
Proteome Analysis of Thyroid Hormone Transporter Mct8/Oatp1c1-Deficient Mice Reveals Novel Dysregulated Target Molecules Involved in Locomotor Function
 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animals
2.2. Protein Extraction and Tryptic Digestion
2.3. High pH Fractionation
2.4. LC-MS/MS Acquisition
2.5. Data Processing
2.6. Transcriptomic Data
2.7. Western Blot
2.8. Immunohistochemistry
2.9. Fluorescence In Situ Hybridization (FISH)
2.10. Data Analysis, Statistics and Visualization
3. Results
3.1. Mass-Spectrometry-Based Proteomic Analysis
3.2. Comparison between Transcriptomic and Proteomic Data
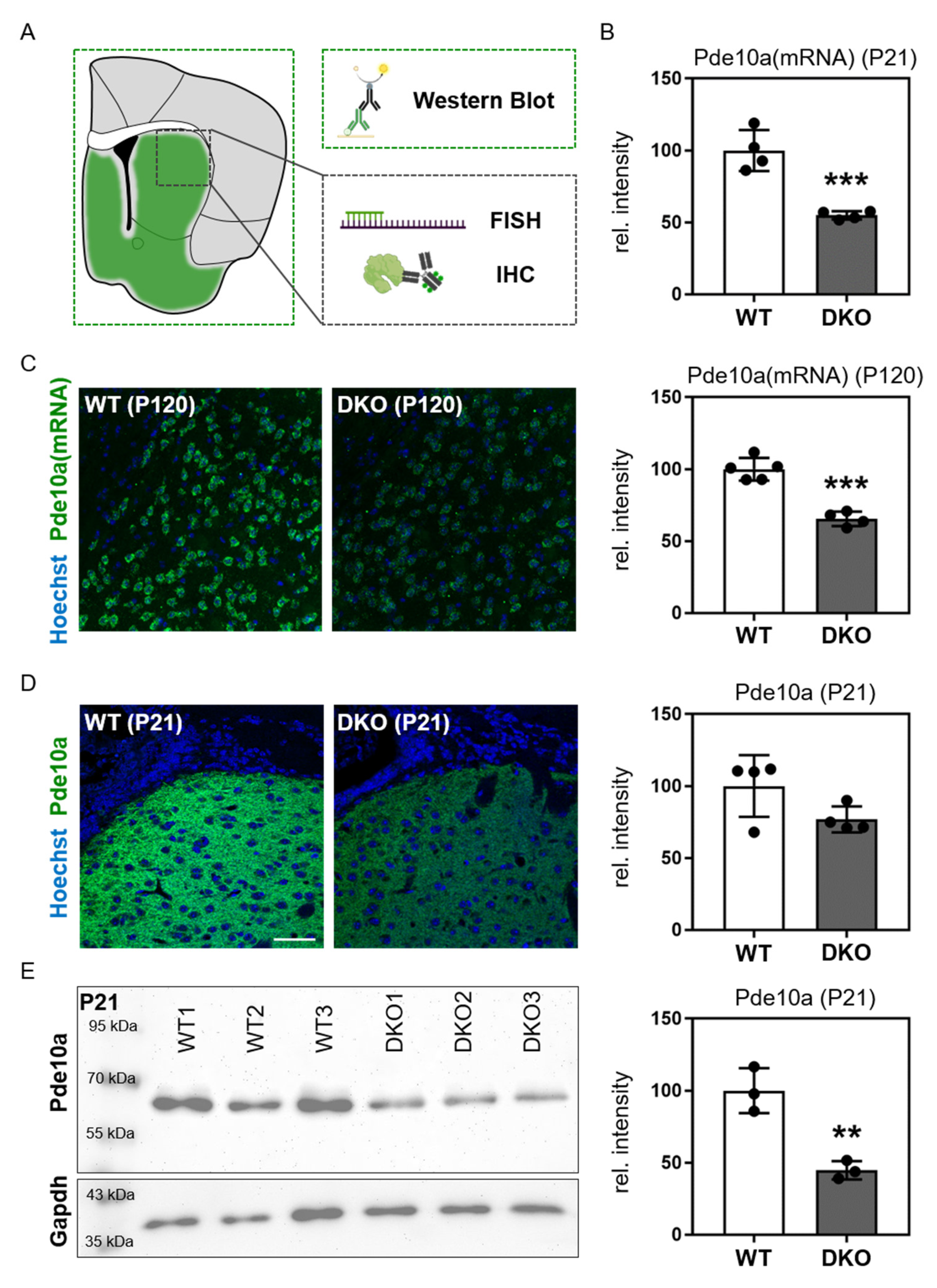
3.3. Candidate Molecule Analysis in the Striatum
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friesema, E.C.H.; Ganguly, S.; Abdalla, A.; Manning Fox, J.E.; Halestrap, A.P.; Visser, T.J. Identification of Monocarboxylate Transporter 8 as a Specific Thyroid Hormone Transporter. J. Biol. Chem. 2003, 278, 40128–40135. [Google Scholar] [CrossRef]
- Dumitrescu, A.M.; Liao, X.-H.; Best, T.B.; Brockmann, K.; Refetoff, S. A Novel Syndrome Combining Thyroid and Neurological Abnormalities Is Associated with Mutations in a Monocarboxylate Transporter Gene. Am. J. Hum. Genet. 2004, 74, 168–175. [Google Scholar] [CrossRef]
- Friesema, E.C.; Grueters, A.; Biebermann, H.; Krude, H.; von Moers, A.; Reeser, M.; Barrett, T.G.; E Mancilla, E.; Svensson, J.; Kester, M.H.; et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004, 364, 1435–1437. [Google Scholar] [CrossRef]
- Schwartz, C.E.; May, M.M.; Carpenter, N.J.; Rogers, R.C.; Martin, J.; Bialer, M.G.; Ward, J.; Sanabria, J.; Marsa, S.; Lewis, J.A.; et al. Allan-Herndon-Dudley Syndrome and the Monocarboxylate Transporter 8 (MCT8) Gene. Am. J. Hum. Genet. 2005, 77, 41–53. [Google Scholar] [CrossRef]
- Groeneweg, S.; van Geest, F.S.; Peeters, R.P.; Heuer, H.; Visser, W.E. Thyroid Hormone Transporters. Endocr. Rev. 2019, 41, 146–201. [Google Scholar] [CrossRef]
- Mayerl, S.; Müller, J.; Bauer, R.; Richert, S.; Kassmann, C.M.; Darras, V.M.; Buder, K.; Boelen, A.; Visser, T.J.; Heuer, H. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J. Clin. Investig. 2014, 124, 1987–1999. [Google Scholar] [CrossRef]
- Mayerl, S.; Chen, J.; Salveridou, E.; Boelen, A.; Darras, V.M.; Heuer, H. Thyroid Hormone Transporter Deficiency in Mice Impacts Multiple Stages of GABAergic Interneuron Development. Cereb. Cortex 2021, 32, 329–341. [Google Scholar] [CrossRef]
