org/10.3390/cells12050736
Vidovic M, Müschen LH, Brakemeier S, Machetanz G, Naumann M, Castro-Gomez S. Current State and Future Directions in the Diagnosis of Amyotrophic Lateral Sclerosis. Cells. 2023; 12(5):736. https://doi.org/10.3390/cells12050736
Chicago/Turabian StyleVidovic, Maximilian, Lars Hendrik Müschen, Svenja Brakemeier, Gerrit Machetanz, Marcel Naumann, and Sergio Castro-Gomez. 2023. "Current State and Future Directions in the Diagnosis of Amyotrophic Lateral Sclerosis" Cells 12, no. 5: 736. https://doi.org/10.3390/cells12050736
APA StyleVidovic, M., Müschen, L. H., Brakemeier, S., Machetanz, G., Naumann, M., & Castro-Gomez, S. (2023). Current State and Future Directions in the Diagnosis of Amyotrophic Lateral Sclerosis. Cells, 12(5), 736. https://doi.org/10.3390/cells12050736