- Reinwald, J.R.; Weber-Fahr, W.; Cosa-Linan, A.; Becker, R.; Sack, M.; Falfan-Melgoza, C.; Gass, N.; Braun, U.; von Hohenberg, C.C.; Chen, J.; et al. TRIAC Treatment Improves Impaired Brain Network Function and White Matter Loss in Thyroid Hormone Transporter Mct8/Oatp1c1 Deficient Mice. Int. J. Mol. Sci. 2022, 23, 15547. [Google Scholar] [CrossRef]
- Mayerl, S.; Heuer, H. Thyroid hormone transporter Mct8/Oatp1c1 deficiency compromises proper oligodendrocyte maturation in the mouse CNS. Neurobiol. Dis. 2023, 184, 106195. [Google Scholar] [CrossRef]
- Espíndola, D.L.; Morales-Bastos, C.; Grijota-Martinez, C.; Liao, X.-H.; Lev, D.; Sugo, E.; Verge, C.F.; Refetoff, S.; Bernal, J.; Guadaño-Ferraz, A. Mutations of the Thyroid Hormone Transporter MCT8 Cause Prenatal Brain Damage and Persistent Hypomyelination. J. Clin. Endocrinol. Metab. 2014, 99, E2799–E2804. [Google Scholar] [CrossRef]
- Remerand, G.; Boespflug-Tanguy, O.; Tonduti, D.; Touraine, R.; Rodriguez, D.; Curie, A.; Perreton, N.; Portes, V.D.; Sarret, C.; RMLX/AHDS Study Group. Expanding the phenotypic spectrum of Allan–Herndon–Dudley syndrome in patients with SLC16A2 mutations. Dev. Med. Child Neurol. 2019, 61, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Valcárcel-Hernández, V.; López-Espíndola, D.; Guillén-Yunta, M.; García-Aldea, Á.; de Toledo Soler, I.L.; Bárez-López, S.; Guadaño-Ferraz, A. Deficient thyroid hormone transport to the brain leads to impairments in axonal caliber and oligodendroglial development. Neurobiol. Dis. 2021, 162, 105567. [Google Scholar] [CrossRef]
- Bernal, J. Thyroid Hormones and Brain Development. Vitam. Horm. 2005, 71, 95–122. [Google Scholar] [CrossRef]
- Bernal, J. Thyroid hormone receptors in brain development and function. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 249–259. [Google Scholar] [CrossRef]
- Wallis, K.; Sjögren, M.; van Hogerlinden, M.; Silberberg, G.; Fisahn, A.; Nordström, K.; Larsson, L.; Westerblad, H.; de Escobar, G.M.; Shupliakov, O.; et al. Locomotor Deficiencies and Aberrant Development of Subtype-Specific GABAergic Interneurons Caused by an Unliganded Thyroid Hormone Receptor α1. J. Neurosci. 2008, 28, 1904–1915. [Google Scholar] [CrossRef]
- Morte, B.; Gil-Ibañez, P.; Heuer, H.; Bernal, J. Brain Gene Expression in Systemic Hypothyroidism and Mouse Models of MCT8 Deficiency: The Mct8-Oatp1c1-Dio2 Triad. Thyroid® 2021, 31, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, Q.; Zhou, X.; Qin, Z.; Yi, S.; Luo, J. Characteristics of Allan-Herndon-Dudley Syndrome in Chinese children: Identification of two novel pathogenic variants of the SLC16A2 gene. Front. Pediatr. 2022, 10, 1050023. [Google Scholar] [CrossRef]
- Heuer, H.; Schäfer, M.K.; O’Donnell, D.; Walker, P.; Bauer, K. Expression of thyrotropin-releasing hormone receptor 2 (TRH-R2) in the central nervous system of rats. J. Comp. Neurol. 2000, 428, 319–336. [Google Scholar] [CrossRef]
- Choi, H.M.T.; Schwarzkopf, M.; Fornace, M.E.; Acharya, A.; Artavanis, G.; Stegmaier, J.; Cunha, A.; Pierce, N.A. Third-generation in situ hybridization chain reaction: Multiplexed, quantitative, sensitive, versatile, robust. Development 2018, 145, dev165753. [Google Scholar] [CrossRef]
- van Geest, F.S.; Gunhanlar, N.; Groeneweg, S.; Visser, W.E. Monocarboxylate Transporter 8 Deficiency: From Pathophysiological Understanding to Therapy Development. Front. Endocrinol. 2021, 12, 723750. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.A.; Mayerl, S. Local Thyroid Hormone Action in Brain Development. Int. J. Mol. Sci. 2023, 24, 12352. [Google Scholar] [CrossRef]
- Diggle, C.P.; Rizzo, S.J.S.; Popiolek, M.; Hinttala, R.; Schülke, J.-P.; Kurian, M.A.; Carr, I.M.; Markham, A.F.; Bonthron, D.T.; Watson, C.; et al. Biallelic Mutations in PDE10A Lead to Loss of Striatal PDE10A and a Hyperkinetic Movement Disorder with Onset in Infancy. Am. J. Hum. Genet. 2016, 98, 735–743. [Google Scholar] [CrossRef]
- Masnada, S.; Sarret, C.; Antonello, C.E.; Fadilah, A.; Krude, H.; Mura, E.; Mordekar, S.; Nicita, F.; Olivotto, S.; Orcesi, S.; et al. Movement disorders in MCT8 deficiency/Allan-Herndon-Dudley Syndrome. Mol. Genet. Metab. 2021, 135, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Vancamp, P.; Demeneix, B.A.; Remaud, S. Monocarboxylate Transporter 8 Deficiency: Delayed or Permanent Hypomyelination? Front. Endocrinol. 2020, 11, 283. [Google Scholar] [CrossRef] [PubMed]
- Bárez-López, S.; Grijota-Martínez, C.; Ausó, E.; Frutos, M.F.-D.; Montero-Pedrazuela, A.; Guadaño-Ferraz, A. Adult Mice Lacking Mct8 and Dio2 Proteins Present Alterations in Peripheral Thyroid Hormone Levels and Severe Brain and Motor Skill Impairments. Thyroid® 2019, 29, 1669–1682. [Google Scholar] [CrossRef]
- Zekri, Y.; Guyot, R.; Flamant, F. An Atlas of Thyroid Hormone Receptors’ Target Genes in Mouse Tissues. Int. J. Mol. Sci. 2022, 23, 11444. [Google Scholar] [CrossRef]
- Frei, J.A.; Andermatt, I.; Gesemann, M.; Stoeckli, E.T. The synaptic cell adhesion molecules SynCAMs are involved in sensory axon pathfinding by regulating axon-axon contacts. J. Cell Sci. 2014, 127, 5288–5302. [Google Scholar] [CrossRef]
- Airola, M.V.; Hannun, Y.A. Sphingolipid Metabolism and Neutral Sphingomyelinases. In Sphingolipids: Basic Science and Drug Development; Handbook of Experimental Pharmacology; Springer: Vienna, Austria, 2013; pp. 57–76. [Google Scholar] [CrossRef]
- Diez, D.; Grijota-Martinez, C.; Agretti, P.; De Marco, G.; Tonacchera, M.; Pinchera, A.; de Escobar, G.M.; Bernal, J.; Morte, B. Thyroid Hormone Action in the Adult Brain: Gene Expression Profiling of the Effects of Single and Multiple Doses of Triiodo-l-Thyronine in the Rat Striatum. Endocrinology 2008, 149, 3989–4000. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and Physiology of Cyclic Nucleotide Phosphodiesterases: Essential Components in Cyclic Nucleotide Signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- Haber, S.N.; Fudge, J.L.; McFarland, N.R. Striatonigrostriatal Pathways in Primates Form an Ascending Spiral from the Shell to the Dorsolateral Striatum. J. Neurosci. 2000, 20, 2369–2382. [Google Scholar] [CrossRef]
- Haber, S.N. Corticostriatal circuitry. Dialog-Clin. Neurosci. 2016, 18, 7–21. [Google Scholar] [CrossRef]
- Menniti, F.S.; Chappie, T.A.; Schmidt, C.J. PDE10A Inhibitors—Clinical Failure or Window into Antipsychotic Drug Action? Front. Neurosci. 2021, 14, 600178. [Google Scholar] [CrossRef]
- Mencacci, N.E.; Kamsteeg, E.-J.; Nakashima, K.; R’bibo, L.; Lynch, D.S.; Balint, B.; Willemsen, M.A.; Adams, M.E.; Wiethoff, S.; Suzuki, K.; et al. De Novo Mutations in PDE10A Cause Childhood-Onset Chorea with Bilateral Striatal Lesions. Am. J. Hum. Genet. 2016, 98, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Trieschmann, G.; Wach, K.; Berweck, S.; Zech, M.; Abel, M.; Tilgner, E. A Novel Homozygous PDE 10A Variant Leading to Infantile-Onset Hyperkinesia. Neuropediatrics 2022, 53, 386–387. [Google Scholar] [CrossRef] [PubMed]
- Siuciak, J.A.; McCarthy, S.A.; Chapin, D.S.; Martin, A.N.; Harms, J.F.; Schmidt, C.J. Behavioral characterization of mice deficient in the phosphodiesterase-10A (PDE10A) enzyme on a C57/Bl6N congenic background. Neuropharmacology 2008, 54, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, Y.; Montero-Pedrazuela, A.; Prensa, L.; Guadaño-Ferraz, A.; Rausell, E. Thyroid Hormone Transporters MCT8 and OATP1C1 Are Expressed in Projection Neurons and Interneurons of Basal Ganglia and Motor Thalamus in the Adult Human and Macaque Brains. Int. J. Mol. Sci. 2023, 24, 9643. [Google Scholar] [CrossRef]
- Kakinuma, H.; Itoh, M.; Takahashi, H. A Novel Mutation in the Monocarboxylate Transporter 8 Gene in a Boy with Putamen Lesions and Low Free T4 Levels in Cerebrospinal Fluid. J. Pediatr. 2005, 147, 552–554. [Google Scholar] [CrossRef]
- Tonduti, D.; Vanderver, A.; Berardinelli, A.; Schmidt, J.L.; Collins, C.D.; Novara, F.; Di Genni, A.; Mita, A.; Triulzi, F.; Brunstrom-Hernandez, J.E.; et al. MCT8 Deficiency. J. Child Neurol. 2012, 28, 795–800. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siemes, D.; Vancamp, P.; Markova, B.; Spangenberg, P.; Shevchuk, O.; Siebels, B.; Schlüter, H.; Mayerl, S.; Heuer, H.; Engel, D.R. Proteome Analysis of Thyroid Hormone Transporter Mct8/Oatp1c1-Deficient Mice Reveals Novel Dysregulated Target Molecules Involved in Locomotor Function. Cells 2023, 12, 2487. https://doi.org/10.3390/cells12202487
Siemes D, Vancamp P, Markova B, Spangenberg P, Shevchuk O, Siebels B, Schlüter H, Mayerl S, Heuer H, Engel DR. Proteome Analysis of Thyroid Hormone Transporter Mct8/Oatp1c1-Deficient Mice Reveals Novel Dysregulated Target Molecules Involved in Locomotor Function. Cells. 2023; 12(20):2487. https://doi.org/10.3390/cells12202487
Chicago/Turabian StyleSiemes, Devon, Pieter Vancamp, Boyka Markova, Philippa Spangenberg, Olga Shevchuk, Bente Siebels, Hartmut Schlüter, Steffen Mayerl, Heike Heuer, and Daniel Robert Engel. 2023. "Proteome Analysis of Thyroid Hormone Transporter Mct8/Oatp1c1-Deficient Mice Reveals Novel Dysregulated Target Molecules Involved in Locomotor Function" Cells 12, no. 20: 2487. https://doi.org/10.3390/cells12202487
APA StyleSiemes, D., Vancamp, P., Markova, B., Spangenberg, P., Shevchuk, O., Siebels, B., Schlüter, H., Mayerl, S., Heuer, H., & Engel, D. R. (2023). Proteome Analysis of Thyroid Hormone Transporter Mct8/Oatp1c1-Deficient Mice Reveals Novel Dysregulated Target Molecules Involved in Locomotor Function. Cells, 12(20), 2487. https://doi.org/10.3390/cells12202487